

In Heart Failure: Congestion Does Not Equal Volume Overload
Most of the current heart failure (HF) prevention or management programs assume that cardiovascular decompensations are primarily driven by volume overload that result in cardiopulmonary congestion. Subsequently, a centerpiece of HF therapies is decongestion of the cardiovascular system. Diuretic drug therapies are known to improve HF symptoms, but more aggressive volume removal strategies such as ultrafiltration have not been proven effective in HF.1, 2 Current strategies for longitudinal HF management often focus on some form of postdischarge surveillance that targets outpatient weight and volume status trends.
Home or remote monitoring efforts commonly rely on daily weight measurements where an acute change in weight would result in a change in medical therapy. The sensitivity of such weight changes prior to hospitalizations were found to be very low (9%), whereas the specificity of such changes is high (97%).3 Notably, many patients do not experience a change in weight before a hospitalization for acute decompensated heart failure (ADHF). In fact, ≈50% of patients gain an insignificant amount of weight (<2 pounds or <2 kg based on the study) in the days preceding a hospitalization.3, 4 Similarly, in the post‐hoc analysis of the ADHF patients in the ASCEND‐HF trial (Acute Study of Clinical Effectiveness of Nesiritide and Decompensated Heart Failure), 26% of patients showed no weight loss (−1 kg ≤ change <1 kg) and 8% actually experienced weight gain (≥1 kg) during the hospitalization.5 Importantly, in the evaluation of intracardiac filling pressures in an ambulatory setting, recent studies found that in ADHF, right‐ and left‐sided pressures generally start to increase before any notable weight changes take place preceding an admission (Figure 1).6, 7 Thus, an increase in right and left heart filling pressures occurs in many cases in the absence of weight gain or total body volume increase.8, 9 Supportive evidence is provided by body volume analysis using the iodohippurate sodium I 131‐labeled human serum albumin indicator–dilution technique. Using this technology, studies found that patients admitted for HF in many cases (34%) were either normovolemic or hypovolemic10 with often normal plasma volume.11 The heterogeneity of clinical presentation suggests the presence of a complementary mechanism to explain increased filling pressures and subsequent ADHF despite a lack of fluid retention. A potential contributing mechanism could be blood volume redistribution between different vascular compartments in the human body. We explore the physiology and rationale for this concept below.
Figure 1.
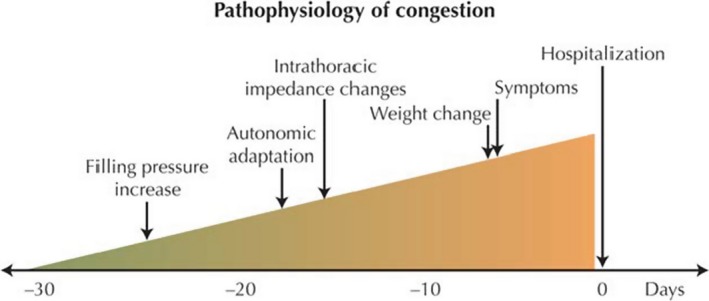
A simplified understanding of heart failure pathophysiology that includes changes in filling pressures using hemodynamic monitoring devices, autonomic adaptation measuring heart rate variability, and alterations in intrathoracic fluid content using thoracic impedance. Reproduced with permission from Adamson.7
Blood Distribution in the Human Body
Blood makes up about 5% of the water in the human body. Most of the blood is held by capacitance vessels, which are generally considered to be veins. Consequently, the venous system contains ≈70% of total blood volume and is roughly 30 times more compliant to store blood than the arterial system.12 The high vascular compliance allows veins to adapt to changes in the blood volume more easily. Blood vessels with a great compliance are able to store large amounts of blood and are called capacitance vessels. However, capacitance vessels do not simply store blood, but are actively involved in the regulation of the preload to the heart and cardiac output via active constrictions of the vessels (vascular capacitance). Interestingly, the venous system does not contribute to the vascular capacitance equally. The veins of the abdomen, otherwise referred to as splanchnic veins, are considerably more compliant than veins of the extremities and skin.13 Unlike many other organs, the visceral organs are marked by a relatively large volume of blood in comparison to the tissue volume. Because of the low vascular resistance and high capacitance, the splanchnic system receives ≈25% of the cardiac output and the splanchnic veins contain anywhere from 20% to 50% of the total blood volume,14, 15 with approximately the following distribution across splanchnic organs: liver 14%; spleen 12%; intestines and stomach about 10%.16
Because of its unique properties, the splanchnic vascular compartment serves as the major blood reservoir (“venous reservoir” or “unstressed volume”) that can take up or release, actively and passively, the major part of any change in circulating blood volume. The “effective circulatory volume” or “stressed volume” refers to a physiologically, but not anatomically separate compartment (Figure 2, Central Compartment illustration), which consists of blood that is present mainly in the arterial system and in nonsplanchnic venous vessels. The “effective circulatory volume” is one of the main determinants of preload to the heart.17, 18
Figure 2.
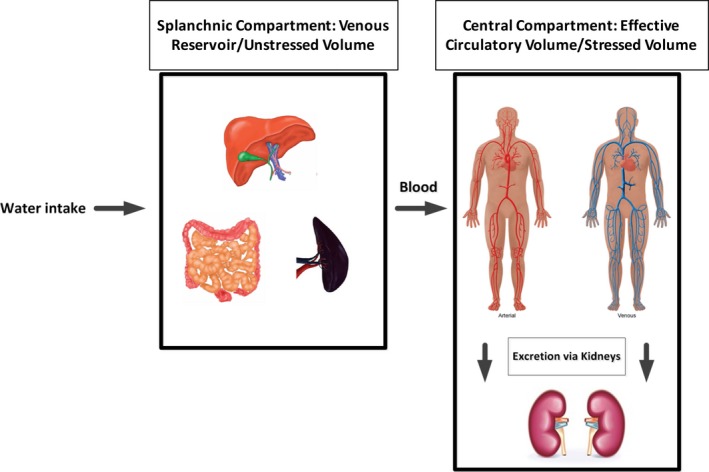
(Central Illustration): Illustrated is the 2‐compartment model of the human blood pool. The splanchnic compartment (left) serves as the venous reservoir/unstressed volume. To the right is the central compartment that contributes to the effective circulation/stressed volume and regulates volume content via the kidneys.
Regulation of Splanchnic Blood Volume
Changes in the blood volume of venous capacitance vessels can be induced via a passive or active mechanism. Passive is the result of transmural pressure changes. Active is the result of a change in the level of contraction of vascular smooth muscle. An example of passive recruitment relevant to HF is “bendopnea,” which refers to a symptom of dyspnea when bending forward.19 One of the main determinants of active recruitment is the sympathetic nervous activity (SNA), which causes venoconstriction through epinephrine and norepinephrine, thereby reducing splanchnic capacitance and recruiting effective circulatory volume. Here cardiopulmonary and arterial baroreflexes appear to play a key role in regulation of SNA,20 where a reduction in vascular or cardiac filling pressures is sensed by baroreceptors and as part of a reflex loop increases the sympathetic tone to recruit blood from the splanchnic and peripheral compartment to the heart. The strong interrelationship between venous vascular tone and the sympathetic nervous system can be explained by a large number of adrenergic receptors on the splanchnic vasculature,12 with a 5 times higher density of adrenergic terminals on veins than arteries.21 The consequence is a more pronounced venous vasomotor response in the splanchnic system compared with other vascular regions.
