

Abstract
Background
Despite recent evidence demonstrating a potent protective effect of adoptively transferred regulatory T cells (Tregs) in ischemic stroke, the mechanism for Treg mobilization and activation in the ischemic brain is, remarkably, unknown. This study determines the role of C‐C chemokine receptor type 5 (CCR5) in mediating the docking and activation of transferred Tregs in their protection of early blood‐brain barrier disruption after stroke.
Methods and Results
Adoptive transfer of CCR5−/− Tregs failed to reduce brain infarct or neurological deficits, indicating an indispensable role of CCR5 in Treg‐afforded protection against cerebral ischemia. Two‐photon live imaging demonstrated that CCR5 was critical for Treg docking at the injured vessel wall, where they interact with blood‐borne neutrophils/macrophages after cerebral ischemic injury. CCR5 deficiency on donor Tregs deprived of their early protection against blood‐brain barrier damage. Using flow cytometry, real‐time polymerase chain reaction, and immunostaining, we confirmed that the expression of CCL5, a CCR5 ligand, was significantly elevated on the injured endothelium after cerebral ischemia, accompanied by CCR5 upregulation on circulating Tregs. In a Treg‐endothelial cell coculture, CCR5 expression was induced on Tregs on their exposure to ischemia‐injured endothelial cells. Furthermore, CCR5 induction on Tregs enhanced expression of the inhibitory molecule programmed death ligand 1, which in turn inhibited neutrophil‐derived matrix metallopeptidase 9.
Conclusions
These results suggest that CCR5 is a critical molecule for Treg‐mediated blood‐brain barrier protection and a potential target to optimize Treg therapy for stroke.
Keywords: blood‐brain barrier, brain ischemia, stroke
Subject Categories: Basic Science Research, Blood-Brain Barrier, Ischemic Stroke
Clinical Perspective
What Is New?
C‐C chemokine receptor type 5 signaling in adoptively transferred regulatory T cells is indispensable for their protection of the blood‐brain barrier on ischemic injury.
Activation of C‐C chemokine receptor type 5 enhances interactions between regulatory T cells and endothelial cells and increases the immune suppressive function of regulatory T cells via upregulating programmed death ligand 1 expression.
What Are the Clinical Implications?
Strategies that enhance C‐C chemokine receptor type 5 signaling may potentiate the therapeutic effect of adoptively transferred regulatory T cells in stroke victims.
Introduction
Both the innate and adaptive immune systems are rapidly activated in response to cerebral ischemia and reperfusion injury,1, 2 which leads to infiltration of various immune cells into the brain parenchyma.3, 4 Accumulating evidence suggests the importance of these immune responses in the pathogenesis of ischemic brain damage.5 Accordingly, immune modulation (immunotherapy) has become a promising concept for stroke treatment.6, 7, 8
Recent studies have revealed that mobilization of CD4+CD25+ regulatory T cells (Tregs), a specialized population of T cells, is an endogenous mechanism of immune modulation and attenuates poststroke inflammation by counterbalancing activation of immune effector cells.9, 10, 11, 12 Depletion of Tregs profoundly augmented the activation of resident and invading inflammatory cells and increased ischemic brain damage.10 Our previous studies have demonstrated that adoptive transfer of Tregs provided acute protection to the ischemic brain and mitigated cerebral inflammation.11, 13 The early anti‐inflammatory effect of adoptively transferred Tregs did not require passage across the blood‐brain barrier (BBB). Rather, these cells inhibited matrix metallopeptidase 9 (MMP‐9) production by peripheral neutrophils, thus preventing proteolytic damage to the BBB.11 The immunosuppressive function of Tregs usually requires local cell proximity or even direct cell‐cell interactions.14, 15, 16 We have found that the crosstalk between Tregs and neutrophils was cell‐cell contact dependent and that inhibitory signaling through programmed death ligand‐1 (PD‐L1) was essential for the protective effect of Tregs.13 However, it is unclear where and how Tregs gain proximity to circulating neutrophils and inhibit neutrophil activation after stroke.
The migration of immune cells toward sites of inflammation is activated through their expression of various chemokine receptors that recognize chemokines released by the inflamed or injured tissue. C‐C chemokine receptor type 5 (CCR5) is a chemokine receptor that is highly expressed on T cells. It mediates T‐cell trafficking out of secondary lymphoid organs toward sites of inflammation.17, 18, 19 CCR5+ T cells have been shown to play important roles in atherosclerosis,20 rheumatoid arthritis,21 and autoimmune encephalomyelitis.22 Interestingly, CCR5 is expressed preferentially on Tregs compared with other CD4+ T cells under certain pathological conditions and exerts multifaceted functions in addition to chemotaxis.23 In particular, Treg CCR5 expression has been associated with their homing to the infarcted heart and their suppressive effects on local inflammation.24 CCR5 knockout in Tregs results in an increased magnitude of adaptive immune reactivity in fungal and parasitic infection.23, 25 The function of CCR5 in Treg responses toward cerebral ischemia is unknown.
In the current study we demonstrate that CCR5 activation is critical for the docking of adoptively transferred Tregs at the ischemia‐injured endothelium where Tregs interact with blood‐borne neutrophils/macrophages. Moreover, the engagement of Treg CCR5 with its ligand CCL5 upregulates the expression of PD‐L1, enhancing the inhibitory effect of Tregs on neutrophil‐derived MMP‐9. Therefore, CCR5 is a critical molecule for Treg‐mediated BBB protection and may represent a novel target to optimize Treg‐based cell therapy for ischemic stroke.
Materials and Methods
Mice
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male 8‐ to 12‐week old C57BL/6J mice, and B6.129P2‐Ccr5tm1Kuz/J mice (C57/BL6 background) were purchased from The Jackson Laboratory (Bar Harbor, ME).
Cerebral Ischemia Model
Mice were anesthetized with 3% isoflurane in 67%:30% N2O/O2 (induction), until they were unresponsive to the tail pinch test and were then fitted with a nose cone providing 1.5% isoflurane for anesthesia maintenance. A monofilament was introduced into the common carotid artery, advanced to the origin of the middle cerebral artery, and left in position for 60 minutes until reperfusion. Cerebral blood flow was measured using laser Doppler flowmeter to confirm the vascular occlusion and reperfusion. The rectal temperature was controlled at 37.0±0.5°C during surgery and middle cerebral artery occlusion (MCAO) via a temperature‐regulated heating pad. Immediately after surgery, animals were randomly assigned to splenocyte or Tregs treatment groups. Investigators were blinded to treatment groups during cell transplantation or vehicle injection and during all outcome assessments. Sham‐operated animals underwent the same anesthesia and surgical procedures except MCAO.
Two‐Dimensional Laser Speckle Imaging Techniques
In selected experiments cerebral blood flow was monitored using the laser speckle imaging technique (PeriCam PSI System; Perimed, Järfälla, Sweden) before MCAO, during ischemia, and after reperfusion. Cerebral blood flow changes were expressed as percentage of pre‐MCAO baseline. Animals that did not show a cerebral blood flow reduction of at least 75% were excluded (fewer than 10% of stroke animals) from further experimentation.
Isolation, Labeling, and Adoptive Transfer of Tregs
Single‐cell suspensions were prepared from inguinal and axillary lymph nodes and spleens. CD4+CD25+ Treg populations were enriched by negative selection and positive selection with regulatory T‐cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Recipient mice received a tail vein injection of 2×106 freshly enriched Tregs or freshly isolated splenocytes in 0.2 mL Dulbecco phosphate‐buffered saline (PBS) at 2 hours after reperfusion. For Treg labeling and tracking, Tregs were incubated with 20 μmol/L CellTracker™ Deep Red Dye (Invitrogen, Carlsbad, CA) at 37°C for 30 minutes before intravenous injection.
Measurement of Infarct Volume
Brains were removed and sliced into 7 coronal sections, each 1 mm thick. Sections were immersed in prewarmed 2% 2,3,5‐triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO) in saline for 10 minutes and then fixed in 4% paraformaldehyde. Animals that developed massive hemorrhage were excluded from further evaluation (about 2% of stroke animals). Infarct volume was determined with National Institutes of Health ImageJ analysis by an observer blinded to experimental group assignment.
Assessments of Neurological Deficit
Neurological deficit was assessed in a blinded fashion using a 5‐point neurological score in mice: 0, no observable deficit; 1, torso flexion to right; 2, spontaneous circling to right; 3, leaning/falling to right; 4, no spontaneous movement; 5, death.
Real‐Time Polymerase Chain Reaction
Total RNA was isolated from ischemic brains 24 hours after stroke using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Five micrograms was used to synthesize the first strand of cDNA using the Superscript First‐Strand Synthesis System for Real‐Time Polymerase Chain Reaction (RT‐PCR) (Invitrogen, Carlsbad, CA). PCR was performed on the Opticon 2 Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA) using corresponding primers (Table S1) and SYBR green PCR Master Mix (Invitrogen). The cycle time values were normalized with glyceraldehyde 3‐phosphate dehydrogenase of the same sample. The expression levels of mRNAs were then calculated and expressed as fold changes versus sham control.
