

Abstract
Although the terms “excessive reactive oxygen species (ROS)” and “oxidative stress” are widely used, the implications of oxidative stress are often misunderstood. ROS are not a single species but a variety of compounds, each with unique biochemical properties and abilities to react with biomolecules. ROS cause activation of growth signals through thiol oxidation and may lead to DNA damage at elevated levels. In this review, we first discuss a conceptual framework for the interplay of ROS and antioxidants. This review then describes ROS signaling using FLT3-mediated growth signaling as an example. We then focus on ROS-mediated DNA damage. High concentrations of ROS result in various DNA lesions, including 8-oxo-7,8-dihydro-guanine, oxazolone, DNA–protein cross-links, and hydantoins, that have unique biological impacts. Here we delve into the biochemistry of nine well-characterized DNA lesions. Within each lesion, the types of repair mechanisms, the mutations induced, and their effects on transcription and replication are discussed. Finally, this review will discuss biochemically inspired implications for cancer therapy. Several teams have put forward designs to harness the excessive ROS and the burdened DNA repair systems of tumor cells for treating cancer. We discuss inhibition of the antioxidant system, the targeting of DNA repair, and ROS-activated prodrugs.
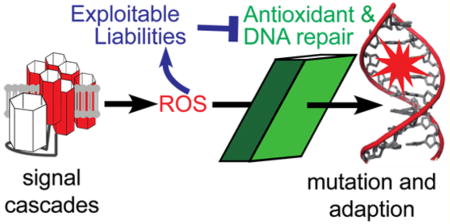
Graphical Abstract
1. REACTIVE OXYGEN SPECIES: OPPOSING ROLES
This review will give the reader a broad overview of reactive oxygen species (ROS) biochemistry. The goal is not to be comprehensive but instead to give concrete examples to serve as a basis for understanding the effects and impacts of ROS within a cell. The major forms of reactive oxygen species (ROS) that we will discuss in this review are superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (HO•). The biochemical and chemical complexity of ROS is often underappreciated.1 Much of this underappreciation results from early research on ROS. Hydrogen peroxide was first produced in the early 1800s and was recognized as a disinfectant that functions by oxidatively damaging biomolecules.2 Hydrogen peroxide is a commonly used ingredient in toothpaste, hair products, bleach-free cleaners, and other household products.3 Because of the ability of hydrogen peroxide to disinfect, it is widely thought to be toxic and thus was long assumed to play no role in “normal” cellular physiology.
In 1933, the existence of the superoxide radical, based on the theory of quantum mechanics, was proposed.4 The discovery of superoxide dismutase (SOD) resulted in the realization that ROS had biological functions.5 Upon protonation, superoxide decomposes to more powerful ROS forms that are highly toxic.6 The hydroxyl radical is a potent oxidizing agent, with a redox potential of 2.40 V. In 1934, Haber and Weiss noted that the highly reactive HO• could be generated from an interaction between O2•− and the less reactive H2O2.7 In biological systems, the iron-catalyzed Haber–Weiss reaction, which makes use of Fenton chemistry, is now considered to be the major mechanism by which HO• is generated.8 In the 1970s and 1980s, two discoveries confirmed that ROS play both positive and negative roles in biology. First, it was shown that phagocytes purposefully produce superoxide via NOX enzymes (NADPH oxidases); this superoxide is important in the immune response against infection.9 Second, signaling peptides that induce production of ROS within cells were characterized. These findings demonstrate the dichotomy that ROS is a deleterious toxin and a beneficial secondary messenger.
2. A CONCEPTUAL FRAMEWORK FOR CELLULAR ROS
Before we enter into a discussion of the chemistry and biochemistry of ROS, a conceptual framework for its effects on cells is needed. Shown in Figure 1 are the three major forms of ROS produced in cells. The differently colored x-axes represent the concentration of each ROS, with OH• having the lowest concentration and narrowest range because it is the most deleterious. Hydrogen peroxide has the highest concentration and allowable range. It has been shown that picomolar concentrations of O2•− are lethal, whereas micromolar concentrations of H2O2 can be tolerated.10 Each ROS is converted to other forms as shown by the gray arrows. Because ROS have essential biochemical functions, cells possess an antioxidant capacity (Figure 1, black arrows) to ensure that the ROS concentration is maintained within a window that allows for these functions without resulting in cytotoxicity (Figure 1, purple region). Antioxidant capacity is comprised of small-molecule antioxidants like glutathione,11 enzymatic antioxidants like catalase,12 and other enzymes that deal with the negative effects of ROS like DNA repair proteins.13 Cells can alter both the ROS levels and/or antioxidant capacity to compensate for different biochemical situations. For example, during tissue repair, binding of cytokine-transforming growth factor β1 to its receptor elevates ROS levels14 while another receptor, FMS-like tyrosine kinase 3 (FLT3) receptor,15 generates ROS during signaling for differentiation. Thus, ROS play a large role in many signaling pathways.
Figure 1.

ROS production and antioxidant capacity of cells. HO• (blue), O2•− (red), and H2O2 (green) levels and cellular antioxidant (black) capacities vary by cell and by biochemical event. There can be three regions for the cell, including normal, damaged, and cell death. HO• is very reactive, while O2•− is more problematic than H2O2. Superoxide is converted to H2O2 by superoxide dismutase (SOD) enzymes and to HO• by Fenton reactions. Cells can display a range of ROS concentrations and still be within the normal range.
If ROS levels are too high and disrupt the balance of ROS, then biomolecular damage ensues. Slightly above the normal concentration range, ROS cause biomolecular damage but not cell death (Figure 1, light purple region). The hydroxyl radical, derived from H2O2 via Fenton reactions or from O2•− at low pH, causes DNA and other biomolecular damage (Figure 1, red region). Too little ROS is also problematic as correct signaling and protein folding do not occur.16 Above thresholds that depend on the species, high concentrations of ROS overwhelm the antioxidant capacity of a cell and lead to cell death. Thus, ROS are beneficial within a narrow concentration range that differs depending on the cell type and on the environmental conditions. Importantly, a ROS concentration causing biomolecular damage to one cell type may be within the normal range for another cell type that has more antioxidant capacity. Therefore, it is the balance of ROS and antioxidant capacity that needs to be derived. The recognition of this phenomenon has led to a revision in the definition of the term “oxidative stress”. The definition now takes into account the fact that this type of stress occurs only in the range of ROS concentration that results in damage to a cell. Thus, oxidative stress is a state in which there is a disruption in ROS signaling, not just an elevation of the ROS concentration.17 This review strives to highlight the recent major focus of research and does not include examples of deleterious protein oxidation18 or DNA modifications that lead to signaling.19 With this conceptual framework, we will begin to delve into how changes in ROS concentration lead to biochemical machinery, first by analyzing how ROS induce signal changes and then by examining the impacts in DNA lesions.
3. ROS MODULATION OF SIGNAL CASCADES
The chemistry of superoxide and hydrogen peroxide is essential for inducing changes in cell signaling. Both superoxide and hydrogen peroxide can cause a myriad of modifications to proteins (Figure 2). Lipid oxidation will not be covered in this review.20,21 The major target of superoxide and hydrogen peroxide is cysteine. Reaction of cysteine on proteins forms sulfenic acids or disulfides, because thiol oxidation is common (Eo ∼ −0.25 V in buffered solutions).22 Other oxidizable amino acids include methionine, tyrosine, and histidine.23
Figure 2.

Protein modification by ROS. Superoxide (path A) can oxidize cysteine thiols within proteins, generating radical sulfenic forms that are converted to a disulfide and regenerate superoxide. Termination via dismutation leads to hydrogen peroxide (path D). Hydrogen peroxide reacts with thiols to directly form sulfenic acid and disulfide forms (path B). Protonation of superoxide leads to Fenton reactions (path C).