The sympathetic nervous system controls the splanchnic compartment via branches from the sympathetic thoracic ganglia (T6 through T11) that converge in the celiac plexus and innervate the splanchnic vasculature.22, 23 When the SNA decreases, the vascular compliance increases, and, subsequently, splanchnic reservoir volume increases. On the other hand, sympathoadrenal stimulation translocates blood volume from the splanchnic reservoir to the central circulation. In animal experiments, targeted splanchnic nerve stimulation can lead to a recruitment of up to 80% of the splanchnic volume, which, despite adrenalectomy,22 results in a blood shift of >20% of the total body blood volume.24, 25 In a human case study, the stimulation of the nerves resulted in an increase in preload (50%) and cardiac output (200%) within 2 minutes.26 Exercise, orthostasis, and hemorrhage are 3 classic examples of an increased demand or a sudden reduction in effective circulatory volume/preload that require rapid recruitment of the unstressed volume via splanchnic bed vasoconstriction.12, 27
Splanchnic Capacitance in HF
In HF, a state of neurohormonal activation, the splanchnic vascular compartment is at the center of volume dysregulation in acute and chronic HF. Current strategies of ADHF management and prevention have focused on the classical paradigm that salt and fluid retention is the culprit of intravascular fluid expansion and cardiac decompensation. However, the concept of disrupted intravascular fluid distribution might play a significant role in the process of chronic HF and/or ADHF even in the absence of increases of total body salt and water17 (Figure 3).
Figure 3.
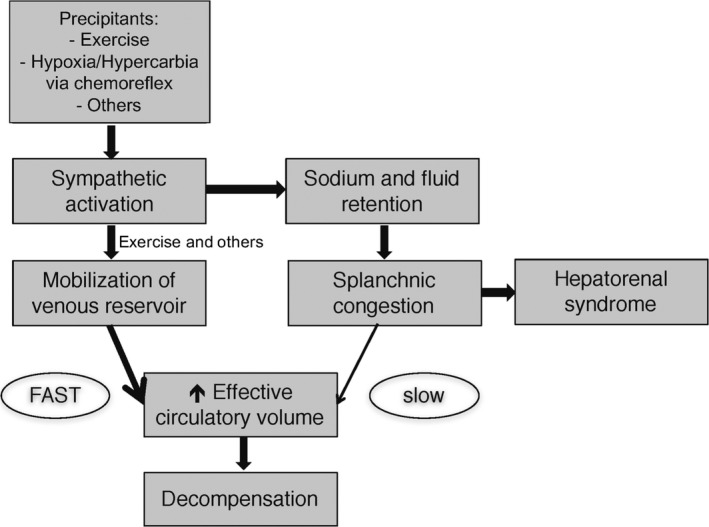
The proposed mechanism of progression from chronic compensated to acute heart failure is summarized in this figure. Sodium retention and fluid expansion result in an increase of unstressed volume and subsequent splanchnic congestion. This process is slow and takes days to weeks. The fast component often observed in the few days before decompensation is driven by autonomic imbalance with overactivity of the sympathetic nervous system. This results in an intercompartmental fluid shift into the central circulation with a subsequent accelerated increase in central filling pressures. Rapid fluid mobilization also occurs with activity and can explain exercise limitations experienced by heart failure patients. Adapted with permission from Fallick et al.17 Promotional and commercial use of the material in print, digital or mobile device format is prohibited without the permission from the publisher Wolters Kluwer.
There appear to be 3 primary mechanisms by which the splanchnic vascular compartment contributes to the inappropriate volume handling in patients with HF:
Passive: Impaired storage capacity of the splanchnic vascular bed that reduces its ability to buffer extra fluid.
Active: Increases in sympathetic tone move fluid out of the splanchnic compartment into the effective circulation that results in increased cardiac preload and extravascular edema.
Cardiorenal/Hepatorenal: Direct interactions between the splanchnic vascular compartment with the kidneys that result in inappropriate fluid retention.
Experimental Evidence
Theoretical modeling by Burkhoff and Tyberg in the 1990s suggested that acute left ventricular dysfunction with associated decrease in cardiac output and increase in peripheral vascular resistance alone cannot explain the rise in pulmonary vascular pressures.28 Evidence suggested that an additional change in venous capacity with associated central volume distribution is a major contributor to the increase in pulmonary venous pressures.28, 29, 30 Next, the contribution of the splanchnic vascular bed to the diminished vascular capacitance in HF was tested in animal models of HF. Cardiac failure induced by rapid right ventricular pacing in dogs resulted in a profound reduction in total vascular capacitance or in other words a reduction in the body's blood storage capacity. Given a more or less stable blood volume over time, the circulatory filling pressures increased.31 These profound effects on total vascular capacitance over several weeks caused enormous changes in cardiac loading conditions. Consequently, decompensation of the HF model following volume loading has been linked to a shift of blood from the splanchnic vascular compartment (unstressed volume) to the central compartment (stressed volume) rather than a primary increase in the stressed volume (for example, by an external fluid bolus).32
Barnes and colleagues provide further evidence for the role of the splanchnic vascular bed as a major contributor to deleterious fluid shifts observed in ADHF. In this study, changes in cardiac output and mean right atrial pressure were evoked at different circulating blood volumes by stimulation of the splanchnic sympathetic nerves. With high circulating volumes, infusion of more volume did not consistently alter output or aortic pressure, but splanchnic nerve stimulation increased peripheral resistance and aortic pressure and commonly evoked a rise in left ventricular stroke volume.22 The study by Barnes and colleagues suggests that constriction of the splanchnic vasculature is more effective in raising cardiac filling pressures than external volume loading alone and thus emphasizes the potential contribution of the splanchnic vascular compartment to cardiac decompensation.
Clinical Evidence for Impaired Splanchnic Capacitance
It is important to note that, because of a lack of simple techniques to study the distribution of intravascular blood volume and measure regional sympathetic tone, there have not been definitive studies to prove the concept of intercompartmental fluid shifts as a mechanism of ADHF in humans.
In one of the few relevant studies, Rapaport and colleagues studied patients with HF and found that, on one side, the splanchnic blood flow was decreased and, on the other side, the splanchnic blood volume was actually elevated when compared with healthy controls.33 These results are not surprising given high right‐sided filling pressures and abdominal congestion, which in some patients is the predominant sign of right‐sided HF. However, it can be speculated that the incremental splanchnic capacity (“remaining storage space”) in HF patients is limited when compared with healthy individuals (Figure 4).
Figure 4.