Two‐Photon In Vivo Imaging
For brain 2‐photon imaging, a 5.0‐mm craniotomy was performed 1.0 mm posterior and 2.5 mm lateral from the bregma.26 The dura mater was left intact. A coverglass at 5.0 mm in diameter was implanted and fixed in position with a combination of silicone (World Precision Instruments, Sarasota, FL) and Cerebond adhesive (MyNeurolab, Richmond, IL) before MCAO surgery. At 24 hours after surgery, the mice were anesthetized, and a catheter prefilled with 1% heparin was placed in the left femoral vein. The mouse was placed in a stereotaxic‐frame for imaging. Blood vessels were labeled by infusion of 200 to 300 μL of Alexa‐Fluora‐488‐conjugated dextran (Invitrogen; 70 kDa MW, 1% solution in PBS). After the baseline image had been taken, 100 μL rhodamine‐6G (Sigma; 0.1 mg/kg diluted in 1% PBS) was injected to label the neutrophils and macrophages,27 and then the cell tracker (CellTracker™ Deep Red Dye, Invitrogen, incubated at 20 μmol/L at 37°C for 30 minutes)‐labeled Tregs (Figure S1) was injected. For simultaneous fluorescence excitation of Alexa‐Fluora‐488, Rhodamine‐6G, and CellTracker™ Deep Red Dye, a 790‐nm wavelength was used. Imaging data were captured using an Olympus multiphoton microscope (FLUOVIEW FV1000, System Version 3.1.1.9, Tokyo, Japan) equipped with a Spectra‐Physics DeepSee Mai Tai Ti‐Sapphire laser (Newport Inc, Mountain View, CA) and a 1.05 NA 25×MPE water immersion objective lens. Images were acquired of the top 100 μm of the cortex including the pial vasculature with a resolution of 508×508 μm (1024×1024 pixels/image). The dwell time was set to 2 μs/pixel. After acquisition, images were processed and analyzed with Fuji and Imaris (Bitplane, Belfast, UK). Mean track velocities (μm/min) were calculated for individual tracks spanning 200 seconds. All track calculations were performed using a Fuji manual tracking plug‐in.
Image Processing and Analysis
Cerebral T‐cell, macrophage, and neutrophil infiltration was quantified in a blinded manner in both the cortex and striatum at 2 coronal levels (0.2 and −0.5 mm relative to bregma). All images were processed with Image J for cell‐based counting of automatically recognized cells. The mean was calculated from 3 fields in the cortex or striatum of each section as mean number of cells per square millimeter. The length of CD31 and CCL5 immunopositive blood vessels was measured using ImageJ in both the cortex and striatum at the same coronal levels described above and adjusted to the total length per square millimeter.
The 2‐photon images were analyzed using Fuji plug‐in. Briefly, image sequences were imported to Fuji, and the manual cell track was initiated. Cells of interest were clicked from the first time frame to the last one, and a cell track route image was constructed. Cell velocity was calculated in micrometers per minute.28 The cell‐cell contact time was determined by manually examining each Treg‐endothelium or Treg‐neutrophil/macrophage interaction; no contact was recorded if black pixels were visualized between the cells.29 Cell displacement was calculated using Microsoft Excel as previously described.30
In Vitro BBB Permeability Assay
Mouse endothelial cells were grown to confluence in the transwell system and subjected to oxygen glucose deprivation (OGD) for 4 hours. Neutrophils and Tregs isolated from donor mice were treated with TNF‐α (100 ng/mL) or CCR5 inhibitor DAPTA (D‐ala‐peptide T‐amide; 100 ng/mL) at 37°C for 2 hours and washed with PBS before being added to the upper chamber (1:1, 105/well). Fluorescence‐labeled dextran (4.4 kDa Alexa‐Fluorer 488 Dextran, Invitrogen) was added into the upper chamber. Culture medium in the lower chamber was collected and measured once an hour after reoxygenation until 6 hours after OGD. The amount of dextran leakage represented the BBB permeability.
Flow Cytometry
For in vivo study, single‐cell suspensions were obtained from blood, spleen, and lymph nodes. Red blood cells were removed from blood and splenocyte suspensions using erythrocyte lysis buffer (Ammonium Chloride Potassium, Invitrogen). Cells were stained and analyzed by flow cytometry for CCR5 expression on CD4+CD25+Foxp3+ Tregs. For in vitro study, cells were collected from a neutrophil‐Treg‐endothelial cell coculture system. MMP‐9 and PD‐L1 were stained and analyzed in neutrophils (Gr‐1+) and Tregs (CD25+), respectively.
Statistical Analysis
Results are presented as mean±SEM. The difference in means between 2 groups was assessed by 2‐tailed Student t test. Differences in means among multiple groups were analyzed using 1‐ or 2‐way ANOVA with time or treatment as independent factors. When ANOVA showed significant differences, pairwise comparisons between means were tested by post hoc Bonferroni tests. In all analyses, P≤0.05 was considered statistically significant.
Results
CCR5 Expression Is Essential for a Neuroprotective Effect of Tregs Against Transient Cerebral Ischemia
Recent studies have reported the protective effect of adoptively transferred Tregs after cerebral ischemia.11, 31 However, it remains unclear where and how these transferred Tregs exert their protection. CCR5, a chemokine receptor that guides T‐cell trafficking, has been shown to preferentially express on Tregs compared with other CD4+ T cells under certain pathological conditions.23 We therefore examined the role of CCR5 in the neuroprotective effect of Tregs against ischemic stroke. Tregs were isolated from CCR5−/− mice or wild type (WT) mice and transferred (2×106/mouse) intravenously into recipient mice within 60 minutes of transient MCAO (tMCAO) followed by 2 hours of reperfusion. Cerebral blood flow was monitored and recorded by speckle laser Doppler. There was no significant difference in cortical blood flow values during MCAO and after reperfusion between mice given WT Tregs or CCR5−/− Tregs (Figure 1A and 1B). Infarct volume determined 3 days after MCAO showed that the brain damage was alleviated by the infusion of WT Tregs but not by CCR5−/− Tregs (Figure 1C and 1D). Brain edema was calculated using the Swanson method32 and revealed that, unlike WT Tregs, CCR5−/− Tregs failed to reduce brain edema (Figure 1E). Consistently, neurological deficits were reduced in WT Treg‐treated stroke mice but not in the CCR5−/− Treg‐treated group (Figure 1F). These data indicate that CCR5 is indispensable for Treg‐afforded neuroprotection against cerebral ischemic injury.
Figure 1.
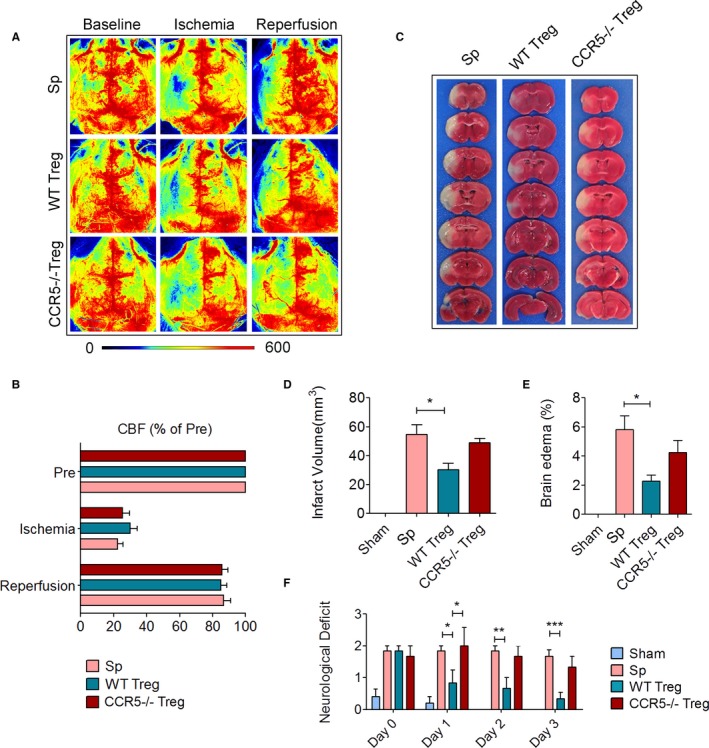
CCR5 expression is essential for the neuroprotective effect of Tregs against transient cerebral ischemia. Cerebral ischemia was induced by transient middle cerebral artery occlusion (tMCAO) for 60 minutes. Mice received 2×106 Tregs isolated from wild type (WT) mice or CCR5−/− donor mice. Control mice received the same number of splenocytes (Sp). Animals were euthanized at 3 days after stroke. A, Representative laser speckle imaging of cerebral blood flow (CBF) before (baseline), during (ischemia), and after MCAO (reperfusion). B, Quantification of CBF as percentages of the preischemia levels. C, Representative images of 2,3,5‐triphenyltetrazolium chloride (TTC) staining for infarct measurement. D, Quantification of infarct volume. E, Quantification of brain edema. F, Neurological score evaluated during the first 3 days after stroke. n=5/group. *P≤0.05, **P≤0.01, ***P≤0.001. CCR5 indicates C‐C chemokine receptor type 5; Treg, regulatory T cell.
