

Abstract
A method is introduced that permits direct observation of the rates at which backbone amide hydrogens become protected from solvent exchange after rapidly dropping the hydrostatic pressure inside the NMR sample cell from denaturing (2.5 kbar) to native (1 bar) conditions. The method is demonstrated for a pressure-sensitized ubiquitin variant that contains two Val to Ala mutations. Increased protection against hydrogen exchange with solvent is monitored as a function of time during the folding process. Results for 53 backbone amides show narrow clustering with protection occurring with a time constant of ca 85 ms, but slower protection is observed around a reverse turn near the C-terminus of the protein. Remarkably, the native NMR spectrum returns with this slower time constant of ca 150 ms, indicating that the almost fully folded protein retains molten globule characteristics with severe NMR line broadening until the final hydrogen bonds are formed. Prior to crossing the transition state barrier, hydrogen exchange protection factors are close to unity, but with slightly elevated values in the β1-β2 hairpin, previously shown to be already lowly populated in the urea-denatured state.
Graphical Abstract
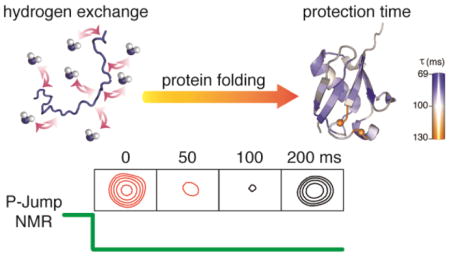
Rapid mixing experiments, where proteins are switched from a denaturing to a folding environment, have long been coupled with studies of the rates at which amide protons exchange with solvent deuterons, or vice versa, to capture information on the time sequence involved in hydrogen bond (H-bond) formation during the protein folding process.1,2 In these so called quenched-flow hydrogen exchange (HX) experiments, samples are harvested and subsequently analyzed by NMR or mass spectrometry (MS) in order to determine the degree to which a given residue (NMR) or group of residues (MS) has undergone hydrogen exchange with solvent during the mixing process.3 These experiments have provided valuable insights into the time course at which individual amides become engaged in H-bond formation, and thereby protected from exchange with solvent, revealing remarkable details on kinetic intermediates that can occur during the folding pathway.2,4
Here, we introduce a more direct NMR method for monitoring the time course of HX protection during the folding process. By unfolding the protein under high hydrostatic pressure inside the NMR sample cell, followed by a rapid drop in pressure to initiate the protein folding process, the change in HX rate can be monitored directly. Conceptually, these experiments are analogous to pressure-jump fluorescence measurements,5 but the combination with 2D NMR spectroscopy provides a separate spectroscopic probe at every backbone amide moiety. Here, we provide the first such example of following the switch between a fully pressure-denatured state to the fully folded state with millisecond resolution, using the change in HX rate during the folding process as the experimentally probed parameter. We note, however, that it is also feasible to probe other parameters, including NOEs, chemical shifts, and relaxation rates.
NMR spectroscopy has been used extensively to explore proteins under high hydrostatic pressure, and has provided site-specific information on folding intermediates and thermodynamic parameters.6 Most of these studies were carried out under static pressure, but more recent studies have also utilized pressure jumps to explore kinetic behavior.7 Here, we show that rapidly and repeatedly switching the pressure in the NMR sample cell between 2.5 kbar and 1 bar allows atomic-level characterization of the protein at millisecond resolution during the actual folding process.
We measure τf, the mean time between the pressure drop (initiation of protein folding) and the time at which individual backbone amides become protected from HX. The ability to denature a protein at high pressure is critical to these experiments and requires a negative change in volume between the folded and unfolded states. This mechanism can be magnified by introducing small hydrophobic cavities in the interior of the protein through mutagenesis. Here, two Val to Ala mutations (V17A/V26A) are used to lower the mid-point of ubiquitin pressure denaturation from >5 kbar to ca 1.4 kbar at 298 K. Very similar values of the structurally sensitive 13Cα and 13C′ chemical shifts indicate that the structure of folded ubiquitin is minimally affected by these mutations (Figure S1).