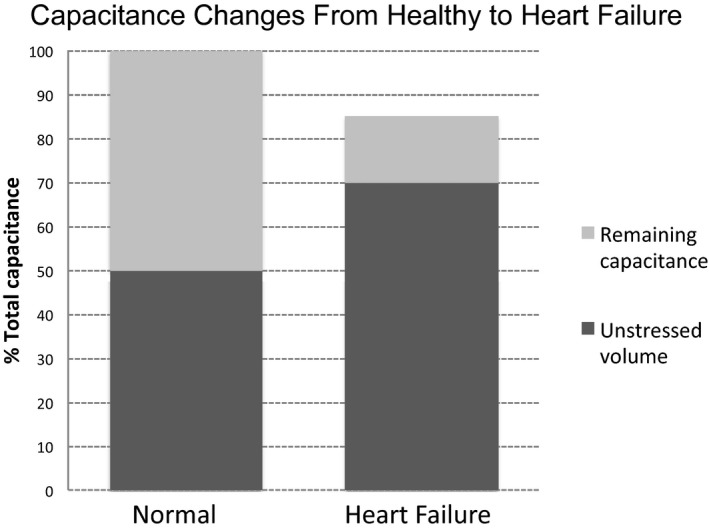
This graphic illustrates the hypothesized variation in splanchnic vascular compliance between healthy and disease states. Heart failure is signified by a decrease in total vascular capacitance of the splanchnic compartment. The remaining storage capacity of the splanchnic compartment depends on the body's volume‐loading condition.
While peripheral vascular capacitance is unchanged in HF with preserved ejection fraction (HFpEF) and HF with reduced ejection fraction (HFrEF) compared with controls,34 the splanchnic vascular capacitance has not been explored in different HF states. The baroreflex normally restrains fluid from shifting away from the splanchnic reservoir by providing a sympatho‐inhibitory influence to this vascular bed.35 The decreased baroreflex function in HF36 inhibits the body's function to buffer eventual rises of effective circulatory volume in chronic, but especially acute disease states.37 Logically, a reduced buffer function (“passive”) of the main storage compartment predisposes the body to decompensate (“overflow”) in the setting of total body fluid volume increase (oral or intravenous fluid intake).
The splanchnic vascular compartment may play a particularly important role in patients with HFpEF. Patients with HFpEF are especially sensitive to volume overload in the setting of exercise, and a striking increase in left atrial pressure often occurs transiently and is rapidly resolved by intravascular volume reduction.38, 39 For example, a passive leg raise in HFpEF patients during a right heart catheterization causes significant increases in wedge pressures that only continue to climb with exercise.40 Such increases in wedge pressure indicate the significance of even small changes in cardiac preload. Although the pathophysiology of HFpEF was initially thought to be caused by left ventricular diastolic dysfunction, recent studies have suggested more complex involvement of multiple abnormalities.41 The diagnosis of HFpEF in many patients at rest would be missed if the diagnosis merely relied on resting clinical, echocardiographic, or even invasive hemodynamic measurements.42 A subset of HFpEF patients experience significant exercise‐related symptoms without evidence of congestion at rest, which suggests a noncompliant cardiopulmonary system as well as a potentially inappropriate volume distribution. With exercise‐induced sympatho‐activation, blood is redirected away from the splanchnic compartment and actively moved out of the abdomen into the central vascular compartment such as the heart and lungs.43 Taken together, the active and passive contribution of the splanchnic vascular compartment can significantly contribute to cardiopulmonary congestion with exercise.
Increased chemosensitivity to hypoxia and hypercarbia could be a mechanism of acute and chronic sympathetic stimulation with subsequent decrease in splanchnic vascular capacitance (Figure 1).44 Patients with HF have an increased sensitivity of the peripheral chemoreceptors,45, 46 leading to exaggerated responses in respiratory drive and sympathetically mediated blood pressure increases to hypoxic stimulation.47, 48 Moreover, peripheral chemoreceptor hypersensitivity was found to be an independent predictor of mortality in patients with HF.49 Activation of the chemoreflex drives activates SNA and further impairs baroreflex function in HF patients,50, 51, 52 which consequently drives the volume intolerance of patients with HFrEF and especially HFpEF.53 Patients with obstructive or central sleep apnea are at an especially high risk of chemoreceptor‐mediated SNA increases, given frequent hypoxic and hypercarbic exposure.54
Splanchnic Compartment: The Link Between HF and Cardiorenal Syndrome
Venous congestion has been found to be the most important hemodynamic factor driving worsening renal function in patients presenting with ADHF.55, 56 A number of mechanisms have been implicated in its development: (1) Global activation of the sympathetic nervous system causes renal vasoconstriction; and (2) Worsening of glomerular filtration rate.57 This effect, combined with the direct effects of elevated venous pressure on the kidney, leads to diuretic resistance and further elevation of filling pressures.58
Moreover, there is evidence that chronic splanchnic congestion by itself can result in renal dysfunction and concomitant diuretic resistance. Local reflex systems such as stretch receptors in the venous wall of the portal vein function as a link between the splanchnic compartment and the kidneys. This local reflex may be involved in an excitatory response to the renal sympathetic nerves, leading to renal vasoconstriction during the portal vein distension (hepatorenal reflex).59 The interaction between liver/spleen and kidneys is complex and implies the presence of osmo‐, chemo‐, and baroreceptors in the liver that sense specific stimuli and react to them through neural‐mediated circuits that directly affect the kidneys and their function.60, 61 Receptor activation results in an increase of sympathetic nervous tone and sodium and water absorption through the kidney. In our opinion, the potentially important role of this reflex for the mechanism of congestion in HF has not received adequate attention.
In animal studies, splanchnic congestion activates hepatic and splenic baroreceptors and, in response, increases the sympathetic efferent nerve activity to the kidneys and cardiopulmonary region. Activation of the hepatic and splenic mechanoreceptors is associated with increases in renal vasoconstriction, renin release, and tubular sodium and water reabsorption or decreases in glomerular filtration rate and renal blood flow.62 Conversely, interruption of the afferent or efferent reflex arm abolishes the hepatorenal or splenorenal reflex,62, 63 as seen with section of the anterior hepatic nerves, which eliminated the reflex increase in renal efferent nerve activity.
The hepatorenal/splenorenal becomes significant in 2 different forms of splanchnic congestion: HF and hepatorenal syndrome. In HF that is characterized by abdominal congestion and increased portal pressures, distension of splanchnic mechanoreceptors leads to the above‐described activation of the hepatorenal and splenorenal reflex with inadvertent sodium and volume retention. Interruption of the reflex arc and offloading those receptors have been shown to eliminate this reflex. A percutaneous nerve block of the lumbar nerves in the clinical setting of hepatorenal syndrome improved renal function with increased urine output.64 Notably, the pathology in liver cirrhosis is comparable in the component of portal hypertension and splanchnic congestion.
Diagnostic and Therapeutic Approaches
The complexity in studying volume shifts explains the lack of attention to the contribution of the splanchnic compartment and intercompartmental fluid shifts to the pathophysiology of HF. Biomarkers such as natriuretic peptides and routine physical exam evaluation using vascular congestion (jugular venous distension), extravascular volume (lower extremity edema and ascites), and change in body weight correlate poorly with total body volume state65, 66, 67 and provide little information on the distribution of intravascular volume between the central and splanchnic vascular compartment. Potentially useful diagnostic tools to measure intercompartmental fluid shifts include radionuclide plethysmography, which allows measurements of the splanchnic blood pool over time.68 However, the technical requirements of this procedure make it unsuitable for clinical practice. Furthermore, bioimpedance69 and bioelectrance70 can provide a less complicated, but yet less accurate estimation of intercompartmental fluid shifts. The bioimpedance and bioelectrance technologies are based on the assumption that human tissue is an inhomogeneous electrical conductor. The properties of electrical current traveling through tissue differ with changes in blood/fluid content; however, the technologies are limited by theirs inability to discriminate intravascular from extravascular volume.