CCR5 Maintains the Transferred Tregs at the Injured Endothelium in the Ischemic Brain
We then explored the mechanism by which CCR5 contributed to the protective effect of Tregs against ischemic stroke. Because CCR5 is known to be important for immune cell recruiting, we first examined whether CCR5 guides the recruitment of the transferred Tregs to the injured barrier. Two‐photon in vivo imaging was performed to observe transferred Tregs in the ischemic area after MCAO. Circulating myeloid cells (mainly neutrophils and macrophages) of the recipient stroke mice were labeled by intravenous injection of rhodamine 6G (red cells in Figures 2 and 3). Dextran‐Alexa Fluoro‐488 (70 kDa) was infused intravenously to visualize the blood vessels. Tregs isolated from CCR5 knockout mice or WT mice were labeled with cell tracker (pseudocolored in blue in Figures 2 and 3) and transferred intravenously after the baseline image was acquired. Time‐lapse imaging was conducted at the ischemic region through a cranial window (Figure 2A) for over 200 seconds. The frequency of Treg appearance during the 200‐second imaging period showed no significant difference between WT Tregs and CCR5−/− Tregs (Figure 2C). However, the migration speed of WT Tregs was significantly lower compared with the CCR5−/− Tregs (Figure 2D). Consistently, the contact time of infiltrated Tregs with the endothelium was significantly elongated by CCR5 expression on the Tregs (Figure 2E through 2H). Meanwhile, the contact time between infused Tregs and endogenous neutrophil/macrophages was significantly longer for WT Tregs than CCR5−/− Tregs (Figure 2I through 2L). These data suggest that although CCR5 expression on Tregs may not increase the recruitment of Tregs to the lesion site significantly, it is important to maintain Tregs at the lesion site and meanwhile increase the interaction among Tregs, endothelium, and neutrophils/macrophages. Interestingly, the migration speed of neutrophils/macrophages was significantly faster in WT Treg‐infused mice compared with splenocyte or CCR5−/− Treg‐infused mice (Figure 3A through 3C).
Figure 2.
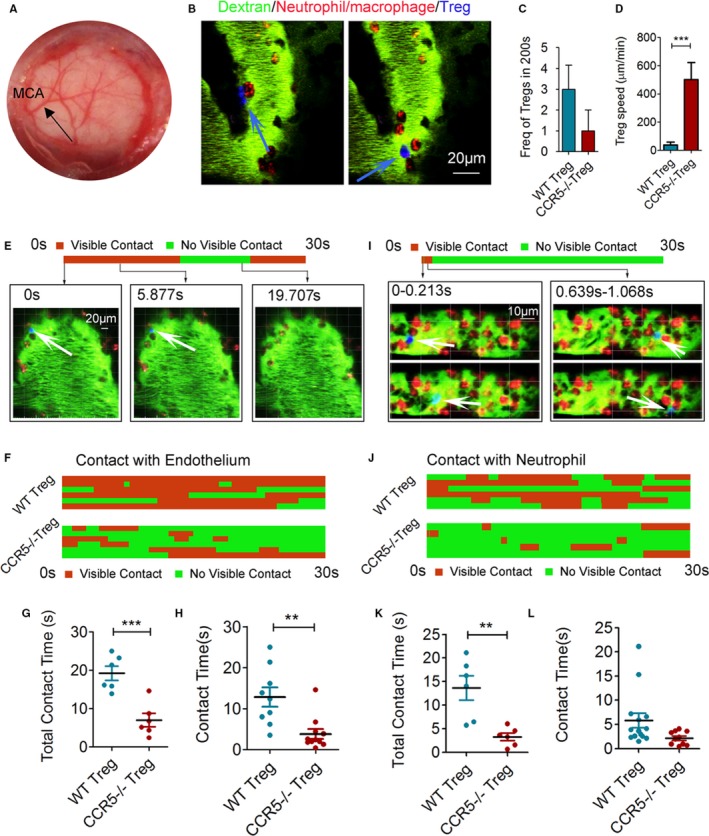
CCR5 maintains the adoptively transferred Tregs at the injured endothelium of the ischemic brain. In vivo 2‐photon images were acquired to observe transferred Tregs in the ischemic area at 1 day after transient middle cerebral artery occlusion (tMCAO). Cranial window and MCAO surgery were performed in the donor mice before imaging. Tregs were isolated from wild type (WT) or CCR5−/− donor mice and stained with CellTracker™ Deep Red Dye before injection (pseudocolored blue). Neutrophils/macrophages were labeled with intravenously injected rhodamine 6G (red). Blood vessels were labeled with intravenously injected fluorescent labeled dextran (green). A, Representative image taken from the cranial window (5 mm in diameter) that was created on the left parietal skull of the recipient mice. The MCA was observed in the middle of the coverslip. B, Representative 2‐photon images of Tregs (blue) and neutrophils or macrophages (red) in the brain vasculature (green). C, Quantification of Tregs frequency in the video recorded over 200 seconds. D, Quantification of Treg migration speed. E through L, Kymographs of Treg‐endothelial interaction and Treg‐neutrophil/macrophage interaction were both generated based on the cell contact time between those cells in videos taken over 30 seconds. E, Representative 2‐photon images and corresponding kymograph of a single Treg (blue, white arrow) interacting with the endothelium (green). F, Kymograph of Treg‐endothelium interactions with multiple Tregs from 6 individual videos of each group. G, Pooled cumulative Treg‐endothelium contact time over the 30 seconds from 6 videos/group. H, Contact time of each Treg interacting with endothelium identified in the videos. I, Representative 2‐photon images and corresponding kymograph of a single Treg (blue) interacting with neutrophils/macrophages (red). J, Kymograph of Treg‐neutrophil/macrophage interactions with multiple Tregs from 6 individual videos of each group. K, Pooled cumulative Treg‐neutrophil/macrophage contact time over 30 seconds from 6 videos/group. L, Contact time of each Treg interacting with neutrophils/macrophages identified in the videos. **P≤0.01, ***P≤0.001. CCR5 indicates C‐C chemokine receptor type 5; Treg, regulatory T cell.
Figure 3.
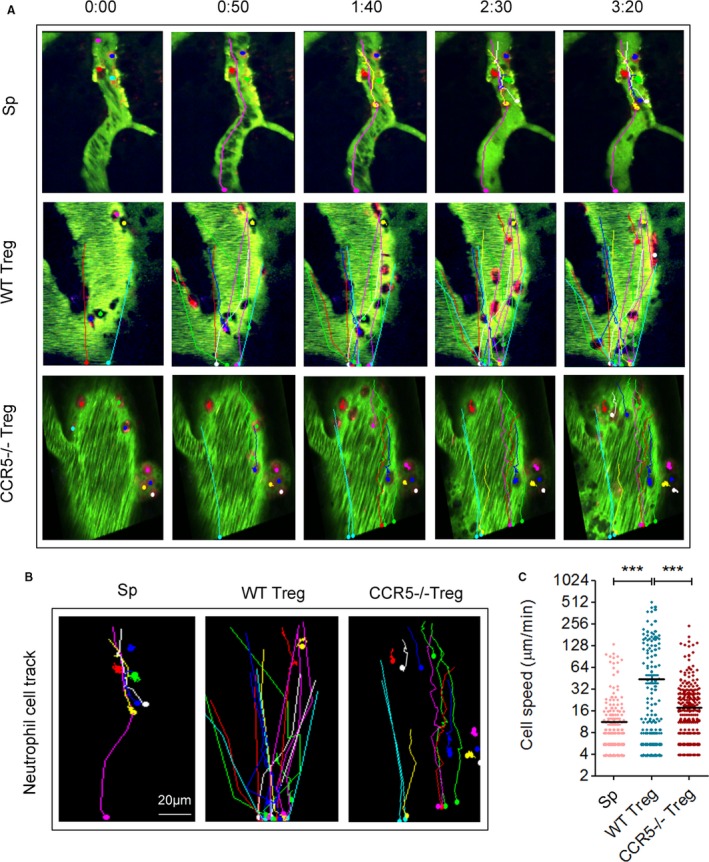
Neutrophils/macrophages in CCR5−/− Treg‐treated stroke mice slow down in the ischemic areas as compared with those in wild type (WT) Treg‐treated stroke mice. A, Representative 2‐photon time lapse images of splenocyte (Sp), WT Treg, or CCR5−/−Treg‐treated stroke animals. Color lines represent the routes of rhodamine 6G‐labeled neutrophils/macrophages. Images are representative of 3 to 5 animals/group. B, Representative images of neutrophil/macrophage routes after intravenous injection of rhodamine 6G. C, Quantification of the moving speed of neutrophils/macrophages during the 2‐photon imaging period. ***P≤0.001. CCR5 indicates C‐C chemokine receptor type 5; Treg, regulatory T cell.
CCR5 Ligands are Upregulated in the Injured Endothelium of the Ischemic Brain
To further confirm a Treg‐endothelium interaction involving CCR5, we then examined the expression of CCR5 ligands, including CCL3, CCL4, and CCL5 in the ischemic brain. As shown in Figure 4A, mRNA expression of CCL3 and CCL5, but not CCL4, increased significantly in the brain 1 day after MCAO. Immunofluorescence staining confirmed an elevation of CCL5 protein expression in CD31+ endothelial cells in the ischemic penumbra 1 day post‐MCAO (Figure 4B). The length of CCL5+CD31+ colocalization along blood vessels increased significantly 1 day after stroke as compared with sham mice (Figure 4C). Consistent with the in vivo data, in vitro cultured endothelial cells exhibited increased expression of CCL5 after 4 hours of OGD (Figure 4D and 4E). The expression of CCL3, another ligand of CCR5, was also elevated in the ischemic brain. However, the Iba‐1+ microglia (Figure 4F and 4H; Figure S2) but not CD31+ endothelial cells (Figure 4F and 4G) are the main cellular source of CCL3 in the ischemic brain.