Following the rapid drop in pressure from 2.5 kbar to 1 bar, and a concomitant 3 K drop in temperature due to adiabatic solvent expansion, the return of the pressure-denatured protein to its native, folded state can be monitored by simply recording 1D NMR spectra (Figure 1). A plot of the intensity of the most downfield shifted amides (I13 and L67) as a function of refolding time shows bi-exponential behavior with about two thirds refolding with a time constant of ca 150 ms, and one third refolding more than an order of magnitude slower (Figure 1C). The same time constants apply to all other well resolved amide resonances, indicating that the I13 and L67 intensities are representative of the final, native state of the protein. The slowly recovering component appears attributable to an off-pathway oligomeric intermediate (see below) and contributes minimally to our measurements of HX protection, which only observe signals from proteins that fold on the faster time scale.
Figure 1.

Recovery of the ubiquitin amide signals after dropping the pressure from 2.5 kbar to 1 bar. Sample: 0.3 mM 15N/13C/2H V17A/V26A ubiquitin in 250 μL H2O/D2O (93/7%), 25 mM phosphate buffer, pH 7.5, 295 K (at 1 bar). (A,B) 1D NMR spectra recorded at different times after the pressure drop. A larger set of such spectra is shown in Figure S6. (C) Plot of the ratio of the time-dependent intensity of the two most downfield shifted amides (I13 and L67) over their equilibrium value (measured 10 s after pressure drop). The 0.04 offset in the exponential fits is the fraction of protein, measured in a separate high S/N HSQC spectrum, that remains folded at 2.5 kbar and 298 K.
In the fully unfolded state, the intrinsic HX rates (kintr) depend strongly on residue type and that of the immediate neighbors.8 Values of kintr at 2.5 kbar were measured using 0.3 mM uniformly 13C/15N/2H-enriched ubiquitin. Perdeuteration avoided any complications from potential NOE transfer of magnetization from Hα protons, and the absence of NOE effects through rapidly exchanging hydroxyl protons was verified by carrying out measurements at three pH values: 6.4, 7.1, and 7.5 (Table S1).
To measure HX with bulk solvent during the folding process, we essentially follow the procedure of Grzesiek and Fitzkee.9 In our experiments, a duration Tex = 125 ms, where water magnetization is either inverted or left unchanged, precedes the start of the 2D 1H-15N heteronuclear single quantum correlation (HSQC) experiment. A refolding delay, τr, separates the start of the Tex period from the time when the pressure drops to 1 bar (Figure 2). Each pulse sequence is executed twice: once with a water-selective inversion pulse and a subsequent water-flip-back pulse immediately prior to the start of the HSQC, and a second, reference experiment where these water inversion pulses are not applied. The ratio between corresponding intensities in the two HSQC spectra then provides a measure for the fraction of any amide hydrogen in the ensemble that exchanged with solvent during Tex. Amides that rapidly exchange with solvent (kex ≥ ~1/Tex) will appear inverted relative to the reference spectrum, while those that exchange slowly will simply be attenuated (Figure 3; Figure S2).
Figure 2.

Schematic diagram of the pressure jump NMR experiment used to measure HX rates during protein folding. Hydrostatic pressure is marked in green. G marks a weak magnetic field gradient (0.5 G/cm), needed to prevent radiation damping of the inverted H2O magnetization. HSQC refers to the standard, gradient-enhanced HSQC experiment,10 including water flip-back to +z. A long delay (12 s at 298 K and 2.5 kbar) between scans was used to unfold the protein prior to the pressure drop.
Figure 3.

Refolding time (τr) dependence of NMR intensities. (A–D) Sections of ubiquitin 2D HSQC NMR spectra (with application of the water inversion pulses), measured at various τr values. Sample conditions are as in Figure 1. The sample temperature was set at 298 K during the high pressure (2.5 kbar) period (12 s). Full HSQC spectra are shown in Figure S2. (E,F) Fits of the Sf(τr,Tex) to Ae−τr/τf + B, for T7, S65, Q40, and Q41. A full set of curves is shown in Figure S3.
Using the experiment of Figure 2 (τr = 0), the difference in intensity of a given amide signal, recorded without and with water inversion, normalized to the signal without water inversion, Sf(Tex), was derived (SI eq 7) by extending the derivations of Grzesiek and Fitzkee9 to include non-equilibrium exchange.11 In the limit that R1u (unfolded state R1), R1f (folded state R1), R1W (water R1), and kf (HX rate in the folded state) are all much smaller than 1/Tex, Sf(Tex) simplifies to:
| (1) |
where Δf denotes the difference in H2O magnetization before and after the selective 180° pulse (Δf ≈1.8); λ is the inverse of τf, the mean time at which a given amide becomes HX-protected; ku is the HX rate at 1 bar, prior to crossing the transition state; and Tex is the duration that the water remains inverted. Note that extracting λ from eq 1 requires knowledge of ku, which may well differ from kintr, the intrinsic, random coil exchange rate (see below).