Superoxide is a radical, and as such, its chemistry involves formation, propagation, and termination: when O2•− is added to a thiol or a double bond, another radical is created. On proteins, O2•− reacts with a cysteine thiol to form a protein-SO•.24 This radical may react with a proximal thiol to produce a disulfide and re-form O2•− (Figure 2, path A), creating a cycle in which one O2•− can form multiple disulfides. Alternatively, protonation of O2•− (pKa ∼ 4.9) at low pH leads to OH• as shown on path C of Figure 2.25 O2•− propagation ceases when metal quenching or diradical quenching reactions occur. O2•−can also be terminated via dismutation to generate H2O2 (Figure 2, path D). For biological processes, the reaction of H2O2 with cysteine leads to a sulfenic acid that can be converted to a disulfide as shown in path B of Figure 2. The oxidative modifications of amino acids may alter protein folding and activity. The reactions to form disulfides may be reversed by exchange with thioredoxins.26 Finally, H2O2 can be converted to more potent hydroxyl radicals by the Fenton reaction.27
In the past two decades, it has become clear that ROS are important signaling molecules.28,29 In a variety of cells, cytokine treatment stimulates production of ROS, and the generation of H2O2 is a common signaling event in response to growth factors.30 For example, in murine embryo fibroblasts, TGFβ1 binding causes an increased level of expression of NOX4 and O2•− generation. The O2•− is converted to H2O2 by superoxide dismutase, leading to oxidation of the nuclear phosphatase MAPK phosphatase 1 (DUSP1) and sustained activation of JNK-mediated signaling.31
Here we will detail a single cascade critical in hematopoietic stem cell renewal and differentiation, to show the beneficial role of ROS and then briefly highlight other known downstream targets. When levels of ROS in stem cells are low, the cells remain in a state of self-renewal, whereas high ROS levels promote differentiation and growth.32 A type III tyrosine kinase FLT3 is a key receptor for ROS-mediated signaling in stem cells (Figure 3, top blue spheres). FLT3 consists of five immunoglobulin-like extracellular domains, a transmembrane domain, a juxtamembrane domain, and two intracellular tyrosine kinase domains linked by a kinase insert domain. In its inactive conformation, FLT3 exists as a monomer in the plasma membrane in a “closed” activation loop conformation that blocks access to the ATP binding site and the active site. The juxtamembrane domain serves as a critical autoinhibitory region sterically preventing dimerization. When FLT3 is stimulated with its ligand FLT3LG, the conformation of FLT3 changes, allowing dimerization to take place. Upon dimerization, FLT3 is phosphorylated in the intracellular domain (top arrow in Figure 3).33 This phosphorylation event activates the receptor and recruits a number of proteins, including SHC and GRB2, to form a complex that activates a number of intracellular mediators, including AKT and MAPK along with STAT5 when an internal tandem duplication is present (FLT3-ITD).34–37
Figure 3.

ROS regulation of FLT3 signaling. FLT3 is a receptor tyrosine kinase that dimerizes upon ligand binding. Autophosphorylation, blocked by PTPRJ, leads to activation of AKT and STAT5 pathways and transcription of NOX4. The complex of NOX4 and CYBA sustains the signal by generating O2•−, which is converted to H2O2 by SOD1, and by oxidatively inactivating PTPRJ and PTEN.
ROS generated by oxidases intersect with this pathway at multiple steps. A key protein is NOX4, a membrane-bound NADPH oxidase (bottom red in Figure 3). NOX4 catalyzes the production of two O2•− and a H+ via oxidation of a NADPH. NOX4 transcription is induced by activation of either AKT or STAT5, and NOX4 is required to sustain the FLT3 signal. NOX4 has six transmembrane domains and an intracellular carboxyl-terminal domain that harbors NADPH binding sites. A loop (between the second and third transmembrane domains) is essential for oxidase activity because it is bound by CYBA, which produces the ROS.38 Thus, during FLT3 signaling, an active NADPH oxidase containing NOX4 and CYBA is required to maintain signaling.39 The O2•− produced is rapidly converted to H2O2 by SOD1.
The resulting H2O2 sustains signaling through the FLT3, AKT, and RAS pathways. Membrane-bound PTPRJ is known to dephosphorylate FLT3,40 and PTPRJ must be inactivated to sustain signaling. PTPRJ possesses an active site cysteine that has been shown to be sensitive to H2O2. The oxidation inactivates PTPRJ, causing more phosphorylation of FLT3, and sustains the signal. There are other levels of ROS regulation in this pathway initiated by FLT3 activation. AKT is activated by binding of phosphatidylinositol 3,4,5-triphosphate.41 The concentration of phosphatidylinositol 3,4,5-triphosphate is negatively regulated by the phosphatase PTEN. H2O2 oxidizes and inactivates PTEN through disulfide bond formation between the Cys-124 and Cys-71 residues in the catalytic domain.42 Once oxidized, PTEN is inactive, and the AKT pathway is activated.43 This highlights a general scheme in which phosphatases are oxidatively inactivated to move the balance of signaling toward kinase action. The problematic side quickly starts to arise. To sustain signaling, ROS are needed, but if the ROS concentration remains elevated, hydrogen peroxide will be bound by metal and form hydroxyl radical. These hydroxyl radicals have sufficient reactivity to modify DNA, causing deleterious lesions.
4. TOO MUCH ROS LEADS TO EXOTIC DNA LESIONS
DNA damage occurs when significant concentrations of potent radicals like OH• are formed and can reach DNA. There are two primary sites of biomolecular damage on DNA: on nucleobases and on the ribose ring. The hydroxyl radical reaction leads to formation of either a ribose-based radical or a nucleobase radical. Two such radicals are shown in Figure 4. All DNA bases can be oxidized, but guanine is the primary target for oxidation because it possesses the most favorable potential (−1.3 V) for oxidation to form a radical compared to the other base.44 Compared to thiol oxidation, DNA is a relatively poor substrate for oxidation, but any modification in DNA not repaired can be mutagenic or lethal. DNA oxidation by ROS has been excessively studied,44–50 and dozens of DNA oxidative structures have been characterized. This review will focus on guanine oxidation. The initial radical formation on guanine, either neutral or as a cation, occurs through reaction with HO• or other potent one-electron oxidants.51 The trapping reaction is slower in a duplex because of solvent accessibility; thus, the radical migrates or can abstract an electron from a proximal base, or from nearby lesions that form double-strand breaks as shown on the right, middle arrow in Figure 4.52 The final, damaged structure results when the radical reacts with O2•− or molecular oxygen.53 Once the two-electron oxidation occurs, a series of reactions occur to stabilize the ring (Figure 4, multiple arrows). Two examples that were chosen because they elicit different types of DNA repair responses, showing alternative outcomes, are shown. When radicals on the ribose are formed, the final lesion produced often requires nucleotide excision repair (NER). Many of the oxidative lesions formed on nucleobases are repaired by glycosylases in the base excision repair (BER) pathway.54,55 Guanine is susceptible to loss of a second pair of electrons44 in forming exotic lesions such as guanidinohydantoin [Gh (Figure 4)].56 Finally, when DNA and protein-centered radicals combine, they can produce a DNA–protein or DNA–DNA cross-link.57,58 Thus, a single oxidative incident can form many different DNA modifications that require various repair responses for the cell to survive.
Figure 4.

DNA oxidation by ROS. In DNA, oxidation at the ribose leads to strand breaks, which are primarily repaired by nucleotide excision repair. Guanine lesions can be formed from a single oxidation or a second oxidation, primarily substrates for BER pathway glycosylases. Termination can lead to double-strand breaks or cross-linking.
The reaction of the hydroxyl radical with guanine follows the one-electron oxidation mechanism, with the initial chemistry taking place either at the C8/N7 double bond or at the C4/5 double bond as shown in the areas shaded in red or green, respectively, in Figure 5A. In each pathway, the reaction is initiated by abstraction of an electron from the double bond or by addition of hydroxyl radical at the double bond, leading to initial formation of a guanine radical cation. The guanine radical cation is a strong electrophile. Trapping of molecular oxygen by this electrophile followed by reduction leads to formation of an 8-oxo-7,8-dihydro-2′-deoxyguanosine lesion [8-oxo-dG (Figure 6)]. The guanine radical cation has a pKa of 3.9 and is deprotonated under biological conditions, but the radical cation is stabilized by the G:C base pair (Figure 5B).59 Thus, in double-stranded DNA, the reaction pathway proceeds via the radical cation, but in single-stranded nucleic acids, nucleosides, or nucleotides, the neutral path is followed. The hydroxyl radical adds at C4 followed by H2O elimination to directly yield a relatively stable guanine radical intermediate.60 Reactions in the C4/5 pathway tend to lead to complex products as regaining aromaticity is challenging. An example is 2,2,4-triamino-5(2H)-oxazolone, which is commonly known as oxazolone or dZ. It is thought that dZ is a major product in nucleoside and nucleotide chemistry that may be subsequently incorporated into a DNA during replication.
Figure 5.

Guanine radical pathways to oxidized products. (A) The initial reaction occurs at either the C8/N7 or the C4/5 bond. Reaction at the C8/N7 bond is favored in duplex DNA. The reaction at the C4/5 bond occurs on free nucleosides, nucleotides, and single-stranded nucleic acids. (B) In a duplex, the radical cation is favored because the positive charge can be shared with the adjacent cytosine. (C) Mispair structure of the two lesion products.
Figure 6.

C8/N7 pathway oxidation products. Radical localization on C8 leads to formation of 8-oxo-dG and Fapy-dG lesions or to exotic lesions such as cross-links or hydantoins.