Figure 4.
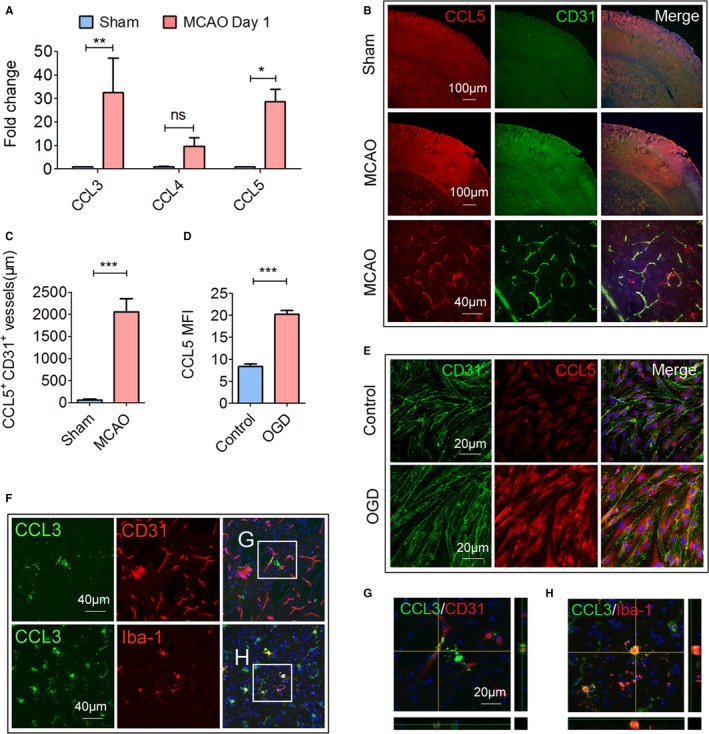
CCR5 ligands are upregulated in the ischemic brain. Cerebral ischemia was induced by 60 minutes of transient middle cerebral artery occlusion MCAO. Animals were euthanized 1 day after MCAO or sham operation. A, The mRNA expression of CCL3, CCL4, and CCL5 was measured by real‐time polymerase chain reaction. B, Representative double immunofluorescence staining of CCL5 and CD31. Images are representative of 3 to 5 animals/group. C, Quantification of CCL5+ CD31+ blood vessel length in sham and tMCAO brains, n=3 to 5/group. D and E, Cultured endothelial cells were challenged with oxygen‐glucose deprivation (OGD) for 4 hours. The cells were fixed and stained for CCL5 and CD31. D, Quantification of mean fluorescence intensity (MFI) of CCL5 in the control or OGD‐challenged endothelial cells. E, Representative CCL5 and CD31 immunofluorescent staining images of control or OGD‐challenged endothelial cells. F, Representative images of CCL3/CD31 and CCL3/Iba‐1 double staining. The white boxes are enlarged (G and H). Images are representative of 3 to 5 animals/group. G, High‐power confocal z‐stack image of CCL3/CD31 double staining, n=5/group. H, High‐power confocal z‐stack image of CCL3/Iba‐1 double staining. *P≤0.05, **P≤0.01, ***P≤0.001. CCL indicates ligand for CCR; CCR5, C‐C chemokine receptor type 5; Treg, regulatory T cell.
Consistent with the increase in CCL5 expression in endothelium, expression of CCR5 on endogenous Tregs increased after cerebral ischemia. We collected peripheral blood, spleen, and lymph nodes from sham or stroke mice at 1 day after MCAO (Figure 5A). We first observed a reduction in the percentage of CD4+CD25+Foxp3+ Tregs among the CD4+ T cells in peripheral blood (Figure 5B). In the presence of previous studies showing reduced number of circulating CD4+ T cells after stroke,33 such reduction of Treg percentage within CD4+ population indicates a dramatic decrease in the absolute number of circulating Tregs. Interestingly, the percentage of CCR5+ cells among Tregs increased significantly in the peripheral blood and spleen 1 day after MCAO (Figure 5C and 5D). The mean fluorescence intensity of CCR5 on blood Tregs and spleen Tregs also significantly increased after stroke (Figure 5E). Cerebral ischemia showed no effect on CCR5 expression on Tregs isolated from lymph nodes (Figure 5C through 5E). Immunostaining further confirmed the expression of CCR5 on Tregs isolated from blood of WT mice at 1 day after MCAO but not on Tregs from CCR5−/− mice (Figure 5F).
Figure 5.
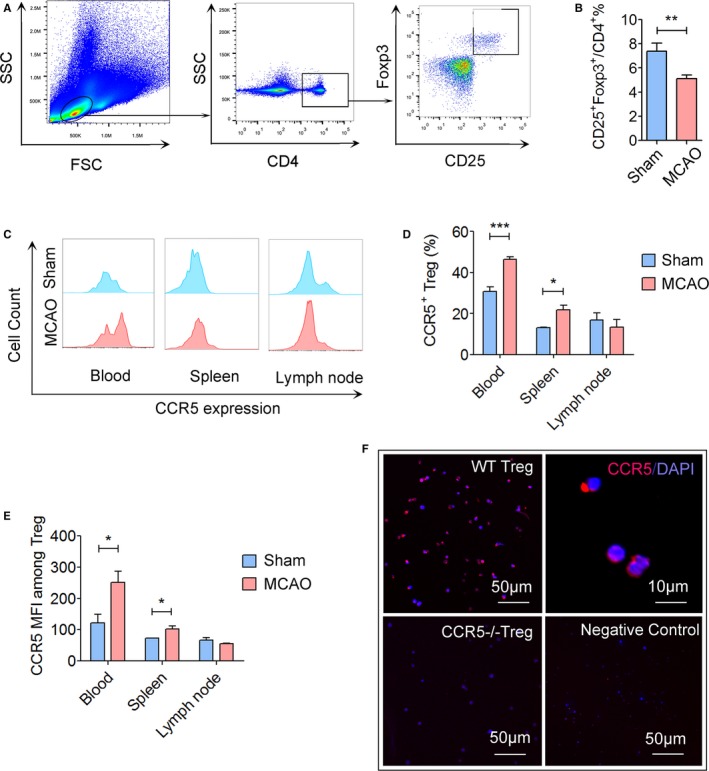
Expression of CCR5 is upregulated on peripheral Tregs after cerebral ischemic stroke. Cerebral ischemia was induced in mice by transient middle cerebral artery occlusion (tMCAO) for 60 minutes. Blood, spleen, and lymph nodes were collected for flow cytometry analysis at 1 day after reperfusion. A, Gating strategy for CD4+ CD25+Foxp3+ Tregs in the blood. B, Quantification of CD4+ CD25+Foxp3+ Tregs among CD4+ lymphocytes in the blood at 1 day after MCAO or sham operation. Cerebral ischemia reduced the percentage of CD4+ CD25+Foxp3+ Tregs. C, Representative flow cytometry plots of CCR5 expression on Tregs in the blood, spleen, and lymph nodes at 1 day after sham or tMCAO. D, Quantification of CCR5+ cells among Tregs. E, Quantification of CCR5 mean fluorescence intensity (MFI) on Tregs at 1 day after sham or tMCAO, n=5 to 6/group. F, Immunostaining of CCR5 on Tregs collected from C57/BL6 wild type (WT) mice or CCR5−/− mice. Representative images are from 2 repeats of independent experiments. *P≤0.05, **P≤0.01, ***P≤0.001. CCR5 indicates C‐C chemokine receptor type 5; Treg, regulatory T cell. FSC, forward scatter; SSC, side scatter.
Taken together, these data suggest that the expression of CCL5 significantly increased on injured endothelium, which could lead to the interaction between endothelium and CCR5‐expressed circulating Tregs. The data also revealed a reduction of circulating Tregs after stroke, which suggest a potential need for Treg supplementation.
CCR5 Expression by Transferred Tregs Plays an Important Role in Their Protective Effect Against BBB Disruption Early After Stroke
With the CCR5‐mediated docking of transferred Tregs on the injured endothelium, Tregs gain proximity to the ischemic barrier. We next tested whether CCR5 was essential for transferred Treg‐afforded protection against BBB disruption after cerebral ischemia.11 We found that loss of CCR5 expression on Treg reversed their protective effect against BBB damage with significantly increased IgG leakage in CCR5−/− Treg‐treated stroke mice as compared to that in the WT Treg‐treated mice (Figure 6A through 6C). Consistently, infiltration of Gr1+ neutrophils (Figure 6D and 6E) was significantly inhibited in WT Treg‐treated but not in CCR5−/− Treg‐treated mice. The infiltration of other immune cells including CD3+ T cells (Figure 6F) and F4/80+ macrophages (Figure 6G) was reduced by WT Treg but not by CCR5−/− Treg treatment. These data support the importance of CCR5 in Treg protection against BBB damage after stroke.
Figure 6.