A very simple diagnostic tool to assess splanchnic vascular capacitance is orthostatic stress. The key component that leads to a drop in preload with orthostasis is a passive blood volume shift into the veins of the abdomen and pelvis more so than peripheral blood vessels (arms and legs).71 The hemodynamic response to tilt‐table testing in chronic HF is atypical; that is, there was no significant peripheral pooling in the upright posture.72 Better tolerance of orthostatic stress in HF patients can be explained by higher filling volumes and decreased splanchnic capacity, which prevents a drop in cardiac preload. In HF patients, decongestion with diuretics73 or blood removal74 results in orthostatic hypotension they previously did not have. Simplified, this means the presence of orthostatic symptoms in a patient with HF suggests normal to high splanchnic (and peripheral) capacitance, whereas a lack of orthostatic symptoms implies the opposite.
A comprehensive approach to the treatment of HF should include a careful assessment of volume status. As we have outlined, ADHF can, in a significant amount of cases, be a problem of volume redistribution instead or in addition to external volume. The challenge that arises is how to reverse the volume shift and direct volume into the splanchnic compartment away from the central compartment and/or decongest both compartments without removing too much volume, thus risking renal dysfunction (Figure 5).
Figure 5.
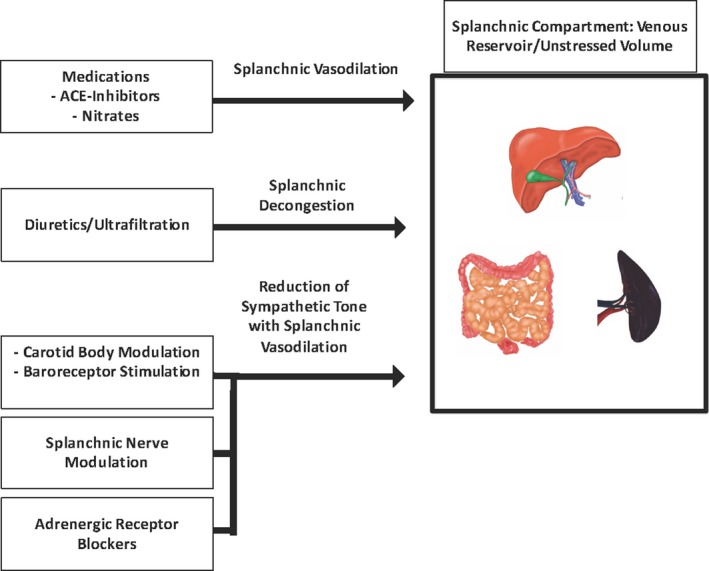
In heart failure, the splanchnic vascular compartment has a decreased vascular storage capacitance. Illustrated are different therapeutic approaches to increase the splanchnic vascular capacitance and allow blood redistribution into the splanchnic compartment. ACE indicates angiotensin‐converting enzyme.
Medications to Increase Splanchnic Vascular Capacitance
Drugs such as angiotensin‐converting enzyme inhibitors and nitrates have an established role in the treatment of HFrEF. Interestingly, both agents were shown to increase splanchnic capacitance.75, 76 Cody and colleagues showed that acute and chronic captopril challenge reestablished orthostatic symptoms in HF with a tilt test.77 Captopril caused a drop in preload, pulmonary pressures, wedge pressure, and cardiac index. These results could not be explained by a change in systemic vascular resistance (as it actually went up), but likely were caused by an acute reduction in preload.
It can be speculated that some of the observed effects of angiotensin‐converting enzyme inhibitors, nitrates, and ganglionic blockers such as trimethaphan78 could stem from an increase in vascular capacitance with a redistribution of blood into the venous reservoir, thereby lowering the left ventricular end diastolic pressure. These effects could be acute or chronic. However, arterial vasodilatory drugs such as hydralazine did not show an effect on splanchnic vascular capacitance.76 To what extent the splanchnic vasodilatory mechanism of a drug reflects its effect on HF morbidity and mortality requires further evaluation. Splanchnic vasodilation could also be achieved via a reduction in sympathetic nervous tone with drugs such as the adrenergic receptor blockers. β‐Blockers such as metoprolol and carvedilol have an established benefit in HFrEF patients,79 but it remains unclear whether global reduction of SNA using adrenergic receptor blockers is sufficient to modify the local SNA in the splanchnic compartment and favorably modify the vascular capacitance. Systemic as opposed to targeted sympathetic blockade might result in the expected splanchnic vasodilatory effect at a potentially high cost of systemic blood pressure reduction and exaggerated negative inotropy, which have been linked to poor clinical outcomes in patients with HF.80, 81
Autonomic Modulation
Autonomic modulation presents an attractive option for the treatment of the described pathomechanism for HF and cardiorenal syndrome. Conceptually, the reduction of splanchnic sympathetic tone may result in an increase in splanchnic vascular capacitance. For patients with HFpEF, more so than HFrEF, an increase in vascular capacitance could improve dyspnea and capacity to exercise. Furthermore, the expected benefits of unloading the central venous and arterial system could lessen the incidence and severity of acute decompensations. This concept has been explored in healthy mongrel dogs, where abdominal sympathetic denervation via the celiac plexus increased splanchnic capacitance.82 Unfortunately, human and animal data are not available for the HF space. Thus, this area holds interesting promise, but requires significantly more work.
Besides a direct modulation of the splanchnic vascular tone, targeted therapies could modify the detrimental influence of the chemoreflex and the impaired baroreflex on the autonomic tone and their role as triggers for HF decompensation. Therapies targeting the carotid body (location of the peripheral chemoreflex)83 and the baroreflex84 are currently under investigation with some initially promising results on outcomes in HF patients. Furthermore, the experimental therapy for renal denervation targets the reflex loops between the kidneys and the central nervous system that appear to contribute to the autonomic imbalance. Despite some early positive results, the intervention failed to meet the set expectations.85, 86 Nevertheless, the evaluation of autonomic modulation is ongoing and holds promise for the treatment of hepatorenal and splenorenal syndrome in the HF setting.
Volume Reduction Therapies
Since the splanchnic and central vascular compartments are connected, selective offloading is not possible. Despite a lack of clear benefit of ultrafiltration over diuretic strategies, ultrafiltration has the ability to provide quicker and higher volume decongestion.1, 2, 87 Ultrafiltration removes intravascular volume and can provide rapid and partial decompression of the renal and splanchnic vasculature,88 which in return offloads portal mechanoreceptors and inhibits the hepatorenal and splenorenal reflex.89 The positive effect of rapid intravascular decongestion was demonstrated in a study by Marenzi and colleagues,90 where ultrafiltration produced a 5‐fold increase in diuresis in ADHF admission with oliguria. This increase could occur even though the output of the heart remained unchanged. In contrast, patients without congestion but with normal urine output and controls did not respond with comparable improvement in diuresis. The UNLOAD (Ultrafiltration versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Congestive Heart Failure) and AVOID‐HF (Aquapheresis Versus Intravenous Diuretics and Hospitalization for Heart Failure) trials showed that ultrafiltration resulted in more weight, net fluid loss, and favorable outcomes compared with usual care.2, 87 Because of concerns about a possible renal impairment from the CARRESS (Cardiorenal Rescue Study in Acute Decompensated Heart Failure) trial, this therapy is currently only the second choice.1 We speculate that the reported association of ultrafiltration with renal side effects is attributable to overaggressive volume removal. Ultrafiltration is possibly more harmful in cases where volume shift is the predominant cause of decompensation rather than an increase in total body fluid volume, since the forceful removal of intravascular volume can lead to vascular underfilling with insufficient time for re‐equilibration with the extravascular space. This question requires further exploration in future studies.