Nevertheless, the scheme of Figure 2 can be used to detect τf, provided that ku » kf. By inserting a refolding delay, τr, prior to the first water inversion pulse, a fraction e−τr/τf of the amides remain in the HX-unprotected state prior to the first water inversion pulse. Sf therefore scales as Sf(τr,Tex) = Sf(0,Tex)exp(−τr/τf) and since it must approach the value obtained at 1 bar for τr → ∞ Sf(τr,Tex) is well described by the function Ae−τr/τf + B.
Spectra were collected in an interleaved manner for six τr values, ranging from 0 to 400 ms. For τr = 0, a number of amide signals appear inverted due to rapid exchange with solvent during the Tex period when water is inverted, whereas others are strongly attenuated (Figure 3A). However, when HX is impeded by protein folding during the τr delay, attenuation due to HX with solvent becomes progressively less (Figure 3B–D), and Sf(τr,Tex) follows the expected Ae−τr/τf + B dependence (Figure 3E,F; Figure S3). Residues that are not significantly protected in the folded protein (e.g., G10-T12) show very little change in intensity when τr is increased.
Values for τf were extracted for 53 amides whose HX rates differ substantially between the unfolded and folded states. Time constants for the entire β-sheet (86±8 ms; N=18) and the long α-helix (88±6 ms; N=11) show narrow clustering (Figure 4). This highly homogeneous set of time constants, gives it the appearance of being a classic two-state folder, in agreement with previous denaturant-based studies.12,13 However, the loop/turn region (I61-T66) shows longer and more heterogeneous time constants, most notably for I61 (τf =126 ms) and S65 (τf =131 ms). The amide of I61 shares an H-bond to the carbonyl of L56, and S65 H-bonds to Q62, stabilizing a type-II β-turn. These observations indicate that protein folding is not fully complete until the final type II β-turn, centered at residues K63-E64, has formed. Remarkably, prior to the formation of this turn, the NMR spectrum of the entire protein is impacted by dynamic processes that cause severe line broadening, essentially rendering it invisible, until this last native element has formed (Figure 1). This penultimate state during the folding process therefore resembles what is sometimes called a “dry molten globule”, where the vast majority of backbone H-bonds are properly in place, but dynamic conformational exchange prevents coalescence to a sharply defined folded structure.14 The NMR resonances are exceptionally sensitive to such dynamic processes, effectively broadening them to an extent that they escape detection, in particular when recording 2D HSQC spectra that involve fixed magnetization transfer delays.
Figure 4.

Protection times, τf = 1/λ, for HX during protein folding. Fitted time constants as a function of residue number in the V17A/V26A ubiquitin mutant at 295 K. Error bars reflect the uncertainty as obtained from the covariance matrix of the fit. A color map of the rates on the ubiquitin ribbon diagram highlights spatial proximity of I61 and S65 (Figure S4).
Besides the fast folding component, the double-mutant ubiquitin also shows a much slower folding component (Figure 1): about two thirds fold at a relatively rapid rate of ca 7 s−1, whereas one third folds at a rate that is more than an order of magnitude slower and is not significantly probed by our HX experiments. A similar biphasic recovery in ubiquitin has previously been attributed to the fraction of unfolded chains for which one or more of the three Pro residues starts in the cis conformation.4 However, the slower time constant may also be caused by an oligomeric off-pathway intermediate.13 Indeed, a pronounced increase in the fraction of slowly recovering protein is observed when the sample concentration is raised from 0.3 mM to 1 mM, confirming the presence of an oligomeric species. This off-pathway oligomerization therefore limits the concentration at which our pressure jump NMR experiments can be carried out. Alternatively, working at a pH where the protein carries a large net charge, or the use of a small volume fraction of a mild organic co-solvent such as ethanol, can mitigate the self-association process.15
Our results provide the residue-specific time constants at which the HX rate switches from that of the unfolded to that of the folded state. Their measurements therefore do not require a high degree of protection in the folded state, and indeed time constants are also obtained for residues that do not engage in H-bonding in the final structure, simply reflecting the change in accessibility to OH− catalyzed hydrogen exchange. For example, the solvent-exposed amides of the β2 strand yield time constants very similar to their immediate H-bonded neighboring residues (Figure 4; Figure S4). However, the smaller difference in HX rate between the folded and unfolded states makes their uncertainty somewhat larger. The pressure jump approach therefore provides access to a larger number of probes than can be evaluated by quenched-flow rapid mixing experiments.