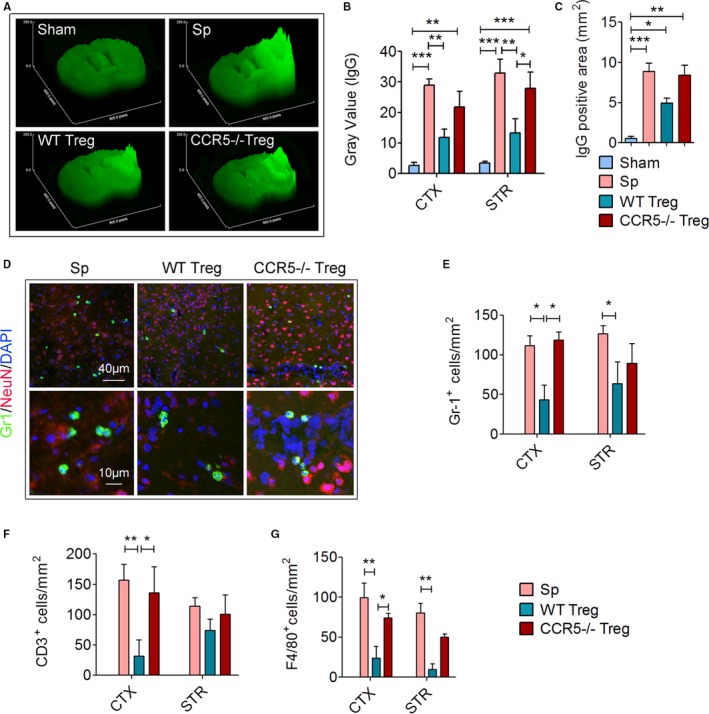
CCR5 expression by transferred Tregs plays an important role in their protective effect against blood‐brain barrier (BBB) disruption early after stroke. Splenocytes (Sp), wild type (WT) Tregs or CCR5−/− Tregs (2×106) were adoptively transferred into stroke mice 2 hours after 60 minutes of transient middle cerebral artery occlusion (tMCAO). Brain sections were collected 1 day after tMCAO. A through C, Brain sections were stained for endogenous plasma IgG to detect BBB leakage (n=6/group). A, Representative surface plot images of endogenous IgG from sham or tMCAO mice treated with WT or CCR5−/− Treg. B, Quantification of gray value of brain sections in A. C, Quantification of positive IgG‐stained area of brain sections in A. D through G, Brain sections were stained with Gr‐1, a neutrophil marker, CD3, a T‐cell marker, and F4/80, a macrophage marker (n=6/group). D, Representative images showing the infiltration of Gr‐1+ neutrophils into the ischemic brain. E, Quantification of the number of Gr‐1+ neutrophils in the ischemic brain (n=5/group). F, Quantification of the number of CD3+ T cells in the ischemic brain. G, Quantification of the number of F4/80+ macrophages in the ischemic brain. *P≤0.05, **P≤0.01, ***P≤0.001. CCR5 indicates C‐C chemokine receptor type 5; CTX, cortex; IgG, immunoglobulin G; STR, striatum; Treg, regulatory T cell.
We further confirmed the role of CCR5 on Treg‐mediated protection against BBB damage using an in vitro endothelial cell‐based model of BBB (Figure 7A). In vitro BBB models have been used for decades to explore the cellular and molecular mechanisms underlying BBB damage or protection.34 BBB integrity changes can be addressed by measuring cell monolayer permeability to different sized tracers. Here, leakage of 4.4‐kDa dextran was measured up to 6 hours of reperfusion after OGD. As shown in Figure 7B, the integrity of the BBB was disrupted progressively from 1 to 6 hours after reperfusion. The addition of TNF‐α–pretreated neutrophils significantly enhanced BBB damage after OGD. Although Tregs showed minimal effect on BBB damage in the absence of neutrophils (not shown), adding Tregs into the endothelial cell‐neutrophil coculture system significantly reduced dextran leakage, suggesting an inhibitory effect of Treg on neutrophil‐enhanced BBB damage (Figure 7C). Interestingly, pretreatment with DAPTA, a CCR5 inhibitor, reduced Treg‐mediated protection against the BBB leakage in the endothelial cell‐neutrophil coculture system (Figure 7C). Similarly, genetic knockout of CCR5 on Tregs significantly removed their protective effect on the BBB (Figure 7C).
Figure 7.
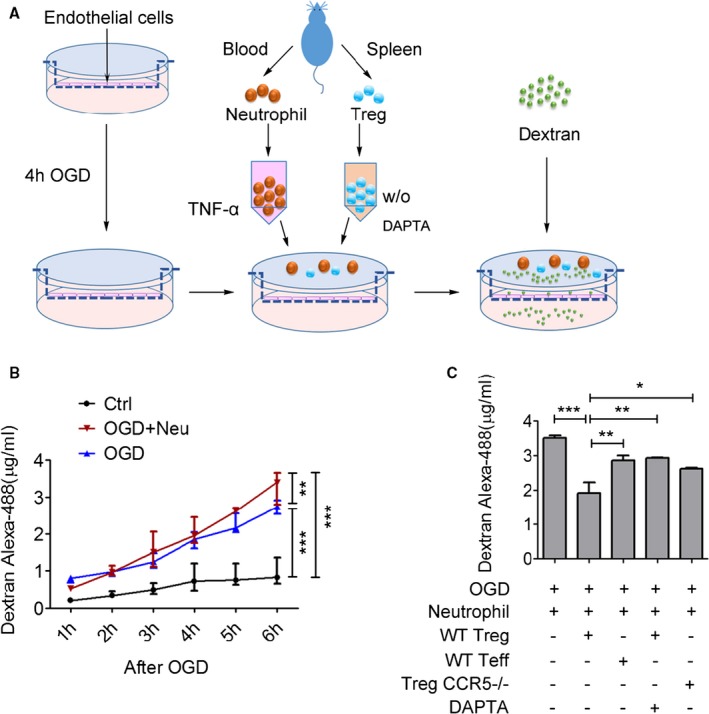
CCR5 is indispensable for Treg‐mediated protection against oxygen‐glucose deprivation (OGD)‐induced blood‐brain barrier (BBB) damage. A, Illustration of in vitro experimental design. A mouse endothelial cell monolayer was seeded on top of a membrane in the cell culture insert and subjected to OGD for 4 hours. After reoxygenation, isolated neutrophils with TNF‐α pretreatment (100 ng/mL) and preactivated Tregs were added into the insert. Then, 4.4‐kDa Dextran‐Alexa‐488 was loaded in the abluminal side. Paracellular permeability was determined by measuring the fluorescence intensity of the culture media in the lower chamber. n=6/group, data are from 2 repeats of independent experiments. B, Dextran‐Alexa‐488 leakage was measured 1 to 6 hours after OGD in different groups. C, BBB integrity was measured by quantifying fluorescence intensity in the culture medium in the lower chamber 6 hours after OGD. Pharmacological inhibition (with DAPTA 100 ng/mL) or genetic depletion of CCR5 abolished Treg‐mediated protection of BBB integrity after OGD. *P≤0.05, **P≤0.01, ***P≤0.001. CCR5 indicates C‐C chemokine receptor type 5; DAPTA, D‐ala‐peptide T‐amide; Treg, regulatory T cell.
Collectively, these findings reveal that CCR5 plays an important role in Tregs’ protection against neutrophil‐enhanced BBB leakage after ischemic challenge.
CCR5 Activation Enhances the Immune Inhibitory Function of Tregs on Neutrophils
It is reported that CCR5 engagement on Tregs may promote other biological functions beyond cell recruiting or docking.35 Therefore, we used CCL5, the chemokine expressed by ischemic brain endothelial cells (Figure 4B and 4C), to activate the CCR5 receptor on enriched Tregs in vitro. Real‐time polymerase chain reaction showed that CCL5 at a concentration of 0.04 μmol/L primed Tregs to express higher mRNA levels of the coinhibitory cell surface molecule PD‐L1 and the immunosuppressive cytokine interleukin‐10 (IL‐10) (Figure 8A). After CCL5 stimulation there was a trend toward increased mRNA expression of CD39, CTLA‐4, and transcription factor FOXp3 (Figure 8A).
Figure 8.