While our review focuses on the role of intravascular fluid compartments for HF, the extravascular volume of the abdominal cavity is of direct relevance to the discussed topic. A large amount of ascites is uncommonly encountered in patients with HF but suggests a high portal pressure. Increased intra‐abdominal pressure (intra‐abdominal pressure ≥12 mm Hg) has been clearly associated with organ dysfunction, especially renal dysfunction91 and conceivably results in an increased splanchnic vascular tone with stimulation of hepatic and splenic baroreceptors that play an integral role in the hepatorenal reflex. Mullens and colleagues demonstrated that HF‐impaired renal function is observed with only small rises in intra‐abdominal pressure, in the range of 8 to 12 mm Hg.92 In contrast, a reduction of ascites via decongestive therapy (possibly also paracentesis) improves renal function,88, 92 suggesting that decongestive therapies need to target not only the intravascular but also the extravascular fluid compartment.
Conclusions
In this review we discussed the concept of a compartment model of intravascular volume distribution and its role in chronic and acute HF. Special attention is paid to the splanchnic vascular compartment and its regulation by the autonomic nervous system. We describe how a decreased vascular capacitance and intercompartmental fluid shift can predispose HF exacerbations regardless of the total body fluid status. This concept is complementary to the established concept of sodium and fluid retention as the main drivers of cardiovascular decompensation. Future studies should focus on confirming the proposed physiology in HF patients and try to identify patients at risk for intercompartmental volume redistribution as a leading component of HF decompensation. Finally, there is a need for more clinically relevant and applicable methods to measure fluid distribution so it can be readily applied in the clinical and research arena.
Sources of Funding
The article has been supported by American Heart Association grant 17MCPRP33460225 and National Institutes of Health T32 grant 5T32HL007101.
Disclosures
Marat Fudim has received consulting fees from AxonTherapeutics, Coridea, Cibiem, and GE Healthcare; Adrian Hernandez has received consulting/speaker fees from: Sanofi, Johnson&Johnson, AstraZeneca, and Corthera; G. Michael Felker has received consulting/speaker fees from: Amgen, Bristol Myers Squibb, GSK, Medtronic, MyoKardia, Novartis, Stealth, Trevena, Amgen, Otsuka, and Roche Diagnostics.
J Am Heart Assoc. 2017;6:e006817 DOI: 10.1161/JAHA.117.006817.28862947
References
- 1. Bart BA, Goldsmith SR, Lee KL, Givertz MM, O'Connor CM, Bull DA, Redfield MM, Deswal A, Rouleau JL, LeWinter MM, Ofili EO, Stevenson LW, Semigran MJ, Felker GM, Chen HH, Hernandez AF, Anstrom KJ, McNulty SE, Velazquez EJ, Ibarra JC, Mascette AM, Braunwald E; Heart Failure Clinical Research N . Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med. 2012;367:2296–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Costanzo MR, Negoianu D, Jaski BE, Bart BA, Heywood JT, Anand IS, Smelser JM, Kaneshige AM, Chomsky DB, Adler ED, Haas GJ, Watts JA, Nabut JL, Schollmeyer MP, Fonarow GC. Aquapheresis versus intravenous diuretics and hospitalizations for heart failure. JACC Heart Fail. 2015;16:69. [DOI] [PubMed] [Google Scholar]
- 3. Lewin J, Ledwidge M, O'Loughlin C, McNally C, McDonald K. Clinical deterioration in established heart failure: what is the value of BNP and weight gain in aiding diagnosis? Eur J Heart Fail. 2005;7:953–957. [DOI] [PubMed] [Google Scholar]
- 4. Chaudhry SI, Wang Y, Concato J, Gill TM, Krumholz HM. Patterns of weight change preceding hospitalization for heart failure. Circulation. 2007;116:1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ambrosy AP, Cerbin LP, Armstrong PW, Butler J, Coles A, DeVore AD, Dunlap ME, Ezekowitz JA, Felker GM, Fudim M, Greene SJ, Hernandez AF, O'Connor CM, Schulte P, Starling RC, Teerlink JR, Voors AA, Mentz RJ. Body weight change during and after hospitalization for acute heart failure: patient characteristics, markers of congestion, and outcomes: findings from the ASCEND‐HF trial. JACC Heart Fail. 2017;5:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zile MR, Bennett TD, St John Sutton M, Cho YK, Adamson PB, Aaron MF, Aranda JM Jr, Abraham WT, Smart FW, Stevenson LW, Kueffer FJ, Bourge RC. Transition from chronic compensated to acute decompensated heart failure: pathophysiological insights obtained from continuous monitoring of intracardiac pressures. Circulation. 2008;118:1433–1441. [DOI] [PubMed] [Google Scholar]
- 7. Adamson PB. Pathophysiology of the transition from chronic compensated and acute decompensated heart failure: new insights from continuous monitoring devices. Curr Heart Fail Rep. 2009;6:287–292. [DOI] [PubMed] [Google Scholar]
- 8. Androne AS, Hryniewicz K, Hudaihed A, Mancini D, Lamanca J, Katz SD. Relation of unrecognized hypervolemia in chronic heart failure to clinical status, hemodynamics, and patient outcomes. Am J Cardiol. 2004;93:1254–1259. [DOI] [PubMed] [Google Scholar]
- 9. Miller WL. Fluid volume overload and congestion in heart failure: time to reconsider pathophysiology and how volume is assessed. Circ Heart Fail. 2016;9:e002922. [DOI] [PubMed] [Google Scholar]
- 10. Strobeck JE, Miller WL. Impact of blood volume quantification on decongestion strategy, readmission rates (RR), and mortality in hospitalized heart failure patients (HHF). Am Coll Cardiol. 2016;67:S1276. [Google Scholar]
- 11. Nijst P, Verbrugge FH, Bertrand PB, Martens P, Dupont M, Drieskens O, Penders J, Tang WH, Mullens W. Plasma volume is normal but heterogeneously distributed, and true anemia is highly prevalent in patients with stable heart failure. J Card Fail. 2017;23:138–144. [DOI] [PubMed] [Google Scholar]
- 12. Gelman S. Venous function and central venous pressure: a physiologic story. Anesthesiology. 2008;108:735–748. [DOI] [PubMed] [Google Scholar]
- 13. Hainsworth R. The importance of vascular capacitance in cardiovascular control. Physiology. 1990;5:250–254. [Google Scholar]