Our pressure jump experiment also yields information on the average degree of HX protection prior to crossing the transition state: to a first approximation, for τr=0 and kf, R1W, R1u, and R1f « 1/Tex, the HX rate of the protein at 1 bar, ku, can be extracted from eq 1, or from SI eq 7 without these assumptions. Comparison of ku with the intrinsic HX rates, kintr, measured at high pressure in the fully unfolded state, shows a close correlation with a slope of ca 1 (Figure S5), indicative of essentially no HX protection prior to the point where the protein crosses the transition state. A possible exception to this observation is seen for residues in the first two β-strands, which account for five of the six highest kintr/ku ratios, ranging from 1.75 to 1.85 (Figure S5B). A caveat in determining these ratios is that kintr values were measured at 2.5 kbar, and conversion to 1 bar requires scaling to account for the pressure dependence of the phosphate buffer pH, lowering it by approximately 1 unit, i.e. 10-fold lower [OH−]. This effect is largely, but not completely, offset by the five-fold increased water dissociation at 2.5 kbar which, combined with the pH effect, results in a net lowering of [OH−], and therefore HX rates, by about 2-fold relative to 1 bar.16
The small degree of HX protection seen for amides involved in formation of the first β1–β2 hairpin is consistent with the transient presence of this structural element even in the urea-denatured protein, albeit to substantially less than 25%.17 Its formation is expected to be essentially instantaneous (~6 μs) on the time scale of our NMR experiment, judging by measurements on the analogous hairpin of the structurally homologous GB1 protein.18 Regardless, our observation indicates that in our ubiquitin mutant, prior to crossing the transition state, dynamic H-bonds are remarkably lowly populated, in agreement with prior work by Gladwin and Evans.19 We are currently exploring NOEs and chemical shifts, which are more precise than HX protection, to unambiguously identify such transient structural elements.
Supplementary Material
Acknowledgments
We thank J. Ying, J.L. Baber, Y. Shen, J. Anderson, B. Howder, J. Louis, and D. Garrett for technical support, and W.A. Eaton, G.M. Clore, and J. Roche for insightful discussions. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and by the Intramural Antiviral Target Program of the Office of the Director, NIH.
Footnotes
Notes
The authors declare no competing financial interests.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and additional data (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Englander SW, Mayne L. Annu Rev Biophys Biomol Struct. 1992;21:243–265. doi: 10.1146/annurev.bb.21.060192.001331. [DOI] [PubMed] [Google Scholar]
- 2.Dyson HJ, Wright PE. Acc Chem Res. 2017;50:105–111. doi: 10.1021/acs.accounts.6b00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miranker A, Robinson CV, Radford SE, Aplin RT, Dobson CM. Science. 1993;262:896–900. doi: 10.1126/science.8235611. [DOI] [PubMed] [Google Scholar]; Bai Y, Sosnick TR, Mayne L, Englander SW. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]; Baldwin RL. Curr Opin Struct Biol. 1993;3:84–91. [Google Scholar]