The best-characterized lesions are 8-oxo-dG and 2,6-diamino-4-hydroxy-5-formamidopyrimidine [Fapy-dG (Figure 6)]. These lesions predominately lead to G → T transversions as these modified bases can stably pair with adenine.61 Oxidation and tautomerization of the guanine radical cation yield 8-oxo-dG, whereas competitive one-electron reduction yields the ring open product Fapy-dG.51,62 In cells, Fapy-dG is produced when oxygen levels are low and the environment is reducing.63 Both 8-oxo-dG and Fapy-dG are highly mutagenic. When 8-oxo-dG adopts a syn conformation, it forms a stable 8-oxo-dG:A base pair, leading to the transversion mentioned above (Figure 4C).64,65
In human cells, OGG1 glycosylase removes 8-oxo-dG when it is opposite dC,66 and MUTYH removes dA that is misincorporated opposite 8-oxo-dG in the template strand.67 In the case of the 8-oxo-dG:dA base pair, in human cells, the MSH2–MSH6 mismatch repair pathway excises the 8-oxo-dG-containing strand.64,65, If 8-oxo-dG escapes the repair machinery, translesion DNA synthesis by translesion polymerases (pol η, pol ι, pol κ, and REV1) may be induced.68,69 These polymerases can replicate strands containing 8-oxo-dG; their error rates vary.70,71 Fapy-dG can adopt either the typical anti conformation or a syn conformation. Both conformations form mispairs with dA. Fapy-dG is more mutagenic than 8-oxo-dG.72,73 Fapy-dG is efficiently recognized by NEIL1, OGG1, and SMUG1 glycosylases.66 8-oxo-dG and Fapy-dG nucleotides are both substrates for MTH1 (or NUDT1), which sanitizes the nucleotide pool to prevent incorporation during replication.74
The fate of the C8/N7 radical is not limited to formation of 8-oxo-dG and Fapy-dG. Radical translocation of the radical cation to the 5′-position followed by cyclization can lead to formation of either of two forms of 8,5′-cyclo-2′-deoxyguanosine [8,5′-cyclo-dG (Figure 6)];75,76 both forms have been detected in vivo.75,77 The additional covalent bond of 8,5′-cyclo-dG greatly alters the DNA structure relative to that of the standard B-form duplex.76 This adduct can be repaired through the NER pathway.65 The efficient repair depends on the sequence context.78 The 8,5′-cyclo-dG lesion is a strong replication block to normal polymerases.78 The lesion can be bypassed by translesion polymerases κ and ζ in an inefficient and highly error-prone fashion,79 and pol η and pol ι can bypass it in a largely error-free fashion. Mutational analysis indicates that the 8,5′-cyclo-dG induces G → T and G → A transformations in human cells.80
One important facet of dG oxidation is the fact that 8-oxo-dG is more readily oxidized than dG. Thus, 8-oxo-dG is thought to reenter the oxidation cascade and form genomic hotspots for additional oxidative damage. Subsequent oxidation of 8-oxo-dG results in formation of either of two hydantoin lesions spiroiminodihydantoin [Sp (Figure 6)] or 5-guanidinohydantoin [Gh (Figure 6)].81 Gh and Sp lesions are detected in the liver and colon tissue of Rag2−/− mice at levels 100-fold lower than that of 8-oxo-dG.82 In addition, these lesions are observed in Neil1−/− and Nth1−/− mice.83 Because these products are found at low levels, the relevance of Gh and Sp lesions is somewhat controversial. Biochemical experiments indicate that these lesions are efficiently recognized by human repair enzymes. Sp and Gh are found to be substrates for human NEIL1 and NEIL3 glycosylases in gene bodies and in telomeric sequences.54,84 Sp and Gh are also recognized by prokaryotic UvrABC nuclease.55
The thermodynamic destabilization caused by these lesions makes them ideal substrates for nucleotide excision repair. A recent study using human cell extracts suggests that Gh and Sp can be excised by either BER or NER mechanisms depending on the competitive binding of the proteins involved in these repair machineries to the lesions.66 When unrepaired, both Gh and Sp are strong replication blocks. A study with RB69 polymerase revealed that Gh is extrahelical and rotated toward the major groove, hindering the incoming nucleotide opposite Gh.85 A crystal structure of polymerase β with an Sp nucleotide in the template strand showed the extent of helix distortion formed by this hydantoin lesion.86 Upon polymerase bypass, both Sp and Gh lesions are highly mutagenic as base pairs with dG and dA, leading to G → C and G → T transversion mutations, respectively.87
DNA–protein cross-links (DPCs) make up another important class of oxidative DNA lesions. These lesions are formed by electrophilic addition of lysine or tyrosine side chains to Sp or Gh lesions or by radical coupling between dG and protein side chains (Figure 6).88,89 Lysine can add to the C5 or C8 position, depending on the initial oxidation of guanine.90 Formation of hydantoin–protein cross-links in the presence of DNA binding proteins at biologically relevant oxidant conditions suggests DPCs are a major type of oxidative lesions.57 DPCs are observed at levels of 0.5–4.5 per 107 bases in human white blood cells.91 Recently, the Greenburg group reported proteinlike radical formation in nucleosome core particles under oxidative stress conditions that initially shields DNA from oxidation; ultimately, DNA is damaged in an oxygen-dependent manner, showing the interplay of DNA and bound proteins in effects of oxidative stress.92 DPCs are large bulky lesions that distort the DNA duplex. NER and homologous repair are the main DNA damage repair pathways responsible for removal of DPCs.93 NER repairs DPCs with protein sizes of less than 12–14 kDa, and cytosolic ATP-dependent proteases are not involved in the processing of DPCs prior to NER. When the cross-linked protein is larger, repair occurs exclusively via homologous recombination involving the RecBCD pathway.94 A recent report identified the yeast metalloprotease WSS1 as a component of the DPC repair pathway that acts on the protein side of the lesion.95 WSS1 has replication-induced proteolytic activity; the proteolytic mechanism has not been characterized in other higher organisms.
The adducts formed via the C5 pathway are not as well characterized as those formed via the C8 pathway. The C5 pathway is thought to be important in damage to nucleotides and single-stranded structures. Three major lesions are formed from the C5 radical intermediate: imidazolones (2,5-diaminoimidazolone-2′-deoxyribonucleoside or dIz), oxazolones (2,2,4-triamino-2H-oxazol-5-one-2′-deoxyribonucleoside or dZ), and imino-hydantion (5-carboxaminido-5-formamido-2-imino-hydantio-2′-deoxyribonucleoside or d2Ih).
dIz (Figure 7) is formed by O2•− addition at the C5 position of a neutral guanine radical followed by ring opening or by C5 oxidation of 8-oxo-dG. This reaction can be initiated by several types of ROS, including the hydroxyl radical.44 dIz is the major oxidation product in single-stranded nucleic acids.96 At physiological pH, dIz is hydrolyzed to form dZ (Figure 7).97 dZ does not degrade readily.98 dZ is ∼10-fold less abundant in rat liver DNA than 8-oxo-dG is, showing this pathway is competitive in cells.99 This dZ adduct is less duplex-distorting than Sp.100 dZ is a substrate for the BER. In human cell systems, NEIL1 and NTHL1 remove dZ in duplex DNA in a manner independent of the base pairing partner, whereas OGG1 cannot excise dZ.101 The dZ:dG base pair is more stable than the dZ base pairs with dA or dT favoring the G → C transversion mutation.102 The translesion polymerase REV1 does efficiently incorporate dC opposite dZ; however, B and Y family polymerases incorporate dG opposite dZ, and polymerase ζ incorporates both dGTP and dATP opposite dZ, suggesting that REV1 can prevent the G → C transversion caused by dZ.103 A recent study states that polymerase ζ efficiently bypasses DNA with two neighboring dZ lesions, whereas polymerases ζ, α, β, ι, REV1, κ, and η all stall or inefficiently bypass contiguous dZ lesions.104
Figure 7.

C4/5 pathway oxidation products. Imidazolnes (dIz), oxazolones (dZ), and imino-hydantoins (d2Ih) are the major proucts of the C4/C5 pathway.
In addition to four-electron oxidation products such as dZ and dIz, the C5 pathway can also result in the two-electron oxidation lesion d2Ih (Figure 7), which is observed in Fenton chemistry in the presence of Pb105 in both duplex and G-quadruplex contexts.56 d2Ih is formed from the C4-localized neutral guanine radical intermediate. Recently, the Burrows group showed that d2Ih is the major product formed from hydroxyl radical under aerobic conditions in a biologically relevant reducing environment.44 Both diastereomers of d2Ih are removed efficiently by both NEIL1 and FPG or by NTH less efficiently. In addition, the excision of d2Ih occurs in the context of all possible base-pairing partners, suggesting that 2Ih is recognized by the BER pathway.44 If left unrepaired, 2Ih is efficiently paired with dGTP and less so with dCTP, leading to G → C or G → T transversion mutations. The (R)-2Ih isomer is a 2-fold more effective than (S)-2Ih as a block to three different polymerases. Interestingly, the R diastereomer of the 2Ih lesion is not as efficiently processed by glycosylase as (S)-2Ih is, suggesting that this lesion could potentially accumulate in the genome and lead to point mutations.106
5. ROS-TARGETED ANTICANCER AGENTS
ROS-targeted anticancer agents are rapidly gaining attention.107 Three common strategies include inhibiting antioxidant systems, targeting DNA repair systems, and prodrug methods (Figure 8). Selected examples of anticancer agents from each strategy are discussed in this section.