- 14. Delorme EJ, Mac PA, Mukherjee SR, Rowlands S. Measurement of the visceral blood volume in dogs. Q J Exp Physiol Cogn Med Sci. 1951;36:219–231. [DOI] [PubMed] [Google Scholar]
- 15. Greenway CV, Lister GE. Capacitance effects and blood reservoir function in the splanchnic vascular bed during non‐hypotensive haemorrhage and blood volume expansion in anaesthetized cats. J Physiol. 1974;237:279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greenway CV, Oshiro G. Comparison of the effects of hepatic nerve stimulation on arterial flow, distribution of arterial and portal flows and blood content in the livers of anaesthetized cats and dogs. J Physiol. 1972;227:487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fallick C, Sobotka PA, Dunlap ME. Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation. Circ Heart Fail. 2011;4:669–675. [DOI] [PubMed] [Google Scholar]
- 18. Rothe CF. Mean circulatory filling pressure: its meaning and measurement. J Appl Physiol (1985). 1993;74:499–509. [DOI] [PubMed] [Google Scholar]
- 19. Thibodeau JT, Turer AT, Gualano SK, Ayers CR, Velez‐Martinez M, Mishkin JD, Patel PC, Mammen PP, Markham DW, Levine BD, Drazner MH. Characterization of a novel symptom of advanced heart failure: bendopnea. JACC Heart Fail. 2014;2:24–31. [DOI] [PubMed] [Google Scholar]
- 20. Rowell LB. Reflex control of regional circulations in humans. J Auton Nerv Syst. 1984;11:101–114. [DOI] [PubMed] [Google Scholar]
- 21. Birch DJ, Turmaine M, Boulos PB, Burnstock G. Sympathetic innervation of human mesenteric artery and vein. J Vasc Res. 2008;45:323–332. [DOI] [PubMed] [Google Scholar]
- 22. Barnes RJ, Bower EA, Rink TJ. Haemodynamic responses to stimulation of the splanchnic and cardiac sympathetic nerves in the anaesthetized cat. J Physiol. 1986;378:417–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greenway CV. Blockade of reflex venous capacitance responses in liver and spleen by hexamethonium, atropine, and surgical section. Can J Physiol Pharmacol. 1991;69:1284–1287. [DOI] [PubMed] [Google Scholar]
- 24. Carneiro JJ, Donald DE. Change in liver blood flow and blood content in dogs during direct and reflex alteration of hepatic sympathetic nerve activity. Circ Res. 1977;40:150–158. [DOI] [PubMed] [Google Scholar]
- 25. Greenway CV. Role of splanchnic venous system in overall cardiovascular homeostasis. Fed Proc. 1983;42:1678–1684. [PubMed] [Google Scholar]
- 26. Fudim M, Yalamuri S, Herbert JT, Liu PR, Patel MR, Sandler A. Raising the pressure: hemodynamic effects of splanchnic nerve stimulation. J Appl Physiol (1985). 2017;23:126–127. jap 00069 2017. [DOI] [PubMed] [Google Scholar]
- 27. Magder S. Volume and its relationship to cardiac output and venous return. Crit Care. 2016;20:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burkhoff D, Tyberg JV. Why does pulmonary venous pressure rise after onset of LV dysfunction: a theoretical analysis. Am J Physiol. 1993;265:H1819–H1828. [DOI] [PubMed] [Google Scholar]
- 29. Andersen MJ, Hwang SJ, Kane GC, Melenovsky V, Olson TP, Fetterly K, Borlaug BA. Enhanced pulmonary vasodilator reserve and abnormal right ventricular: pulmonary artery coupling in heart failure with preserved ejection fraction. Circ Heart Fail. 2015;8:542–550. [DOI] [PubMed] [Google Scholar]
- 30. Fujimoto N, Borlaug BA, Lewis GD, Hastings JL, Shafer KM, Bhella PS, Carrick‐Ranson G, Levine BD. Hemodynamic responses to rapid saline loading: the impact of age, sex, and heart failure. Circulation. 2013;127:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ogilvie RI, Zborowska‐Sluis D. Effect of chronic rapid ventricular pacing on total vascular capacitance. Circulation. 1992;85:1524–1530. [DOI] [PubMed] [Google Scholar]
- 32. Ogilvie RI, Zborowska‐Sluis D. Acute effect of rapid ventricular pacing and volume loading on total vascular capacitance. Can J Cardiol. 1992;8:1071–1078. [PubMed] [Google Scholar]
- 33. Rapaport E, Weisbart MH, Levine M. The splanchnic blood volume in congestive heart failure. Circulation. 1958;18:581–587. [DOI] [PubMed] [Google Scholar]
- 34. Balmain S, Padmanabhan N, Ferrell WR, Morton JJ, McMurray JJ. Differences in arterial compliance, microvascular function and venous capacitance between patients with heart failure and either preserved or reduced left ventricular systolic function. Eur J Heart Fail. 2007;9:865–871. [DOI] [PubMed] [Google Scholar]
- 35. Hainsworth R. Vascular capacitance: its control and importance. Rev Physiol Biochem Pharmacol. 1986;105:101–173. [DOI] [PubMed] [Google Scholar]
- 36. Floras JS. Sympathetic nervous system activation in human heart failure: clinical implications of an updated model. J Am Coll Cardiol. 2009;54:375–385. [DOI] [PubMed] [Google Scholar]
- 37. Creager MA, Hirsch AT, Dzau VJ, Nabel EG, Cutler SS, Colucci WS. Baroreflex regulation of regional blood flow in congestive heart failure. Am J Physiol. 1990;258:H1409–H1414. [DOI] [PubMed] [Google Scholar]
- 38. Verhaert D, Mullens W, Borowski A, Popovic ZB, Curtin RJ, Thomas JD, Tang WH. Right ventricular response to intensive medical therapy in advanced decompensated heart failure. Circ Heart Fail. 2010;3:340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Borlaug BA. Mechanisms of exercise intolerance in heart failure with preserved ejection fraction. Circ J. 2014;78:20–32. [DOI] [PubMed] [Google Scholar]
- 40. Borlaug BA, Nishimura RA, Sorajja P, Lam CS, Redfield MM. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail. 2010;3:588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burkhoff D, Maurer MS, Packer M. Heart failure with a normal ejection fraction: is it really a disorder of diastolic function? Circulation. 2003;107:656–658. [DOI] [PubMed] [Google Scholar]
- 42. Obokata M, Kane GC, Reddy YN, Olson TP, Melenovsky V, Borlaug BA. The role of diastolic stress testing in the evaluation for HFpEF: a simultaneous invasive‐echocardiographic study. Circulation. 2016;135:825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McAllister RM. Adaptations in control of blood flow with training: splanchnic and renal blood flows. Med Sci Sports Exerc. 1998;30:375–381. [DOI] [PubMed] [Google Scholar]