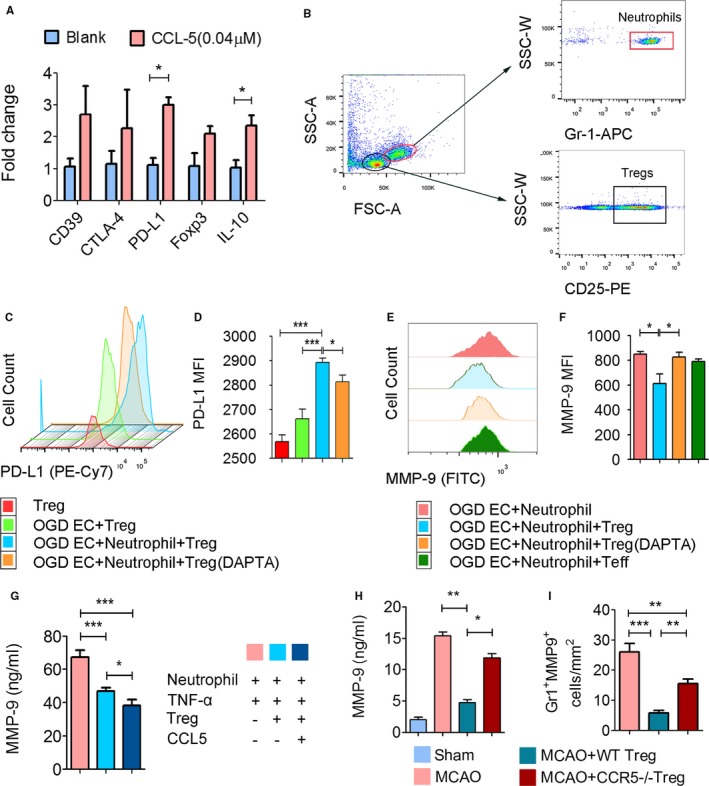
CCL5 expression by Tregs enhances their immune inhibitory function on neutrophils. A, RNA exacts were prepared from Tregs treated with CCL5 (0.04 μmol/L at 37°C for 2 hours). mRNA levels of PD‐L1, CD39, CTLA‐4, Foxp3, and IL‐10 were detected by real‐time polymerase chain reaction and expressed as fold change compared to PBS‐treated controls (blank). n=3 to 4/group. B through F, Tregs and neutrophils were seeded into in vitro cultured mouse endothelial cells challenged with 4 hours of oxygen glucose deprivation (OGD). After another 4 hours of coculture, cells were collected for flow cytometry. Pharmacological inhibition of CCR5 on Tregs was achieved by pretreatment with DAPTA (100 ng/mL) at 37°C for 2 hours. B, Gating strategy of neutrophils and Tregs. n=5 to 7/group. C, Representative histogram of PD‐L1+ cells among Tregs gated from the cocultured cells. D, Mean fluorescence intensity (MFI) of PD‐L1 on Tregs. E, Representative histogram of MMP‐9+ cells among Gr1+ neutrophils gated from the coculture cells. F, MFI of MMP‐9 on neutrophils. G, Tregs treated with or without CCL5 were cocultured with TNF‐α–challenged neutrophils. MMP‐9 level in the culture medium was quantified by enzyme‐linked immunosorbent assay (ELISA). n=6/group. H, Peripheral plasma was collected from sham or transient middle cerebral artery occlusion MCAO mice treated with wild type (WT) or CCR5−/− Tregs for MMP‐9 ELISA measurement. n=6/group. CCR5−/− Tregs failed to inhibit the MMP‐9 rise in MCAO mice. I, Quantification of MMP‐9+Gr1+ neutrophils in the brain slices collected from WT Treg‐ or CCR5−/− Treg‐treated stroke mice at 3 days after MCAO. Adoptive transfer of wild type Tregs, but not CCR5−/− Tregs, significantly reduced MMP‐9 expression in infiltrated neutrophils. *P≤0.05, **P≤0.01, ***P≤0.001. CCR5 indicates C‐C chemokine receptor type 5; DAPTA, D‐ala‐peptide T‐amide; Treg, regulatory T cell, FSC‐A, forward scatter area; SSC‐A, side scatter area; SSC‐W, side scatter width.
PD‐L1 has been shown to be a critical molecule that mediates Tregs’ inhibition of neutrophils.11 We therefore further assessed the influence of CCR5 on the expression and function of PD‐L1 in a Treg‐neutrophil‐endothelial coculture system. Tregs were cocultured with OGD‐challenged endothelial cells and TNF‐α–activated neutrophils for 24 hours. Flow cytometry was used to identify CD25+ Tregs and Gr1+ neutrophils (Figure 8B). As shown in Figure 8C and 8D, the mean fluorescence intensity of PD‐L1 on Tregs increased significantly after being cocultured with OGD‐challenged endothelial cells and neutrophils as compared with the naive Tregs. Blocking CCR5 on Tregs (by pretreating Tregs with DAPTA at a concentration of 100 ng/mL) significantly attenuated the increase in PD‐L1 expression (Figure 8C and 8D), suggesting the critical role of CCR5 in PD‐L1 expression on Tregs. Meanwhile, we measured neutrophil MMP‐9 expression by flow cytometry in the coculture system. Blocking CCR5 on Tregs with DAPTA significantly impaired the inhibitory effect of Tregs on neutrophil‐derived MMP‐9 in the coculture system (Figure 8E and 8F).
We then tested the influence of CCL5 priming of Tregs on inhibition of MMP‐9 production from neutrophils in a neutrophil‐Treg coculture system. MMP‐9 production by neutrophils cocultured with CCL5‐primed Tregs was lower than that from those cocultured with the unprimed Tregs (Figure 8G). Finally, we confirmed in vivo that WT Tregs significantly inhibited MMP‐9 elevation in the plasma after ischemic stroke, whereas CCR5−/− Tregs failed to do so (Figure 8H). Furthermore, adoptive transfer of WT Tregs, but not CCR5−/− Tregs, significantly reduced MMP‐9 expression in infiltrated neutrophils (Figure 8I). Collectively, these data suggest that CCL5‐primed Tregs exhibit stronger immune inhibitory function against neutrophils.
Taken together, our data suggest that CCR5 signaling mediates the upregulation of PD‐L1 on Tregs once recruited to the ischemic BBB, which subsequently inhibits MMP‐9 production by neutrophils through enhanced (Treg)/PD‐1(neutrophil) signaling.12
Discussion
Previous preclinical studies have revealed a potent protective effect of Tregs in ischemic stroke.9, 11 Moreover, several recent clinical trials using adoptive Treg therapy in human inflammatory diseases have reported promising results,36, 37, 38, 39, 40, 41 arousing interest in Treg therapy of stroke. The current study elucidates the importance of CCR5 for the protective effect of adoptively transferred Tregs in ischemic stroke. The results reveal that CCR5 is important for the accumulation of Tregs at the ischemic BBB. Moreover, CCR5 signaling enhances the inhibitory effect of transferred Tregs on neutrophil‐derived MMP‐9 (Figure 9). These 2 mechanisms endorse CCR5 as a critical molecule for Treg‐afforded BBB protection early after ischemic stroke. This discovery may provide new strategies to enhance the therapeutic effect of Tregs in ischemic stroke.
Figure 9.
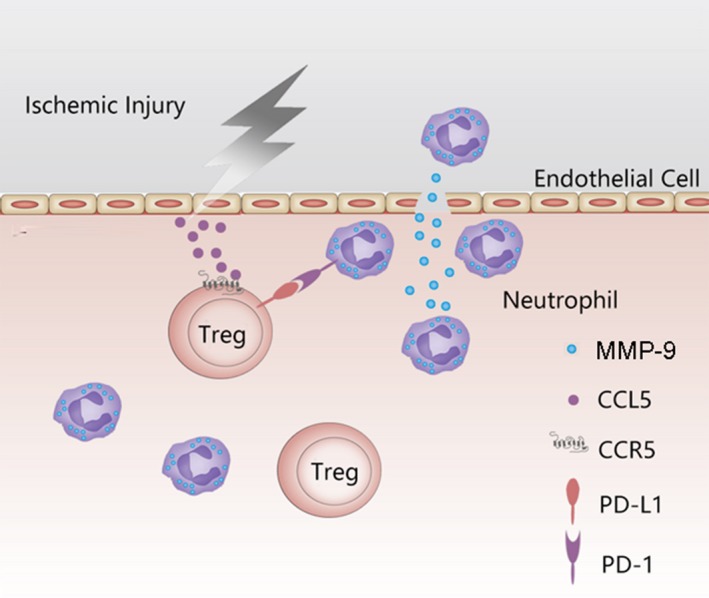
Schematic overview of the proposed mechanism of CCR5‐mediated Treg docking and activation at the ischemic blood‐brain barrier. In response to cerebral ischemic stroke, the endothelial cells express CCL5, which mediates the adoptively transferred Tregs to dock at the ischemic blood‐brain barrier. Once engaged with CCL5, CCR5 is activated and upregulates the expression of PD‐L1 on Tregs, which inhibits the production of MMP‐9 from neutrophils. As a result, CCR5 signaling contributes to Treg‐afforded protection of the blood‐brain barrier after ischemic stroke. CCL5 indicates ligand for CCR5; CCR5, C‐C chemokine receptor type 5; Treg, regulatory T cell.
CCR5 is well known to be responsible for the chemotaxis of immune cells. It is responsive to different chemokines, including CCL3, CCL4, and CCL5. Our data showed that the expression of CCR5 on endogenous Tregs increased significantly after ischemia, which was accompanied by elevated CCL5 expression on endothelial cells and increased CCL3 expression on microglia. These data suggest a potential role of CCR5 in Treg function in the ischemic brain. Considering that adoptively transferred Tregs do not enter the brain parenchyma until 3 to 5 days after stroke,10, 11 CCL5+ endothelial cells might play more important roles than CCL3+ microglia in the initial recruitment and/or docking of Tregs in early response after ischemia. Actually, CCR5‐CCL5 interaction has been reported previously to be important for Treg recruitment to tumors.42, 43 Interestingly, our studies using 2‐photon in vivo imaging revealed that the loss of CCR5 expression resulted in a slight, but not significant, decrease in the number of Tregs in the observed area. This Treg CCR5 deficiency resulted in prominently increased traveling velocity of the Tregs. This result indicates that CCR5 expression on Tregs can slow down the adoptively transferred Tregs at the site of BBB injury, allowing more time for interaction between Tregs and the injured BBB. In support of this notion, loss of CCR5 expression reduced the contact time between Tregs and endothelial cells.