Figure 8.

ROS-based anticancer strategies. Cells generate superoxide and peroxide to sustain signaling. Sustained signaling leads to growth and adaption (green). Cellular antioxidants are essential to limit the damaging effects of each ROS, especially hydroxyl radical. Without antioxidants, ROS accumulate, leading to cell damage and cell death (brown). Three strategies (red) for exploiting elevated levels of ROS in cancer are shown in red and include inhibition of antioxidants, inhibition of repair, and novel ROS-activated prodrugs.
The first strategy being utilized is the inhibition of antioxidant enzymes (red 1 in Figure 8). Because cancer cells utilize high ROS concentrations to sustain signaling, antioxidants are needed for compensation to ensure survival; thus, depletion of antioxidants elevates the ROS level to pathogenic levels. Buthionine sulfoximine is a potent and specific inhibitor of γ-glutamylcysteine ligase. The enzyme γ-glutamylcysteine ligase is the rate-limiting enzyme of de novo synthesis of glutathione, which catalyzes the synthesis of intermediate dipeptide γ-glutamylcysteine by ligating glutamic acid and cysteine.108 Inhibition of γ-glutamylcysteine ligase by Buthionine sulfoximine causes a large drop in the amount of cellular glutathione and increases the level of ROS. Many types of cancer show elevation of glutathione levels and resistance to oxidative stress. The high level of glutathione in some tumor cells is typically associated with higher activity of γ-glutamylcysteine ligase. Therefore, Buthionine sulfoximine is more cytotoxic to certain types of cancer cells than it is to healthy cells.109 Thus, disruption of antioxidant systems is a potential target in several cancer cells that rely on antioxidants to compensate for elevated concentrations of ROS.110,111
Another means of disrupting the antioxidant system is to overwhelm it with even more ROS using pro-oxidants. This reduces the level of antioxidant below a sustainable level. A quinone compound, β-lapachone, a unique NAD(P)H quinone oxidoreductase 1 (NQO1) bioactivatable drug, undergoes two-electron reduction to a hydroquinone form, utilizing NAD(P)H or NADH as an electron source. Its cellular mechanism of action is to produce ROS once reduced by forming semiquinone radicals. Lapachone depletes cells of NAD(P)H and NADH as a result of cycling between the oxidized and reduced forms.112 Recently, it was reported that β-lapachone synergizes with ionizing radiation.113 Piperlongumine is another pro-oxidant agent with anticancer activity. Piperlongumine induces ROS production in cells by acting as a suicide inhibitor of critical ROS homeostatic regulator proteins such as glutathione S-transferase 1.114,115 A derivative of piperlonguimine can also inhibit thioredoxin reductase, which has recently been reported.116
The next strategy is to interfere with the repair of DNA (red 2 in Figure 8). The cellular nucleotide pool is a major target of ROS. Incorporation of oxidized nucleotides into DNA can lead to mutations and cell death. For example, incorporation of 8-oxo-dG triphosphate will prime the DNA for formation of exotic lesions that are more difficult to replicate and bypass. Thus, cells have defense systems for sanitizing the nucleotide pool. One such example is MTH1 (or NUDT1), which hydrolyses oxidized purine nucleotide triphosphates such as 8-oxo-dG triphosphate into the monophosphate and pyrophosphate.65 This renders the oxidized nucleotide unable to be added to the growing DNA chain. It has been found that MTH1 activity is low in healthy cells, but many cancer cells rely on MTH1 activity for survival.74 Therefore, it has been proposed that inhibition of MTH1 in cancer cells results in unsustainable levels of oxidized nucleotide triphosphates being incorporated into DNA. Several MTH1 inhibitors with anticancer properties have been reported.117,118 Similarly, poly(ADP-ribose) polymerase (PARP) enzymes are necessary to repair strand breaks within cancer cells.119,120 PARP inhibition results in persistent single-strand breaks. Trapping of PARP1 on DNA, thereby preventing downstream repair proteins from access, also occurs.121,122 The PARP inhibitors synergize with many traditional chemotherapeutics and are especially effective against cells with BRCA1 deficient and activating PTEN.123–125 Niraparib (MK-4827) is a PARP-1 and PARP-2 inhibitor that induces cell cycle arrest at the G2/M phase in BRCA1 deficient cells.126 Combinational treatment of niraparib with ROS induction, like ionizing radiation that requires more DNA repair activity, showed strong synergy. Several other PARP inhibitors are in late stage clinical development.127
ROS-activated prodrug strategies are attracting an increased level of attention (red 3 in Figure 8). These strategies exploit the high levels of hydrogen peroxide in cancer cells. For example, increased levels of hydrogen peroxide were found in prostate cancer cells with aggressive phenotypes and AML cells with FLT3 ITD mutations, as measured by fluorescent probes.128,129 It should be noted that the quantification of ROS, especially hydrogen peroxide, is a nontrival task.130 Different ROS-accepting/sensitive groups have been used in prodrug strategies. Most current prodrug strategies rely on Chan-Lam type coupling (Scheme 1), in which a phenol in a bioactive molecule is replaced with a boronic ester. The bioactive molecule is nonfunctional when the boron ester is present as the hydroxyl is essential for anticancer activity. In high-ROS concentration environments, like in cancer cells, formation of the active phenol is catalyzed by hydrogen peroxide.131 Several groups have shown that this strategy is effective against leukemia cells.131,132 Recently, boronic acid-substituted analogues of topoisomerase inhibitor camptothecin were reported as highly selective antitumor alkaloids.133 Additionally, prodrug matrix metalloproteinase inhibitors have been developed using self-immolative boronic ester-protected methyl salicylates and metal binding groups.134
Scheme 1.

Boronic Acid-Based Prodrugsa
aActivation of the prodrug by H2O2 oxidation and release of an active drug.
Our group develops novel ROS-activated cytotoxic agents (RACs) that include a hydroquinone moiety and a nucleophile connected via a linker.135,136 We have shown that one agent is highly active and selective toward leukemia cells. Biochemical evaluation of these leukemia cells showed that levels of the catalase antioxidant enzyme are reduced compared to those of untransformed cells. These cells also showed larger amounts of 8-oxo-dG measured by an enzyme-linked immunosorbent assay. The agent is unusual because ROS activation leads to formation of a reactive electrophile that reacts with three of the four DNA bases. The reaction leads to addition of a bulky aromatic ring on the nucleobase. Especially interesting is the reaction with dG, which adds the ring to both N2 and N3 in the minor groove. This lesion leads to DNA strand breaks, as shown by a COMET assay, and apoptosis. Cells attempt to survive by specifically activating homologous recombination as visualized by ATM, ATR, and BRCA1 phosphorylation shortly after treatment. Simultaneous treatment with the RAC and an inhibitor of homologous recombination leads to strong synergy that kills cancer cells and spares healthy cells (Figure 9).137 Other new ROS-activated strategies that are more selective and generate a novel reactive electrophile are being designed.138 These new strategies lead to novel DNA lesions being formed and will force cells to use alternative survival mechanisms that can lead to differential sensitivity compared to that of current RAC agents.
Figure 9.

Mechanism of ROS-activated cytotoxic agents. Several cancers are thought to be addicted to elevated levels of ROS; thus, we design agents that are activated by ROS leading to strand breaks only in cancerous cells. ROS activation leads to formation of a bulky lesion requiring homologous recombination repair for survival.
Acknowledgments
Funding
This study was supported by National Institutes of Health Grant R21A185370 to E.J.M.
Footnotes
Author Contributions
S.F.A. and F.S.T. are co-first authors.
The authors declare no competing financial interest.
References
- 1.Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–5826. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- 2.Constantine MT, Cain EF. Hydrogen peroxide handbook. Rocketdyne; Canoga Park, CA: 1967. [Google Scholar]
- 3. [accessed May 27, 2016];Household Products Database: Health and Safety Information on Household Products. https://hpd.nlm.nih.gov/cgi-bin/household/search?tbl=TblChemicals&queryx=7722-84-1.