- 44. Burchell AE, Sobotka PA, Hart EC, Nightingale AK, Dunlap ME. Chemohypersensitivity and autonomic modulation of venous capacitance in the pathophysiology of acute decompensated heart failure. Curr Heart Fail Rep. 2013;10:139–146. [DOI] [PubMed] [Google Scholar]
- 45. Narkiewicz K, Pesek CA, van de Borne PJ, Kato M, Somers VK. Enhanced sympathetic and ventilatory responses to central chemoreflex activation in heart failure. Circulation. 1999;100:262–267. [DOI] [PubMed] [Google Scholar]
- 46. Chua TP, Ponikowski P, Webb‐Peploe K, Harrington D, Anker SD, Piepoli M, Coats AJ. Clinical characteristics of chronic heart failure patients with an augmented peripheral chemoreflex. Eur Heart J. 1997;18:480–486. [DOI] [PubMed] [Google Scholar]
- 47. Cutler MJ, Swift NM, Keller DM, Wasmund WL, Burk JR, Smith ML. Periods of intermittent hypoxic apnea can alter chemoreflex control of sympathetic nerve activity in humans. Am J Physiol Heart Circ Physiol. 2004;287:H2054–H2060. [DOI] [PubMed] [Google Scholar]
- 48. Niewinski P, Engelman ZJ, Fudim M, Tubek S, Paleczny B, Jankowska EA, Banasiak W, Sobotka PA, Ponikowski P. Clinical predictors and hemodynamic consequences of elevated peripheral chemosensitivity in optimally treated men with chronic systolic heart failure. J Card Fail. 2013;19:408–415. [DOI] [PubMed] [Google Scholar]
- 49. Ponikowski P, Chua TP, Anker SD, Francis DP, Doehner W, Banasiak W, Poole‐Wilson PA, Piepoli MF, Coats AJ. Peripheral chemoreceptor hypersensitivity: an ominous sign in patients with chronic heart failure. Circulation. 2001;104:544–549. [DOI] [PubMed] [Google Scholar]
- 50. Nazare Nunes Alves MJ, dos Santos MR, Nobre TS, Martinez DG, Pereira Barretto AC, Brum PC, Rondon MU, Middlekauff HR, Negrao CE. Mechanisms of blunted muscle vasodilation during peripheral chemoreceptor stimulation in heart failure patients. Hypertension. 2012;60:669–676. [DOI] [PubMed] [Google Scholar]
- 51. Ponikowski P, Chua TP, Piepoli M, Ondusova D, Webb‐Peploe K, Harrington D, Anker SD, Volterrani M, Colombo R, Mazzuero G, Giordano A, Coats AJ. Augmented peripheral chemosensitivity as a potential input to baroreflex impairment and autonomic imbalance in chronic heart failure. Circulation. 1997;96:2586–2594. [DOI] [PubMed] [Google Scholar]
- 52. Despas F, Lambert E, Vaccaro A, Labrunee M, Franchitto N, Lebrin M, Galinier M, Senard JM, Lambert G, Esler M, Pathak A. Peripheral chemoreflex activation contributes to sympathetic baroreflex impairment in chronic heart failure. J Hypertens. 2012;30:753–760. [DOI] [PubMed] [Google Scholar]
- 53. Funakoshi K, Hosokawa K, Kishi T, Ide T, Sunagawa K. Striking volume intolerance is induced by mimicking arterial baroreflex failure in normal left ventricular function. J Card Fail. 2014;20:53–59. [DOI] [PubMed] [Google Scholar]
- 54. Narkiewicz K, van de Borne PJ, Montano N, Dyken ME, Phillips BG, Somers VK. Contribution of tonic chemoreflex activation to sympathetic activity and blood pressure in patients with obstructive sleep apnea. Circulation. 1998;97:943–945. [DOI] [PubMed] [Google Scholar]
- 55. Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, Young JB, Tang WH. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Damman K, van Deursen VM, Navis G, Voors AA, van Veldhuisen DJ, Hillege HL. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J Am Coll Cardiol. 2009;53:582–588. [DOI] [PubMed] [Google Scholar]
- 57. Ammons WS, Koyama S, Manning JW. Neural and vascular interaction in renin response to graded renal nerve stimulation. Am J Physiol. 1982;242:R552–R562. [DOI] [PubMed] [Google Scholar]
- 58. Firth JD, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet. 1988;1:1033–1035. [DOI] [PubMed] [Google Scholar]
- 59. Koyama S, Nishida K, Terada N, Shiojima Y, Takeuchi T. Reflex renal vasoconstriction on portal vein distension. Jpn J Physiol. 1986;36:441–450. [DOI] [PubMed] [Google Scholar]
- 60. DiBona GF, Sawin LL. Role of renal nerves in sodium retention of cirrhosis and congestive heart failure. Am J Physiol. 1991;260:R298–R305. [DOI] [PubMed] [Google Scholar]
- 61. Rzouq F, Alahdab F, Olyaee M. New insight into volume overload and hepatorenal syndrome in cirrhosis, “the hepatorenal reflex hypothesis”. Am J Med Sci. 2014;348:244–248. [DOI] [PubMed] [Google Scholar]
- 62. Kostreva DR, Castaner A, Kampine JP. Reflex effects of hepatic baroreceptors on renal and cardiac sympathetic nerve activity. Am J Physiol. 1980;238:R390–R394. [DOI] [PubMed] [Google Scholar]
- 63. Hamza SM, Kaufman S. Role of spleen in integrated control of splanchnic vascular tone: physiology and pathophysiology. Can J Physiol Pharmacol. 2009;87:1–7. [DOI] [PubMed] [Google Scholar]
- 64. Solis‐Herruzo JA, Duran A, Favela V, Castellano G, Madrid JL, Munoz‐Yague MT, Morillas JD, Estenoz J. Effects of lumbar sympathetic block on kidney function in cirrhotic patients with hepatorenal syndrome. J Hepatol. 1987;5:167–173. [DOI] [PubMed] [Google Scholar]
- 65. Kouw PM, Kooman JP, Cheriex EC, Olthof CG, de Vries PM, Leunissen KM. Assessment of postdialysis dry weight: a comparison of techniques. J Am Soc Nephrol. 1993;4:98–104. [DOI] [PubMed] [Google Scholar]
- 66. Lauster F, Gerzer R, Weil J, Fulle HJ, Schiffl H. Assessment of dry body‐weight in haemodialysis patients by the biochemical marker cGMP. Nephrol Dial Transplant. 1990;5:356–361. [DOI] [PubMed] [Google Scholar]
- 67. Frank Peacock W, Soto KM. Current technique of fluid status assessment. Congest Heart Fail. 2010;16(suppl 1):S45–S51. [DOI] [PubMed] [Google Scholar]
- 68. Schmitt M, Blackman DJ, Middleton GW, Cockcroft JR, Frenneaux MP. Assessment of venous capacitance. Radionuclide plethysmography: methodology and research applications. Br J Clin Pharmacol. 2002;54:565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tang WH, Tong W. Measuring impedance in congestive heart failure: current options and clinical applications. Am Heart J. 2009;157:402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Engineer RS, Benoit JL, Hicks CW, Kolattukudy SJ, Burkhoff D, Peacock WF. Hemodynamic changes as a diagnostic tool in acute heart failure—a pilot study. Am J Emerg Med. 2012;30:174–180. [DOI] [PubMed] [Google Scholar]