- 4.Briggs MS, Roder H. Proc Natl Acad Sci U S A. 1992;89:2017–2021. doi: 10.1073/pnas.89.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panick G, Vidugiris GJA, Malessa R, Rapp G, Winter R, Royer CA. Biochemistry. 1999;38:4157–4164. doi: 10.1021/bi982608e. [DOI] [PubMed] [Google Scholar]; Liu YX, Prigozhin MB, Schulten K, Gruebele M. J Am Chem Soc. 2014;136:4265–4272. doi: 10.1021/ja412639u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samarasinghe SD, Campbell DM, Jonas A, Jonas J. Biochemistry. 1992;31:7773–7778. doi: 10.1021/bi00149a005. [DOI] [PubMed] [Google Scholar]; Royer CA, Hinck AP, Loh SN, Prehoda KE, Peng XD, Jonas J, Markley JL. Biochemistry. 1993;32:5222–5232. doi: 10.1021/bi00070a034. [DOI] [PubMed] [Google Scholar]; Yamaguchi T, Yamada H, Akasaka K. J Mol Biol. 1995;250:689–694. doi: 10.1006/jmbi.1995.0408. [DOI] [PubMed] [Google Scholar]; Nash DP, Jonas J. Biochem Biophys Res Commun. 1997;238:289–291. doi: 10.1006/bbrc.1997.7308. [DOI] [PubMed] [Google Scholar]; Inoue K, Yamada H, Akasaka K, Hermann C, Kremer W, Maurer T, Doker R, Kalbitzer HR. Nature Structural Biology. 2000;7:547–550. doi: 10.1038/76764. [DOI] [PubMed] [Google Scholar]; Silva JL, Foguel D, Royer CA. Trends Biochem Sci. 2001;26:612–618. doi: 10.1016/s0968-0004(01)01949-1. [DOI] [PubMed] [Google Scholar]; Kranz JK, Flynn PF, Fuentes EJ, Wand AJ. Biochemistry. 2002;41:2599–2608. doi: 10.1021/bi011818f. [DOI] [PubMed] [Google Scholar]
- 7.Kremer W, Arnold M, Munte CE, Hartl R, Erlach MB, Koehler J, Meier A, Kalbitzer HR. J Am Chem Soc. 2011;133:13646–13651. doi: 10.1021/ja2050698. [DOI] [PubMed] [Google Scholar]; Roche J, Dellarole M, Caro JA, Norberto DR, Garcia AE, Garcia-Moreno B, Roumestand C, Royer CA. J Am Chem Soc. 2013;135:14610–14618. doi: 10.1021/ja406682e. [DOI] [PubMed] [Google Scholar]
- 8.Bai Y, Milne JS, Englander SW. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grzesiek S, Bax A. J Biomol NMR. 1993;3:627–638. doi: 10.1007/BF00198368. [DOI] [PubMed] [Google Scholar]; Fitzkee NC, Torchia DA, Bax A. Protein Sci. 2011;20:500–512. doi: 10.1002/pro.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kay LE, Keifer P, Saarinen T. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 11.Helgstrand M, Hard T, Allard P. J Biomol NMR. 2000;18:49–63. doi: 10.1023/a:1008309220156. [DOI] [PubMed] [Google Scholar]
- 12.Krantz BA, Sosnick TR. Biochemistry. 2000;39:11696–11701. doi: 10.1021/bi000792+. [DOI] [PubMed] [Google Scholar]
- 13.Went HM, Benitez-Cardoza CG, Jackson SE. FEBS Lett. 2004;567:333–338. doi: 10.1016/j.febslet.2004.04.089. [DOI] [PubMed] [Google Scholar]
- 14.Shakhnovich EI, Finkelstein AV. Biopolymers. 1989;28:1667–1680. doi: 10.1002/bip.360281003. [DOI] [PubMed] [Google Scholar]; Baldwin RL, Frieden C, Rose GD. Proteins. 2010;78:2725–2737. doi: 10.1002/prot.22803. [DOI] [PMC free article] [PubMed] [Google Scholar]; Neumaier S, Kiefhaber T. J Mol Biol. 2014;426:2520–2528. doi: 10.1016/j.jmb.2014.04.022. [DOI] [PubMed] [Google Scholar]
- 15.Eliezer D, Yao J, Dyson HJ, Wright PE. Nature Structural Biology. 1998;5:148–155. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]; Redfield C. Methods. 2004;34:121–132. doi: 10.1016/j.ymeth.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Peng XD, Jonas A, Jonas J. Biochemistry. 1995;34:8631–8641. doi: 10.1021/bi00027a012. [DOI] [PubMed] [Google Scholar]; Fuentes EJ, Wand AJ. Biochemistry. 1998;37:9877–9883. doi: 10.1021/bi980894o. [DOI] [PubMed] [Google Scholar]
- 17.Meier S, Strohmeier M, Blackledge M, Grzesiek S. J Am Chem Soc. 2007;129:754–755. doi: 10.1021/ja067522k. [DOI] [PubMed] [Google Scholar]
- 18.Munoz V, Thompson PA, Hofrichter J, Eaton WA. Nature. 1997;390:196–199. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 19.Gladwin ST, Evans PA. Folding & Design. 1996;1:407–417. doi: 10.1016/S1359-0278(96)00057-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.