- 4.Pauling L. The discovery of the superoxide radical. Trends Biochem. Sci. 1979;4:N270–N271. [Google Scholar]
- 5.Bannister WH, Bannister JV. Isolation and characterization of superoxide dismutase: A personal history and tribute to Joe McCord and Irwin Fridovich. Free Radical Biol. Med. 1988;5:371–376. doi: 10.1016/0891-5849(88)90110-4. [DOI] [PubMed] [Google Scholar]
- 6.Sawyer DT, Gibian MJ, Morrison MM, Seo ET. On the chemical reactivity of superoxide ion. J. Am. Chem. Soc. 1978;100:627–628. [Google Scholar]
- 7.Haber F, Weiss J. The Catalytic Decomposition of Hydrogen Peroxide by Iron Salts. Proc. R. Soc. London, Ser. A. 1934;147:332–351. [Google Scholar]
- 8.Kehrer JP. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology. 2000;149:43–50. doi: 10.1016/s0300-483x(00)00231-6. [DOI] [PubMed] [Google Scholar]
- 9.Auclair C, Cramer E, Hakim J, Boivin P. Studies on the mechanism of NADPH oxidation by the granule fraction isolated from human resting polymorphonuclear blood cells. Biochimie. 1977;58:1359–1366. doi: 10.1016/s0300-9084(77)80020-5. [DOI] [PubMed] [Google Scholar]
- 10.Miwa S, Beckman KB, Muller FL. Oxidative stress in aging: From model systems to human diseases. Humana Press; Totowa, NJ: 2008. [Google Scholar]
- 11.Dannenmann B, Lehle S, Hildebrand DG, Kübler A, Grondona P, Schmid V, Holzer K, Fröschl M, Essmann F, Rothfuss O, Schulze-Osthoff K. High Glutathione and Glutathione Peroxidase-2 Levels Mediate Cell-Type-Specific DNA Damage Protection in Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2015;4:886–898. doi: 10.1016/j.stemcr.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, Riet T, Abken H, Kiessling R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016;196:759–766. doi: 10.4049/jimmunol.1401710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fridlich R, Annamalai D, Roy R, Bernheim G, Powell SN. BRCA1 and BRCA2 protect against oxidative DNA damage converted into double-strand breaks during DNA replication. DNA Repair. 2015;30:11–20. doi: 10.1016/j.dnarep.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Davies KJA, Forman HJ. TGFβ1 rapidly activates Src through a non-canonical redox signaling mechanism. Arch. Biochem. Biophys. 2015;568:1–7. doi: 10.1016/j.abb.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woolley JF, Naughton R, Stanicka J, Gough DR, Bhatt L, Dickinson BC, Chang CJ, Cotter TG. H2O2 production downstream of FLT3 is mediated by p22phox in the endoplasmic reticulum and is required for STAT5 signalling. PLoS One. 2012;7:e34050. doi: 10.1371/journal.pone.0034050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 17.Schieber M, Chandel NS. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, Xie S, Laurenti E, Hermans K, Eppert K, Marciniak SJ, Goodall JC, Green AR, Wouters BG, Wienholds E, Dick JE. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature. 2014;510:268–272. doi: 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- 19.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015;116:531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nordgren M, Fransen M. Peroxisomal metabolism and oxidative stress. Biochimie. 2014;98:56–62. doi: 10.1016/j.biochi.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 21.Ayala A, Munoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longevity. 2014;2014:360438. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta V, Carroll KS. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta, Gen. Subj. 2014;1840:847–875. doi: 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lizama-Manibusan B, McLaughlin B. Redox modification of proteins as essential mediators of CNS autophagy and mitophagy. FEBS Lett. 2013;587:2291–2298. doi: 10.1016/j.febslet.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinha RK, Maitre P, Piccirillo S, Chiavarino B, Crestoni ME, Fornarini S. Cysteine radical cation: A distonic structure probed by gas phase IR spectroscopy. Phys. Chem. Chem. Phys. 2010;12:9794–9800. doi: 10.1039/c003576a. [DOI] [PubMed] [Google Scholar]
- 25.Fridovich I. Superoxide Dismutases. Annu. Rev. Biochem. 1975;44:147–159. doi: 10.1146/annurev.bi.44.070175.001051. [DOI] [PubMed] [Google Scholar]
- 26.Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015;33:8–13. doi: 10.1016/j.ceb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas C, Mackey MM, Diaz AA, Cox DP. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Redox Rep. 2009;14:102–108. doi: 10.1179/135100009X392566. [DOI] [PubMed] [Google Scholar]
- 28.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 29.Rhee SG. Redox signaling: hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999;31:53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- 30.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 31.Liu RM, Choi J, Wu JH, Gaston Pravia KA, Lewis KM, Brand JD, Mochel NS, Krzywanski DM, Lambeth JD, Hagood JS, Forman HJ, Thannickal VJ, Postlethwait EM. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J. Biol. Chem. 2010;285:16239–16247. doi: 10.1074/jbc.M110.111732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, Lu XJ, Ledergor G, Kollet O, Lapidot T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signaling. 2014;21:1605–1619. doi: 10.1089/ars.2014.5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meshinchi S, Appelbaum FR. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin. Cancer Res. 2009;15:4263–4269. doi: 10.1158/1078-0432.CCR-08-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dosil M, Wang S, Lemischka IR. Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Mol. Cell. Biol. 1993;13:6572–6585. doi: 10.1128/mcb.13.10.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavagna-Sevenier C, Marchetto S, Birnbaum D, Rosnet O. FLT3 signaling in hematopoietic cells involves CBL, SHC and an unknown P115 as prominent tyrosine-phosphorylated substrates. Leukemia. 1998;12:301–310. doi: 10.1038/sj.leu.2400921. [DOI] [PubMed] [Google Scholar]
- 36.Zhang S, Broxmeyer HE. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem. Biophys. Res. Commun. 2000;277:195–199. doi: 10.1006/bbrc.2000.3662. [DOI] [PubMed] [Google Scholar]
- 37.Marchetto S, Fournier E, Beslu N, Aurran-Schleinitz T, Dubreuil P, Borg JP, Birnbaum D, Rosnet O. SHC and SHIP phosphorylation and interaction in response to activation of the FLT3 receptor. Leukemia. 1999;13:1374–1382. doi: 10.1038/sj.leu.2401527. [DOI] [PubMed] [Google Scholar]
- 38.Jackson HM, Kawahara T, Nisimoto Y, Smith SM, Lambeth JD. Nox4 B-loop creates an interface between the transmembrane and dehydrogenase domains. J. Biol. Chem. 2010;285:10281–10290. doi: 10.1074/jbc.M109.084939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stasia MJ. CYBA encoding p22(phox), the cytochrome b558 alpha polypeptide: gene structure, expression, role and physiopathology. Gene. 2016;586:27–35. doi: 10.1016/j.gene.2016.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Godfrey R, Arora D, Bauer R, Stopp S, Muller JP, Heinrich T, Bohmer SA, Dagnell M, Schnetzke U, Scholl S, Ostman A, Bohmer FD. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/PTPRJ. Blood. 2012;119:4499–4511. doi: 10.1182/blood-2011-02-336446. [DOI] [PubMed] [Google Scholar]