In contrast to the increased migration speed of CCR5−/− Tregs, the speed of neutrophils/macrophages in the infarct area of CCR5−/− Treg‐treated animals was much slower than that in the WT Treg‐treated animals. Mounting evidence has suggested the critical role of neutrophils and macrophages in BBB disruption after cerebral ischemic stroke.44, 45, 46, 47 After cerebral ischemia, the neutrophil flow in the blood vessels slows down prominently, and this is accompanied by the progressive adherence of neutrophils to the inflamed vasculature of the ischemic brain.47, 48 Therefore, the reduced speed of neutrophil/macrophage movement that we observed in CCR5−/− Treg‐ or PBS‐treated stroke mice may allow the infiltration of these peripheral proinflammatory cells. This result is consistent with the immunostaining data showing increased infiltration of neutrophils and macrophages into the ischemic brain 1 day (Figure 6) after stroke. WT Treg treatment, but not the CCR5−/− Treg treatment, reduced the influx of neutrophils after stroke. Taken together, Treg CCR5 expression increases Tregs’ contact with injured endothelium and enhances interaction with other immune cells, preventing these immune cells from lingering in the ischemic area.
In addition to the docking of Tregs, the current study shows that CCR5 signaling plays an important role in promoting Tregs’ postischemia immunosuppressive function. Tregs primed with CCL5 expressed higher levels of PD‐L1, a suppressive coregulatory protein. We previously have identified PD‐L1 as an essential signaling molecule between Tregs and neutrophils in BBB protection following ischemic stroke.13 CCR5 signaling in Tregs may further upregulate the expression of PD‐L1 and subsequently enhance the inhibitory effect of Tregs on MMP‐9 production by neutrophils. In addition to PD‐L1, CCR5 expression also influences the expression of CTLA‐4 and CD39, both of which are shown to be critical for the immunosuppressive functions of Tregs.49, 50 It is noted that CCR5 activation also significantly increased IL‐10 expression by Tregs. IL‐10 has been shown to be important for endogenous Treg‐afforded protection of the ischemic brain.10 However, our previous study suggested that although adoptive Treg transfer elevated the number of IL‐10‐producing Tregs in the blood and spleen of stroke mice, IL‐10 was not critical for Treg‐afforded BBB protection.11 Whether CCL5‐induced upregulation of IL‐10 is important for other beneficial effects of Tregs warrants further exploration. Endothelial cells are also important sources of MMP‐9 in response to ischemic challenge.51, 52 However, Tregs do not show inhibition on MMP‐9 production in endothelia‐Treg coculture without neutrophils (data not shown).
It is noted that CCR5 is not specifically expressed on Tregs but is also expressed on other types of T cells including the recently characterized CCR5+CD4+Foxp3+CD25− T cells. These CCR5+CD4+Foxp3+CD25− cells are not regulatory cells, and adoptive transfer of these cells exacerbates atherosclerosis.20 In our study we focused on the importance of CCR5 in adoptively transferred Tregs and the underlying mechanism of protection. Based on our results, CCR5 expression specifically on adoptively transferred Tregs is essential for their therapeutic effects. How CCR5 influences the brain targeting and function of endogenous Tregs is another important question that warrants further study in the future. However, because CCR5 is widely expressed in almost all types of CD4+ T cells, it is challenging to specifically target CCR5 expression on endogenous Tregs for their therapeutic purpose.
In summary, this study demonstrates that CCR5 signaling is indispensable for the ability of adoptively transferred Tregs to protect the ischemic BBB. Activation of CCR5 enhances interactions between Tregs and endothelial cells and increases the immune suppressive function of Tregs by upregulating PD‐L1 expression. In light of the importance of CCR5 in Treg function, strategies that enhance CCR5 signaling may potentiate the therapeutic effect of adoptively transferred Tregs. Future studies are warranted to evaluate the application of these approaches to optimize Treg treatment after stroke.
Author Contributions
Li and Wang contributed equally. Li, Wang, Zhu, and Jiang analyzed data and prepared the figures. Xia, Gan, and Zhou isolated and cultured the cells and performed the in vivo experiments. Jiang, Vazquez, and Watkins performed the 2‐photon and confocal imaging. Hu, Chen, and Yu conceptualized and supervised the project and designed and conducted experiments. Li, Gan, Thomson, and Hu prepared the article.
Sources of Funding
This work was supported by the grants from NIH/National Institute of Neurological Disorders and Stroke (NINDS) (NS094573 and NS092618 to Hu; NS089534 and NS095671 to Chen). Hu is also supported by a Chinese Natural Science Foundation (NCSF) grant 81571152. Chen is also supported by a Chinese Natural Science Foundation (NCSF) grant 81529002. Li is supported by the Shanghai Rising‐Star Program (16QA1402600), the Shanghai Natural Science Foundation (13ZR1452200), and NCSF (81400956). Gan is supported by the NCSF (81371306 and 81571285) and the Shanghai Committee of Science and Technology Support Program (14431907002). Yu is supported by the NCSF (81370513 and 81571048).
Disclosures
None.
Supporting information
Table S1. Primers for Real‐Time Polymerase Chain Reaction
Figure S1. Treg labeling for 2‐photon in vivo imaging. A, Tregs were isolated from spleens and lymph nodes of donor mice and incubated with CellTracker™ Deep Red Dye (Invitrogen, Carlsbad, CA) at a concentration of 20 μmol/L at 37°C for 30 minutes. Representative images were obtained from confocal microscopy. Treg indicates regulatory T cell.
Figure S2. CCL3 expression in the ischemic brain at different time points after MCAO. Cerebral ischemia was induced in wild type mice by 60 minutes of transient MCAO. Animals were euthanized at 6 hours and 1, 3, and 7 days after reperfusion. Immunofluorescence staining shows that CCL3 expression was markedly increased at 1 day after MCAO, and its expression was colocalized with Iba‐1, a microglial cell marker. Expression of CCL3 in the ischemic brain was diminished 3 days after stroke. Images are representative of 3 to 5 independent animals. CCL3 indicates ligand for C‐C chemokine receptor type 3; MCAO, middle cerebral artery occlusion.
(J Am Heart Assoc. 2017;6:e006387 DOI: 10.1161/JAHA.117.006387.)28768648
Contributor Information
Peiying Li, Email: peiyingli.md@gmail.com.
Xiaoming Hu, Email: hux2@upmc.edu.
References
- 1. Subramanian S, Zhang B, Kosaka Y, Burrows GG, Grafe MR, Vandenbark AA, Hurn PD, Offner H. Recombinant T cell receptor ligand treats experimental stroke. Stroke. 2009;40:2539–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Becker KJ. Modulation of the postischemic immune response to improve stroke outcome. Stroke. 2010;41:S75–S78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, Arumugam TV, Orthey E, Gerloff C, Tolosa E, Magnus T. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 4. Fumagalli S, Coles JA, Ejlerskov P, Ortolano F, Bushell TJ, Brewer JM, De Simoni MG, Dever G, Garside P, Maffia P, Carswell HV. In vivo real‐time multiphoton imaging of T lymphocytes in the mouse brain after experimental stroke. Stroke. 2011;42:1429–1436. [DOI] [PubMed] [Google Scholar]
- 5. Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T‐ and B‐cell‐deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27:1798–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yenari MA, Kunis D, Sun GH, Onley D, Watson L, Turner S, Whitaker S, Steinberg GK. Hu23F2G, an antibody recognizing the leukocyte CD11/CD18 integrin, reduces injury in a rabbit model of transient focal cerebral ischemia. Exp Neurol. 1998;153:223–233. [DOI] [PubMed] [Google Scholar]
- 7. Connolly ES Jr, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, Stern DM, Solomon RA, Gutierrez‐Ramos JC, Pinsky DJ. Cerebral protection in homozygous null ICAM‐1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996;97:209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soriano SG, Coxon A, Wang YF, Frosch MP, Lipton SA, Hickey PR, Mayadas TN. Mice deficient in Mac‐1 (CD11B/CD18) are less susceptible to cerebral ischemia/reperfusion injury. Stroke. 1999;30:134–139. [DOI] [PubMed] [Google Scholar]
- 9. Wang J, Xie L, Yang C, Ren C, Zhou K, Wang B, Zhang Z, Wang Y, Jin K, Yang GY. Activated regulatory T cell regulates neural stem cell proliferation in the subventricular zone of normal and ischemic mouse brain through interleukin 10. Front Cell Neurosci. 2015;9:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liesz A, Suri‐Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192–199. [DOI] [PubMed] [Google Scholar]
- 11. Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, Liang W, Thomson AW, Chen J, Hu X. Adoptive regulatory T‐cell therapy protects against cerebral ischemia. Ann Neurol. 2013;74:458–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mao L, Li P, Zhu W, Cai W, Liu Z, Wang Y, Luo W, Stetler RA, Leak RK, Yu W, Gao Y, Chen J, Chen G, Hu X. Regulatory T cells ameliorate tissue plasminogen activator‐induced brain haemorrhage after stroke. Brain. 2017;140:1914–1931. DOI: 10.1093/brain/awx111. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li P, Mao L, Liu X, Gan Y, Zheng J, Thomson AW, Gao Y, Chen J, Hu X. Essential role of program death 1‐ligand 1 in regulatory T‐cell‐afforded protection against blood‐brain barrier damage after stroke. Stroke. 2014;45:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miska J, Abdulreda MH, Devarajan P, Lui JB, Suzuki J, Pileggi A, Berggren PO, Chen Z. Real‐time immune cell interactions in target tissue during autoimmune‐induced damage and graft tolerance. J Exp Med. 2014;211:441–456.24567447 [Google Scholar]