- 71. Diedrich A, Biaggioni I. Segmental orthostatic fluid shifts. Clin Auton Res. 2004;14:146–147. [DOI] [PubMed] [Google Scholar]
- 72. Abelmann WH, Fareeduddin K. Increased tolerance of orthostatic stress in patients with heart disease. Am J Cardiol. 1969;23:354–363. [DOI] [PubMed] [Google Scholar]
- 73. Ramirez A, Abelmann WH. Hemodynamic effects of diuresis by ethacrynic acid in normal subjects and in patients with congestive heart failure. Arch Intern Med. 1968;121:320–327. [DOI] [PubMed] [Google Scholar]
- 74. Duke M, Herbert VD, Abelmann WH. Hemodynamic effects of blood transfusion in chronic anemia. N Engl J Med. 1964;271:975–980. [DOI] [PubMed] [Google Scholar]
- 75. Manyari DE, Wang Z, Cohen J, Tyberg JV. Assessment of the human splanchnic venous volume‐pressure relation using radionuclide plethysmography. Effect of nitroglycerin. Circulation. 1993;87:1142–1151. [DOI] [PubMed] [Google Scholar]
- 76. Wang SY, Manyari DE, Scott‐Douglas N, Smiseth OA, Smith ER, Tyberg JV. Splanchnic venous pressure‐volume relation during experimental acute ischemic heart failure. Differential effects of hydralazine, enalaprilat, and nitroglycerin. Circulation. 1995;91:1205–1212. [DOI] [PubMed] [Google Scholar]
- 77. Cody RJ, Franklin KW, Kluger J, Laragh JH. Mechanisms governing the postural response and baroreceptor abnormalities in chronic congestive heart failure: effects of acute and long‐term converting‐enzyme inhibition. Circulation. 1982;66:135–142. [DOI] [PubMed] [Google Scholar]
- 78. Okamoto LE, Gamboa A, Shibao C, Arnold AC, Celedonio JE, Diedrich A, Farley G, Paranjape SY, Biaggioni I. Abstract 018: sympathetic contribution to obesity hypertension: differential hemodynamic mechanisms between prehypertension and hypertension. Hypertension. 2014;64:A018. [Google Scholar]
- 79. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C, Levine GN, O'Gara PT, Halperin JL, Al‐Khatib SM, Birtcher KK, Brindis RG, Cigarroa JE, Curtis LH, Fleisher LA, Gentile F, Gidding S, Hlatky MA, Ikonomidis J, Joglar J, Pressler SJ, Wijeysundera DN. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Card Fail. 2017;Apr 25. pii: S1071‐9164(17)30107‐0. [DOI] [PubMed] [Google Scholar]
- 80. Cohn JN, Pfeffer MA, Rouleau J, Sharpe N, Swedberg K, Straub M, Wiltse C, Wright TJ; Investigators M . Adverse mortality effect of central sympathetic inhibition with sustained‐release moxonidine in patients with heart failure (MOXCON). Eur J Heart Fail. 2003;5:659–667. [DOI] [PubMed] [Google Scholar]
- 81. Swedberg K, Bristow MR, Cohn JN, Dargie H, Straub M, Wiltse C, Wright TJ; Moxonidine S and Efficacy I . Effects of sustained‐release moxonidine, an imidazoline agonist, on plasma norepinephrine in patients with chronic heart failure. Circulation. 2002;105:1797–1803. [DOI] [PubMed] [Google Scholar]
- 82. Fujita Y. Splanchnic circulation following coeliac plexus block. Acta Anaesthesiol Scand. 1988;32:323–327. [DOI] [PubMed] [Google Scholar]
- 83. Niewinski P, Janczak D, Rucinski A, Jazwiec P, Sobotka PA, Engelman ZJ, Fudim M, Tubek S, Jankowska EA, Banasiak W, Hart EC, Paton JF, Ponikowski P. Carotid body removal for treatment of chronic systolic heart failure. Int J Cardiol. 2013;168:2506–2509. [DOI] [PubMed] [Google Scholar]
- 84. Abraham WT, Zile MR, Weaver FA, Butter C, Ducharme A, Halbach M, Klug D, Lovett EG, Muller‐Ehmsen J, Schafer JE, Senni M, Swarup V, Wachter R, Little WC. Baroreflex activation therapy for the treatment of heart failure with a reduced ejection fraction. JACC Heart Fail. 2015;3:487–496. [DOI] [PubMed] [Google Scholar]
- 85. Bhatt DL, Kandzari DE, O'Neill WW, D'Agostino R, Flack JM, Katzen BT, Leon MB, Liu M, Mauri L, Negoita M, Cohen SA, Oparil S, Rocha‐Singh K, Townsend RR, Bakris GL; Investigators SH‐ . A controlled trial of renal denervation for resistant hypertension. N Engl J Med. 2014;370:1393–1401. [DOI] [PubMed] [Google Scholar]
- 86. Patel HC, Rosen SD, Hayward C, Vassiliou V, Smith GC, Wage RR, Bailey J, Rajani R, Lindsay AC, Pennell DJ, Underwood SR, Prasad SK, Mohiaddin R, Gibbs JS, Lyon AR, Di Mario C. Renal denervation in heart failure with preserved ejection fraction (RDT‐PEF): a randomized controlled trial. Eur J Heart Fail. 2016;18:703–712. [DOI] [PubMed] [Google Scholar]
- 87. Costanzo MR, Guglin ME, Saltzberg MT, Jessup ML, Bart BA, Teerlink JR, Jaski BE, Fang JC, Feller ED, Haas GJ, Anderson AS, Schollmeyer MP, Sobotka PA; Investigators UT . Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J Am Coll Cardiol. 2007;49:675–683. [DOI] [PubMed] [Google Scholar]
- 88. Mullens W, Abrahams Z, Francis GS, Taylor DO, Starling RC, Tang WH. Prompt reduction in intra‐abdominal pressure following large‐volume mechanical fluid removal improves renal insufficiency in refractory decompensated heart failure. J Card Fail. 2008;14:508–514. [DOI] [PubMed] [Google Scholar]
- 89. Niijima A. The Reflex Effects of Hepatic and Mesenteric Afferents. In Zucker IH, Gilmore JP. (eds): Reflex Control of the Circulation. Boca Raton: CRC Press; 1991, pp 529–549. [Google Scholar]
- 90. Marenzi G, Grazi S, Giraldi F, Lauri G, Perego G, Guazzi M, Salvioni A, Guazzi MD. Interrelation of humoral factors, hemodynamics, and fluid and salt metabolism in congestive heart failure: effects of extracorporeal ultrafiltration. Am J Med. 1993;94:49–56. [DOI] [PubMed] [Google Scholar]
- 91. Malbrain ML, Chiumello D, Pelosi P, Wilmer A, Brienza N, Malcangi V, Bihari D, Innes R, Cohen J, Singer P, Japiassu A, Kurtop E, De Keulenaer BL, Daelemans R, Del Turco M, Cosimini P, Ranieri M, Jacquet L, Laterre PF, Gattinoni L. Prevalence of intra‐abdominal hypertension in critically ill patients: a multicentre epidemiological study. Intensive Care Med. 2004;30:822–829. [DOI] [PubMed] [Google Scholar]
- 92. Mullens W, Abrahams Z, Skouri HN, Francis GS, Taylor DO, Starling RC, Paganini E, Tang WH. Elevated intra‐abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol. 2008;51:300–306. [DOI] [PubMed] [Google Scholar]