- 41.Jo H, Lo PK, Li Y, Loison F, Green S, Wang J, Silberstein LE, Ye K, Chen H, Luo HR. Deactivation of Akt by a small molecule inhibitor targeting pleckstrin homology domain and facilitating Akt ubiquitination. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6486–6491. doi: 10.1073/pnas.1019062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signalling. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Josephs DH, Sarker D. Pharmacodynamic Biomarker Development for PI3K Pathway Therapeutics. Transl. Oncogenomics. 2015;7:33–49. doi: 10.4137/TOG.S30529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alshykhly OR, Fleming AM, Burrows CJ. 5-Carboxamido-5-formamido-2-iminohydantoin, in Addition to 8-oxo-7,8-Dihydroguanine, Is the Major Product of the Iron-Fenton or X-ray Radiation-Induced Oxidation of Guanine under Aerobic Reducing Conditions in Nucleoside and DNA Contexts. J. Org. Chem. 2015;80:6996–7007. doi: 10.1021/acs.joc.5b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cadet J, Douki T, Ravanat JL. Oxidatively generated base damage to cellular DNA. Free Radical Biol. Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 46.Zhu J, Fleming AM, Orendt AM, Burrows CJ. pH-Dependent Equilibrium between 5-Guanidinohydantoin and Iminoallantoin Affects Nucleotide Insertion Opposite the DNA Lesion. J. Org. Chem. 2016;81:351–359. doi: 10.1021/acs.joc.5b02180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghosh S, Greenberg MM. Correlation of Thermal Stability and Structural Distortion of DNA Interstrand Cross-Links Produced from Oxidized Abasic Sites with Their Selective Formation and Repair. Biochemistry. 2015;54:6274–6283. doi: 10.1021/acs.biochem.5b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gruppi F, Hejazi L, Christov PP, Krishnamachari S, Turesky RJ, Rizzo CJ. Characterization of nitrogen mustard formamidopyrimidine adduct formation of bis(2-chloroethyl)ethylamine with calf thymus DNA and a human mammary cancer cell line. Chem. Res. Toxicol. 2015;28:1850–1860. doi: 10.1021/acs.chemrestox.5b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang J, Yennie CJ, Delaney S. Klenow Fragment Discriminates against the Incorporation of the Hyperoxidized dGTP Lesion Spiroiminodihydantoin into DNA. Chem. Res. Toxicol. 2015;28:2325–2333. doi: 10.1021/acs.chemrestox.5b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cui L, Ye W, Prestwich EG, Wishnok JS, Taghizadeh K, Dedon PC, Tannenbaum SR. Comparative Analysis of Four Oxidized Guanine Lesions from Reactions of DNA with Peroxynitrite, Singlet Oxygen, and γ-Radiation. Chem. Res. Toxicol. 2013;26:195–202. doi: 10.1021/tx300294d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cadet J, Wagner JR. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 2014;557:47–54. doi: 10.1016/j.abb.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 52.Arnold AR, Grodick MA, Barton JK. DNA Charge Transport: from Chemical Principles to the Cell. Cell Chem. Biol. 2016;23:183–197. doi: 10.1016/j.chembiol.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pratviel G, Meunier B. Guanine oxidation: one-and two-electron reactions. Chem. - Eur. J. 2006;12:6018–6030. doi: 10.1002/chem.200600539. [DOI] [PubMed] [Google Scholar]
- 54.Zhou J, Fleming AM, Averill AM, Burrows CJ, Wallace SS. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015;43:4039–4054. doi: 10.1093/nar/gkv252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKibbin PL, Fleming AM, Towheed MA, Van Houten B, Burrows CJ, David SS. Repair of Hydantoin Lesions and Their Amine Adducts in DNA by Base and Nucleotide Excision Repair. J. Am. Chem. Soc. 2013;135:13851–13861. doi: 10.1021/ja4059469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fleming AM, Burrows CJ. G-Quadruplex Folds of the Human Telomere Sequence Alter the Site Reactivity and Reaction Pathway of Guanine Oxidation Compared to Duplex DNA. Chem. Res. Toxicol. 2013;26:593–607. doi: 10.1021/tx400028y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Solivio MJ, Nemera DB, Sallans L, Merino EJ. Biologically Relevant Oxidants Cause Bound Proteins To Readily Oxidatively Cross-Link at Guanine. Chem. Res. Toxicol. 2012;25:326–336. doi: 10.1021/tx200376e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quiñones JL, Thapar U, Yu K, Fang Q, Sobol RW, Demple B. Enzyme mechanism-based, oxidative DNA–protein cross-links formed with DNA polymerase β in vivo. Proc. Natl. Acad. Sci. U. S. A. 2015;112:8602–8607. doi: 10.1073/pnas.1501101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Candeias LP, Steenken S. Structure and acid-base properties of one-electron-oxidized deoxyguanosine, guanosine, and 1-methylguanosine. J. Am. Chem. Soc. 1989;111:1094–1099. [Google Scholar]
- 60.Fleming AM, Muller JG, Ji I, Burrows CJ. Characterization of 2[prime or minute]-deoxyguanosine oxidation products observed in the Fenton-like system Cu(ii)/H2O2/reductant in nucleoside and oligodeoxynucleotide contexts. Org. Biomol. Chem. 2011;9:3338–3348. doi: 10.1039/c1ob05112a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gehrke TH, Lischke U, Gasteiger KL, Schneider S, Arnold S, Müller HC, Stephenson DS, Zipse H, Carell T. Unexpected non-Hoogsteen-based mutagenicity mechanism of FaPy-DNA lesions. Nat. Chem. Biol. 2013;9:455–461. doi: 10.1038/nchembio.1254. [DOI] [PubMed] [Google Scholar]
- 62.Cadet J, Douki T, Ravanat JL. Oxidatively generated base damage to cellular DNA. Free Radical Biol. Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 63.Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harbor Perspect. Biol. 2013;5:a012559. doi: 10.1101/cshperspect.a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Markkanen E, Dorn J, Hübscher U. MUTYH DNA glycosylase: the rationale for removing undamaged bases from the DNA. Front. Genet. 2013;4:18. doi: 10.3389/fgene.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakabeppu Y. Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci. 2014;15:12543–12557. doi: 10.3390/ijms150712543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shafirovich V, Kropachev K, Anderson T, Liu Z, Kolbanovskiy M, Martin BD, Sugden K, Shim Y, Chen X, Min JH, Geacintov NE. Base and Nucleotide Excision Repair of Oxidatively Generated Guanine Lesions in DNA. J. Biol. Chem. 2016;291:5309–5319. doi: 10.1074/jbc.M115.693218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chatterjee A. Reduced Glutathione: A Radioprotector or a Modulator of DNA-Repair Activity? Nutrients. 2013;5:525–542. doi: 10.3390/nu5020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kamiya H, Yamaguchi A, Suzuki T, Harashima H. Roles of specialized DNA polymerases in mutagenesis by 8-hydroxyguanine in human cells. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2010;686:90–95. doi: 10.1016/j.mrfmmm.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 70.Patra A, Nagy LD, Zhang Q, Su Y, Muller L, Guengerich FP, Egli M. Kinetics, structure, and mechanism of 8-Oxo-7,8-dihydro-2′-deoxyguanosine bypass by human DNA polymerase eta. J. Biol. Chem. 2014;289:16867–16882. doi: 10.1074/jbc.M114.551820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su Y, Patra A, Harp JM, Egli M, Guengerich FP. Roles of Residues Arg-61 and Gln-38 of Human DNA Polymerase in Bypass of Deoxyguanosine and 7,8-Dihydro-8-oxo-2-deoxyguanosine. J. Biol. Chem. 2015;290:15921–15933. doi: 10.1074/jbc.M115.653691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kalam MA, Haraguchi K, Chandani S, Loechler EL, Moriya M, Greenberg MM, Basu AK. Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 2006;34:2305–2315. doi: 10.1093/nar/gkl099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pande P, Haraguchi K, Jiang Y-L, Greenberg MM, Basu AK. Unlike Catalyzing Error-Free Bypass of 8-oxodGuo, DNA Polymerase λ Is Responsible for a Significant Part of Fapy•dG-Induced G→T Mutations in Human Cells. Biochemistry. 2015;54:1859–1862. doi: 10.1021/acs.biochem.5b00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gad H, Koolmeister T, Jemth A-S, Eshtad S, Jacques SA, Strom CE, Svensson LM, Schultz N, Lundback T, Einarsdottir BO, Saleh A, Gokturk C, Baranczewski P, Svensson R, Berntsson RPA, Gustafsson R, Stromberg K, Sanjiv K, Jacques-Cordonnier M-C, Desroses M, Gustavsson A-L, Olofsson R, Johansson F, Homan EJ, Loseva O, Brautigam L, Johansson L, Hoglund A, Hagenkort A, Pham T, Altun M, Gaugaz FZ, Vikingsson S, Evers B, Henriksson M, Vallin KSA, Wallner OA, Hammarstrom LGJ, Wiita E, Almlof I, Kalderen C, Axelsson H, Djureinovic T, Puigvert JC, Haggblad M, Jeppsson F, Martens U, Lundin C, Lundgren B, Granelli I, Jensen AJ, Artursson P, Nilsson JA, Stenmark P, Scobie M, Berglund UW, Helleday T. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508:215–221. doi: 10.1038/nature13181. [DOI] [PubMed] [Google Scholar]
- 75.Chatgilialoglu C, Ferreri C, Terzidis MA. Purine 5′,8-cyclonucleoside lesions: chemistry and biology. Chem. Soc. Rev. 2011;40:1368–1382. doi: 10.1039/c0cs00061b. [DOI] [PubMed] [Google Scholar]
- 76.Chatgilialoglu C, D’Angelantonio M, Kciuk G, Bobrowski K. New Insights into the Reaction Paths of Hydroxyl Radicals with 2′-Deoxyguanosine. Chem. Res. Toxicol. 2011;24:2200–2206. doi: 10.1021/tx2003245. [DOI] [PubMed] [Google Scholar]