- 15. Thauland TJ, Koguchi Y, Dustin ML, Parker DC. CD28‐CD80 interactions control regulatory T cell motility and immunological synapse formation. J Immunol. 2014;193:5894–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. [DOI] [PubMed] [Google Scholar]
- 18. Geginat J, Sallusto F, Lanzavecchia A. Cytokine‐driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. J Exp Med. 2001;194:1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukada K, Sobao Y, Tomiyama H, Oka S, Takiguchi M. Functional expression of the chemokine receptor CCR5 on virus epitope‐specific memory and effector CD8+ T cells. J Immunol. 2002;168:2225–2232. [DOI] [PubMed] [Google Scholar]
- 20. Li J, McArdle S, Gholami A, Kimura T, Wolf D, Gerhardt T, Miller J, Weber C, Ley K. CCR5+T‐bet+FoxP3+ effector CD4 T cells drive atherosclerosis. Circ Res. 2016;118:1540–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang CR, Liu MF. Regulation of CCR5 expression and MIP‐1α production in CD4+ T cells from patients with rheumatoid arthritis. Clin Exp Immunol. 2003;132:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bagaeva LV, Williams LP, Segal BM. IL‐12 dependent/IFN‐γ independent expression of CCR5 by myelin‐reactive T cells correlates with encephalitogenicity. J Neuroimmunol. 2003;137:109–116. [DOI] [PubMed] [Google Scholar]
- 23. Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA. CCR5‐dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. J Exp Med. 2006;203:2451–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dobaczewski M, Xia Y, Bujak M, Gonzalez‐Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kroetz DN, Deepe GS Jr. An aberrant thymus in CCR5‐/‐ mice is coupled with an enhanced adaptive immune response in fungal infection. J Immunol. 2011;186:5949–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kozai TD, Vazquez AL, Weaver CL, Kim SG, Cui XT. In vivo two‐photon microscopy reveals immediate microglial reaction to implantation of microelectrode through extension of processes. J Neural Eng. 2012;9:066001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niu K, Cong S, Lee SY. Femtosecond stimulated Raman scattering for polyatomics with harmonic potentials: application to rhodamine 6G. J Chem Phys. 2009;131:054311. [DOI] [PubMed] [Google Scholar]
- 28. Coelho FM, Natale D, Soriano SF, Hons M, Swoger J, Mayer J, Danuser R, Scandella E, Pieczyk M, Zerwes HG, Junt T, Sailer AW, Ludewig B, Sharpe J, Figge MT, Stein JV. Naive B‐cell trafficking is shaped by local chemokine availability and LFA‐1‐independent stromal interactions. Blood. 2013;121:4101–4109. [DOI] [PubMed] [Google Scholar]
- 29. Mitchell JS, Burbach BJ, Srivastava R, Fife BT, Shimizu Y. Multistage T cell‐dendritic cell interactions control optimal CD4 T cell activation through the ADAP‐SKAP55‐signaling module. J Immunol. 2013;191:2372–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD‐1 and PD‐L1 promote tolerance by blocking the TCR‐induced stop signal. Nat Immunol. 2009;10:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brea D, Agulla J, Rodriguez‐Yanez M, Barral D, Ramos‐Cabrer P, Campos F, Almeida A, Davalos A, Castillo J. Regulatory T cells modulate inflammation and reduce infarct volume in experimental brain ischaemia. J Cell Mol Med. 2014;18:1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Swanson RA, Morton MT, Tsao‐Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. [DOI] [PubMed] [Google Scholar]
- 33. Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke‐induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1‐like immunostimulation. J Exp Med. 2003;198:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helms HC, Abbott NJ, Burek M, Cecchelli R, Couraud PO, Deli MA, Forster C, Galla HJ, Romero IA, Shusta EV, Stebbins MJ, Vandenhaute E, Weksler B, Brodin B. In vitro models of the blood‐brain barrier: an overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab. 2016;36:862–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ding Y, Xu J, Bromberg JS. Regulatory T cell migration during an immune response. Trends Immunol. 2012;33:174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Theil A, Tuve S, Oelschlagel U, Maiwald A, Dohler D, Ossmann D, Zenkel A, Wilhelm C, Middeke JM, Shayegi N, Trautmann‐Grill K, von Bonin M, Platzbecker U, Ehninger G, Bonifacio E, Bornhauser M. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft‐versus‐host disease. Cytotherapy. 2015;17:473–486. [DOI] [PubMed] [Google Scholar]
- 37. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P, McGlave PB, Blazar BR, Wagner JE. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117:1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, Del Papa B, Zei T, Ostini RI, Cecchini D, Aloisi T, Perruccio K, Ruggeri L, Balucani C, Pierini A, Sportoletti P, Aristei C, Falini B, Reisner Y, Velardi A, Aversa F, Martelli MF. Tregs prevent GVHD and promote immune reconstitution in HLA‐haploidentical transplantation. Blood. 2011;117:3921–3928. [DOI] [PubMed] [Google Scholar]
- 39. Marek‐Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, Wujtewicz MA, Witkowski P, Mlynarski W, Balcerska A, Mysliwska J, Trzonkowski P. Administration of CD4+CD25highCD127– regulatory T cells preserves β‐cell function in type 1 diabetes in children. Diabetes Care. 2012;35:1817–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7:315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, Watanabe M, Aoyagi T, Suzuki T, Shimamura T, Kamiyama T, Sato N, Sugita J, Hatanaka K, Bashuda H, Habu S, Demetris AJ, Okumura K. A pilot study of operational tolerance with a regulatory T‐cell‐based cell therapy in living donor liver transplantation. Hepatology. 2016;64:632–643. [DOI] [PubMed] [Google Scholar]
- 42. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, Eberlein TJ, Hsieh CS, Linehan DC. Disruption of CCR5‐dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ward ST, Li KK, Hepburn E, Weston CJ, Curbishley SM, Reynolds GM, Hejmadi RK, Bicknell R, Eksteen B, Ismail T, Rot A, Adams DH. The effects of CCR5 inhibition on regulatory T‐cell recruitment to colorectal cancer. Br J Cancer. 2015;112:319–328.25405854 [Google Scholar]
- 44. Ludewig P, Sedlacik J, Gelderblom M, Bernreuther C, Korkusuz Y, Wagener C, Gerloff C, Fiehler J, Magnus T, Horst AK. Carcinoembryonic antigen‐related cell adhesion molecule 1 inhibits MMP‐9‐mediated blood‐brain‐barrier breakdown in a mouse model for ischemic stroke. Circ Res. 2013;113:1013–1022. [DOI] [PubMed] [Google Scholar]
- 45. Bao Dang Q, Lapergue B, Tran‐Dinh A, Diallo D, Moreno JA, Mazighi M, Romero IA, Weksler B, Michel JB, Amarenco P, Meilhac O. High‐density lipoproteins limit neutrophil‐induced damage to the blood‐brain barrier in vitro. J Cereb Blood Flow Metab. 2013;33:575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rosell A, Cuadrado E, Ortega‐Aznar A, Hernandez‐Guillamon M, Lo EH, Montaner J. MMP‐9‐positive neutrophil infiltration is associated to blood‐brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–1126. [DOI] [PubMed] [Google Scholar]
- 47. Stowe AM, Adair‐Kirk TL, Gonzales ER, Perez RS, Shah AR, Park TS, Gidday JM. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis. 2009;35:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Neumann J, Riek‐Burchardt M, Herz J, Doeppner TR, Konig R, Hutten H, Etemire E, Mann L, Klingberg A, Fischer T, Gortler MW, Heinze HJ, Reichardt P, Schraven B, Hermann DM, Reymann KG, Gunzer M. Very‐late‐antigen‐4 (VLA‐4)‐mediated brain invasion by neutrophils leads to interactions with microglia, increased ischemic injury and impaired behavior in experimental stroke. Acta Neuropathol. 2015;129:259–277. [DOI] [PubMed] [Google Scholar]
- 49. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen‐specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA‐4. Immunity. 2014;41:1013–1025. [DOI] [PubMed] [Google Scholar]
- 50. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Hopner S, Centonze D, Bernardi G, Dell'Acqua ML, Rossini PM, Battistini L, Rotzschke O, Falk K. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. [DOI] [PubMed] [Google Scholar]
- 51. Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood‐brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xing C, Arai K, Park KP, Lo EH. Induction of vascular endothelial growth factor and matrix metalloproteinase‐9 via CD47 signaling in neurovascular cells. Neurochem Res. 2010;35:1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers for Real‐Time Polymerase Chain Reaction
Figure S1. Treg labeling for 2‐photon in vivo imaging. A, Tregs were isolated from spleens and lymph nodes of donor mice and incubated with CellTracker™ Deep Red Dye (Invitrogen, Carlsbad, CA) at a concentration of 20 μmol/L at 37°C for 30 minutes. Representative images were obtained from confocal microscopy. Treg indicates regulatory T cell.
Figure S2. CCL3 expression in the ischemic brain at different time points after MCAO. Cerebral ischemia was induced in wild type mice by 60 minutes of transient MCAO. Animals were euthanized at 6 hours and 1, 3, and 7 days after reperfusion. Immunofluorescence staining shows that CCL3 expression was markedly increased at 1 day after MCAO, and its expression was colocalized with Iba‐1, a microglial cell marker. Expression of CCL3 in the ischemic brain was diminished 3 days after stroke. Images are representative of 3 to 5 independent animals. CCL3 indicates ligand for C‐C chemokine receptor type 3; MCAO, middle cerebral artery occlusion.