- 77.Belmadoui N, Boussicault F, Guerra M, Ravanat J-L, Chatgilialoglu C, Cadet J. Radiation-induced formation of purine 5[prime or minute],8-cyclonucleosides in isolated and cellular DNA: high stereospecificity and modulating effect of oxygen. Org. Biomol. Chem. 2010;8:3211–3219. doi: 10.1039/c004531d. [DOI] [PubMed] [Google Scholar]
- 78.Pande P, Das RS, Sheppard C, Kow YW, Basu AK. Repair efficiency of (5′S)-8,5′-cyclo-2′-deoxyguanosine and (5′S)-8,5′-cyclo-2′-deoxyadenosine depends on the complementary base. DNA Repair. 2012;11:926–931. doi: 10.1016/j.dnarep.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.You C, Swanson AL, Dai X, Yuan B, Wang J, Wang Y. Translesion synthesis of 8,5′-cyclopurine-2′-deoxynucleosides by DNA polymerases eta, iota, and zeta. J. Biol. Chem. 2013;288:28548–28556. doi: 10.1074/jbc.M113.480459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pednekar V, Weerasooriya S, Jasti VP, Basu AK. Mutagenicity and genotoxicity of (5′S)-8,5′-cyclo-2′-deoxyadenosine in Escherichia coli and replication of (5′S)-8,5′-cyclopurine-2′-deoxynucleosides in vitro by DNA polymerase IV, exo-free Klenow fragment, and Dpo4. Chem. Res. Toxicol. 2014;27:200–210. doi: 10.1021/tx4002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fleming AM, Muller JG, Dlouhy AC, Burrows CJ. Structural Context Effects in the Oxidation of 8-Oxo-7,8-dihydro-2′-deoxyguanosine to Hydantoin Products: Electrostatics, Base Stacking, and Base Pairing. J. Am. Chem. Soc. 2012;134:15091–15102. doi: 10.1021/ja306077b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye W, Prestwich E, Cui L, McFaline JL, Mobley M, Ge Z, Taghizadeh K, Wishnok JS, Wogan GN, Fox JG, Tannenbaum SR, Dedon PC. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E1820–E1829. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chan MK, Ocampo-Hafalla MT, Vartanian V, Jaruga P, Kirkali G, Koenig KL, Brown S, Lloyd RS, Dizdaroglu M, Teebor GW. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair. 2009;8:786–794. doi: 10.1016/j.dnarep.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krokeide SZ, Laerdahl JK, Salah M, Luna L, Cederkvist FH, Fleming AM, Burrows CJ, Dalhus B, Bjoras M. Human NEIL3 is mainly a monofunctional DNA glycosylase removing spiroimindiohydantoin and guanidinohydantoin. DNA Repair. 2013;12:1159–1164. doi: 10.1016/j.dnarep.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aller P, Ye Y, Wallace SS, Burrows CJ, Doublie S. Crystal Structure of a Replicative DNA Polymerase Bound to the Oxidized Guanine Lesion Guanidinohydantoin. Biochemistry. 2010;49:2502–2509. doi: 10.1021/bi902195p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eckenroth BE, Fleming AM, Sweasy JB, Burrows CJ, Doublié S. Crystal Structure of DNA Polymerase β with DNA Containing the Base Lesion Spiroiminodihydantoin in a Templating Position. Biochemistry. 2014;53:2075–2077. doi: 10.1021/bi500270e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Henderson PT, Delaney JC, Muller JG, Neeley WL, Tannenbaum SR, Burrows CJ, Essigmann JM. The Hydantoin Lesions Formed from Oxidation of 7,8-Dihydro-8-oxoguanine Are Potent Sources of Replication Errors in Vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 88.Uvaydov Y, Geacintov NE, Shafirovich V. Generation of guanine-amino acid cross-links by a free radical combination mechanism. Phys. Chem. Chem. Phys. 2014;16:11729–11736. doi: 10.1039/c4cp00675e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ide H, Shoulkamy MI, Nakano T, Miyamoto-Matsubara M, Salem AM. Repair and biochemical effects of DNA-protein crosslinks. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2011;711:113–122. doi: 10.1016/j.mrfmmm.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 90.Xu X, Muller JG, Ye Y, Burrows CJ. DNA-protein cross-links between guanine and lysine depend on the mechanism of oxidation for formation of C5 vs C8 guanosine adducts. J. Am. Chem. Soc. 2008;130:703–709. doi: 10.1021/ja077102a. [DOI] [PubMed] [Google Scholar]
- 91.Voitkun V, Zhitkovich A. Analysis of DNA-protein crosslinking activity of malondialdehyde in vitro. Mutat. Res., Fundam. Mol. Mech. Mutagen. 1999;424:97–106. doi: 10.1016/s0027-5107(99)00011-1. [DOI] [PubMed] [Google Scholar]
- 92.Zhou C, Greenberg MM. DNA damage by histone radicals in nucleosome core particles. J. Am. Chem. Soc. 2014;136:6562–6565. doi: 10.1021/ja501285s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baker DJ, Wuenschell G, Xia L, Termini J, Bates SE, Riggs AD, O’Connor TR. Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J. Biol. Chem. 2007;282:22592–22604. doi: 10.1074/jbc.M702856200. [DOI] [PubMed] [Google Scholar]
- 94.Nakano T, Morishita S, Katafuchi A, Matsubara M, Horikawa Y, Terato H, Salem AM, Izumi S, Pack SP, Makino K, Ide H. Nucleotide excision repair and homologous recombination systems commit differentially to the repair of DNA-protein crosslinks. Mol. Cell. 2007;28:147–158. doi: 10.1016/j.molcel.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 95.Stingele J, Schwarz MS, Bloemeke N, Wolf PG, Jentsch S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 2014;158:327–338. doi: 10.1016/j.cell.2014.04.053. [DOI] [PubMed] [Google Scholar]
- 96.Morikawa M, Kino K, Oyoshi T, Suzuki M, Kobayashi T, Miyazawa H. Product analysis of photooxidation in isolated quadruplex DNA; 8-oxo-7,8-dihydroguanine and its oxidation product at 3[prime or minute]-G are formed instead of 2,5-diamino-4H–imidazol-4-one. RSC Adv. 2013;3:25694–25697. [Google Scholar]
- 97.Jena NR, Mishra PC. Formation of ring-opened and rearranged products of guanine: Mechanisms and biological significance. Free Radical Biol. Med. 2012;53:81–94. doi: 10.1016/j.freeradbiomed.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 98.Shao J, Geacintov NE, Shafirovich V. Oxidative Modification of Guanine Bases Initiated by Oxyl Radicals Derived from Photolysis of Azo Compounds. J. Phys. Chem. B. 2010;114:6685–6692. doi: 10.1021/jp100686j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matter B, Malejka-Giganti D, Csallany AS, Tretyakova N. Quantitative analysis of the oxidative DNA lesion, 2,2-diamino-4-(2-deoxy-β-d-erythro-pentofuranosyl)amino]-5(2H)-oxazolone (oxazolone), in vitro and in vivo by isotope dilution-capillary HPLC-ESI-MS/MS. Nucleic Acids Res. 2006;34:5449–5460. doi: 10.1093/nar/gkl596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Suzuki M, Kino K, Morikawa M, Kobayashi T, Miyazawa H. Calculating Distortions of Short DNA Duplexes with Base Pairing Between an Oxidatively Damaged Guanine and a Guanine. Molecules. 2014;19:11030–11044. doi: 10.3390/molecules190811030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kino K, Takao M, Miyazawa H, Hanaoka F. A DNA oligomer containing 2,2,4-triamino-5(2H)-oxazolone is incised by human NEIL1 and NTH1. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2012;734:73–77. doi: 10.1016/j.mrfmmm.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 102.Suzuki M, Ohtsuki K, Kino K, Kobayashi T, Morikawa M, Kobayashi T, Miyazawa H. Effects of stability of base pairs containing an oxazolone on DNA elongation. J. Nucleic Acids. 2014;2014:178350. doi: 10.1155/2014/178350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Suzuki M, Kino K, Kawada T, Morikawa M, Kobayashi T, Miyazawa H. Analysis of nucleotide insertion opposite 2,2,4-triamino-5(2H)-oxazolone by eukaryotic B- and Y-family DNA polymerases. Chem. Res. Toxicol. 2015;28:1307–1316. doi: 10.1021/acs.chemrestox.5b00114. [DOI] [PubMed] [Google Scholar]
- 104.Suzuki M, Kino K, Kawada T, Oyoshi T, Morikawa M, Kobayashi T, Miyazawa H. Contiguous 2,2,4-triamino-5(2H)-oxazolone obstructs DNA synthesis by DNA polymerases alpha, beta, eta, iota, kappa, REV1 and Klenow Fragment exo-, but not by DNA polymerase zeta. J. Biochem. 2016;159:323–329. doi: 10.1093/jb/mvv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Banu L, Blagojevic V, Bohme DK. Lead(II)-Catalyzed Oxidation of Guanine in Solution Studied with Electrospray Ionization Mass Spectrometry. J. Phys. Chem. B. 2012;116:11791–11797. doi: 10.1021/jp302720z. [DOI] [PubMed] [Google Scholar]
- 106.Alshykhly OR, Fleming AM, Burrows CJ. Guanine Oxidation Product 5-Carboxamido-5-formamido-2-iminohydantoin Induces Mutations When Bypassed by DNA Polymerases and Is a Substrate for Base Excision Repair. Chem. Res. Toxicol. 2015;28:1861–1871. doi: 10.1021/acs.chemrestox.5b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discovery. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 108.Ferguson G, Bridge W. Glutamate cysteine ligase and the age-related decline in cellular glutathione: The therapeutic potential of γ-glutamylcysteine. Arch. Biochem. Biophys. 2016;593:12–23. doi: 10.1016/j.abb.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 109.Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxid. Med. Cell. Longevity. 2013;2013:10. doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liaudat AC, Bohl LP, Tolosa de Talamoni NG, Maletto B, Pistoresi-Palencia MC, Picotto G. Oxidative stress, cell cycle arrest and differentiation contribute toward the antiproliferative action of BSO and calcitriol on Caco-2 cells. Anti-Cancer Drugs. 2014;25:810–818. doi: 10.1097/CAD.0000000000000109. [DOI] [PubMed] [Google Scholar]
- 111.Bailey HH, Ripple G, Tutsch KD, Arzoomanian RZ, Alberti D, Feierabend C, Mahvi D, Schink J, Pomplun M, Mulcahy RT, Wilding G. Phase I study of continuousinfusion L-S,R-buthionine sulfoximine with intravenous melphalan. J. Natl. Cancer Inst. 1997;89:1789–1796. doi: 10.1093/jnci/89.23.1789. [DOI] [PubMed] [Google Scholar]
- 112.Ahn KJ, Lee HS, Bai SK, Song CW. Enhancement of radiation effect using beta-lapachone and underlying mechanism. Radiat. Oncol. J. 2013;31:57–65. doi: 10.3857/roj.2013.31.2.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li LS, Reddy S, Lin ZH, Liu S, Park H, Chun SG, Bornmann WG, Thibodeaux J, Yan J, Chakrabarti G, Xie XJ, Sumer BD, Boothman DA, Yordy JS. NQO1-Mediated Tumor-Selective Lethality and Radiosensitization for Head and Neck Cancer. Mol. Cancer Ther. 2016;15:1757–1767. doi: 10.1158/1535-7163.MCT-15-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, Stern AM, Mandinova A, Schreiber SL, Lee SW. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 115.Adams DJ, Boskovic ZV, Theriault JR, Wang AJ, Stern AM, Wagner BK, Shamji AF, Schreiber SL. Discovery of Small-Molecule Enhancers of Reactive Oxygen Species That are Nontoxic or Cause Genotype-Selective Cell Death. ACS Chem. Biol. 2013;8:923–929. doi: 10.1021/cb300653v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yan WJ, Wang Q, Yuan CH, Wang F, Ji Y, Dai F, Jin XL, Zhou B. Designing piperlongumine-directed anticancer agents by an electrophilicity-based prooxidant strategy: A mechanistic investigation. Free Radical Biol. Med. 2016;97:109–123. doi: 10.1016/j.freeradbiomed.2016.05.021. [DOI] [PubMed] [Google Scholar]
- 117.Petrocchi A, Leo E, Reyna NJ, Hamilton MM, Shi X, Parker CA, Mseeh F, Bardenhagen JP, Leonard P, Cross JB, Huang S, Jiang Y, Cardozo M, Draetta G, Marszalek JR, Toniatti C, Jones P, Lewis RT. Identification of potent and selective MTH1 inhibitors. Bioorg. Med. Chem. Lett. 2016;26:1503–1507. doi: 10.1016/j.bmcl.2016.02.026. [DOI] [PubMed] [Google Scholar]
- 118.Kettle JG, Alwan H, Bista M, Breed J, Davies NL, Eckersley K, Fillery S, Foote KM, Goodwin L, Jones DR, Käck H, Lau A, Nissink JWM, Read J, Scott JS, Taylor B, Walker G, Wissler L, Wylot M. Potent and Selective Inhibitors of MTH1 Probe Its Role in Cancer Cell Survival. J. Med. Chem. 2016;59:2346–2361. doi: 10.1021/acs.jmedchem.5b01760. [DOI] [PubMed] [Google Scholar]
- 119.Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J. Clin. Oncol. 2015;33:1397–1406. doi: 10.1200/JCO.2014.58.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gojo I, Beumer JH, Pratz KW, McDevitt MA, Baer MR, Blackford AL, Smith BD, Gore SD, Carraway H, Showel MM, Levis MJ, Dezern A, Gladstone DE, Ji JJ, Wang L, Kinders R, Pouquet M, Ali-Walbi I, Rudek MA, Poh W, Herman JG, Karnitz L, Kaufmann SH, Chen A, Karp J. A phase 1 study of the PARP inhibitor veliparib in combination with Temozolomide in acute myeloid leukemia. Clin. Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Young K, Starling N, Cunningham D. Targeting deficient DNA damage repair in gastric cancer. Expert Opin. Pharmacother. 2016;17:1757. doi: 10.1080/14656566.2016.1217992. [DOI] [PubMed] [Google Scholar]
- 122.Liu JF, Matulonis UA. What Is the Place of PARP Inhibitors in Ovarian Cancer Treatment? Curr. Oncol. Rep. 2016;18:29. doi: 10.1007/s11912-016-0515-z. [DOI] [PubMed] [Google Scholar]
- 123.Zaremba T, Curtin NJ. PARP inhibitor development for systemic cancer targeting. Anti-Cancer Agents Med. Chem. 2007;7:515–523. doi: 10.2174/187152007781668715. [DOI] [PubMed] [Google Scholar]
- 124.Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, Lamartina S, Monteagudo E, Ontoria JM, Orsale MV, Palumbi MC, Pesci S, Roscilli G, Scarpelli R, Schultz-Fademrecht C, Toniatti C, Rowley M. Discovery of 2-{4-[(3S)-Piperidin-3-yl]phenyl}-2H–indazole-7-carboxamide (MK-4827): A Novel Oral Poly(ADP-ribose)polymerase (PARP) Inhibitor Efficacious in BRCA-1 and −2 Mutant Tumors. J. Med. Chem. 2009;52:7170–7185. doi: 10.1021/jm901188v. [DOI] [PubMed] [Google Scholar]
- 125.Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, Kreischer N, Thway K, Gevensleben H, Sun L, Loughney J, Chatterjee M, Toniatti C, Carpenter CL, Iannone R, Kaye SB, de Bono JS, Wenham RM. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–892. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 126.Jones P, Wilcoxen K, Rowley M, Toniatti C. Niraparib: A Poly(ADP-ribose) Polymerase (PARP) Inhibitor for the Treatment of Tumors with Defective Homologous Recombination. J. Med. Chem. 2015;58:3302–3314. doi: 10.1021/jm5018237. [DOI] [PubMed] [Google Scholar]
- 127.Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ, Webber SE, Curtin NJ. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol. Cancer Ther. 2007;6:945–956. doi: 10.1158/1535-7163.MCT-06-0552. [DOI] [PubMed] [Google Scholar]
- 128.Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68:1777–1785. doi: 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- 129.Sallmyr A, Fan J, Datta K, Kim K-T, Grosu D, Shapiro P, Small D, Rassool F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111:3173–3182. doi: 10.1182/blood-2007-05-092510. [DOI] [PubMed] [Google Scholar]
- 130.Forman HJ, Bernardo A, Davies KJ. What is the concentration of hydrogen peroxide in blood and plasma? Arch. Biochem. Biophys. 2016;603:48–53. doi: 10.1016/j.abb.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 131.Peng X, Gandhi V. ROS-activated anticancer prodrugs: a new strategy for tumor-specific damage. Ther. Delivery. 2012;3:823–833. doi: 10.4155/tde.12.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cao S, Wang Y, Peng X. ROS-inducible DNA cross-linking agent as a new anticancer prodrug building block. Chem. -Eur. J. 2012;18:3850–3854. doi: 10.1002/chem.201200075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang L, Xie S, Ma L, Chen Y, Lu W. 10-Boronic acid substituted camptothecin as prodrug of SN-38. Eur. J. Med. Chem. 2016;116:84–89. doi: 10.1016/j.ejmech.2016.03.063. [DOI] [PubMed] [Google Scholar]
- 134.Jourden JLM, Daniel KB, Cohen SM. Investigation of self-immolative linkers in the design of hydrogen peroxide activated metalloprotein inhibitors. Chem. Commun. 2011;47:7968–7970. doi: 10.1039/c1cc12526e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bell-Horwath TR, Vadukoot AK, Thowfeik FS, Li G, Wunderlich M, Mulloy JC, Merino EJ. Novel ROS-activated agents utilize a tethered amine to selectively target acute myeloid leukemia. Bioorg. Med. Chem. Lett. 2013;23:2951–2954. doi: 10.1016/j.bmcl.2013.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jones AR, Bell-Horwath TR, Li G, Rollmann SM, Merino EJ. Novel Oxidatively Activated Agents Modify DNA and Are Enhanced by Ercc1 Silencing. Chem. Res. Toxicol. 2012;25:2542–2552. doi: 10.1021/tx300337j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thowfeik FS, AbdulSalam SF, Wunderlich M, Wyder M, Greis KD, Kadekaro AL, Mulloy JC, Merino EJ. A ROS-Activatable Agent Elicits Homologous Recombination DNA Repair and Synergizes with Pathway Compounds. ChemBioChem. 2015;16:2513–2521. doi: 10.1002/cbic.201500304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vadukoot AK, AbdulSalam SF, Wunderlich M, Pullen ED, Landero-Figueroa J, Mulloy JC, Merino EJ. Design of a hydrogen peroxide-activatable agent that specifically targets cancer cells. Bioorg. Med. Chem. 2014;22:6885–6892. doi: 10.1016/j.bmc.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]