

Abstract
Corticotropin-releasing factor (CRF) signaling in the central nucleus of the amygdala (CeA) is hypothesized to drive the development of alcohol dependence, as it regulates ethanol intake and several anxiogenic behaviors linked to withdrawal. Excitatory glutamatergic neurotransmission contributes to alcohol reinforcement, tolerance and dependence. Therefore, in this study we used in vitro slice electrophysiology to investigate the effects of CRF and its receptor subtype (CRF1 and CRF2) antagonists on both evoked and spontaneous action potential-independent glutamatergic transmission in the CeA of naive and ethanol-dependent Sprague-Dawley rats. We found that CRF (25–200 nM) concentration-dependently diminished evoked compound excitatory postsynaptic potentials (EPSPs), but increased miniature excitatory postsynaptic current (mEPSC) frequencies similarly in CeA neurons of both naïve and ethanol-dependent rats, indicating reduced evoked glutamatergic responses and enhanced vesicular glutamate release, respectively. This CRF-induced vesicular glutamate release was prevented by the CRF1/2 antagonist (Astressin B) and the CRF1 antagonist (R121919), but not by the CRF2 antagonist (Astressin 2B). Similarly, CRF’s effects on evoked glutamatergic responses were completely blocked by CRF1 antagonism, but only slightly decreased in the presence of the CRF2 antagonist. Moreover, CRF1 antagonism reveals a tonic facilitation of vesicular glutamate, whereas the CRF2 antagonism revealed a tonic inhibition of vesicular glutamate release. Collectively our data show that CRF primarily acts at presynaptic CRF1 to produce opposite effects on CeA evoked and spontaneous glutamatergic release and that the CRF system modulates CeA glutamatergic synapses throughout the development of alcohol dependence.
Keywords: amygdala, glutamate, alcohol/ethanol, CRF, CRF receptor, electrophysiology
1. INTRODUCTION
Alcohol dependence is a chronic relapsing disorder, defined by the emergence of a negative emotional state that is mediated by the recruitment of brain stress systems, including the central nucleus of the amygdala (CeA) (Koob, 2008; Koob and Le Moal, 2008; Roberto et al., 2012). The CeA is the major output relay of the amygdala complex, and serves as a neuropeptidergic hub of anxiety, stress, and addiction-related functioning. In particular, the stress peptide corticotropin-releasing factor (CRF) plays a critical role in several CeA-driven addiction-related processes, including ethanol consumption and the anxiogenic effects of drug withdrawal (Breese et al., 2005; Eckardt et al., 1998; Heinrichs et al., 1995; Koob, 1998; Koob and Le Moal, 2001; Pich et al., 1995; Rassnick et al., 1993; Roberts et al., 1996).
CRF and its receptors (CRF1 and CRF2) are widely distributed in several stress-responsive brains regions of many species (e.g., human (Charlton et al., 1997), rat (Fischman and Moldow, 1982), and mouse (Justice et al., 2008)), playing a critical role in integrating the body’s overall response to stress (Koob, 1999). The peptide can be synthesized and stored at specific GABAergic and glutamatergic synapses (Cain et al., 1991; Valentino et al., 2001a; Valentino et al., 2001b), and is usually co-released with classical neurotransmitters (Partridge et al., 2016) in response to neuronal firing to modulate their synaptic effects (Rainnie et al., 1992; Yu and Shinnick-Gallagher, 1998). The CRF system can produce a variety of region-, cell type- and synapse-specific effects depending on the distribution of its signaling elements within local and regional circuits (Henckens et al., 2016a). Both CRF1 and CRF2 mRNA expression have been detected in the CeA (Van Pett et al., 2000), and their pre- vs. postsynaptic distribution is critical for the CRF-induced inhibition and facilitation of glutamatergic transmission within this nucleus (Liu et al., 2004; Liu et al., 2005). As CRF signaling is critical to the role of the CeA in anxiety and alcohol use disorders (Gilpin et al., 2015), understanding its regulation of CeA glutamatergic transmission and its potential neuroadaptation with ethanol dependence are important.
Glutamate is the major excitatory neurotransmitter in the central nervous system and its ethanol-induced dysregulation contributes to several addiction-related behaviors, including drug reinforcement, tolerance and dependence (Lovinger and Roberto, 2013). Although the CeA mainly (95%) comprises γ-aminobutyric acid (GABA) neurons, it receives numerous glutamatergic afferents from other important addiction-related regions, including the basolateral amygdala, thalamus and cortex (Krettek and Price, 1978; Pitkanen et al., 1995; Savander et al., 1995). We recently reported on the effects of CRF and a CRF1 antagonist on spontaneous and evoked glutamatergic transmission in CeA neurons of Wistar rats and Marchigian Sardinian Preferring (msP) rats, a line genetically selected for excessive ethanol drinking and characterized by heightened activity of the CRF1 system that mimics the post-dependent phenotype (Ciccocioppo et al., 2006; Herman et al., 2016; Natividad et al., 2017). We found that CRF had mixed effects on spontaneous action potential-dependent and -independent glutamate release in the CeA of Wistar rats (with mainly a CRF-induced increase in action potential-independent glutamate release). However, in the msP rats, CRF had mixed effects on action potential-independent glutamate release, but only increased action potential-dependent glutamate release. CRF also decreased evoked CeA glutamatergic transmission in Wistar, but not in msP rats, though its site(s) of action (CRF1 vs. CRF2) remain unknown. We and others have also previously shown that ethanol dependence is associated with increased CRF influence over GABA release in the CeA (Roberto et al., 2010), and that systemic and intra-CeA CRF1 blockade both prevent the development of excessive ethanol consumption (Roberto et al., 2010; Varodayan et al., 2017) and withdrawal-associated anxiety-like behavior (Rassnick et al., 1993). However, to our knowledge, CRF’s regulation of CeA glutamatergic synapses has yet to be investigated after ethanol dependence.
In this study, we used an in vitro slice preparation to assess the effects of CRF and its receptor antagonists on both evoked and spontaneous action potential-independent glutamatergic transmission in the CeA of naive and ethanol-dependent Sprague-Dawley rats. We found that CRF acts predominantly at CRF1 to differentially modulate both forms of glutamatergic transmission at CeA synapses, and that ethanol dependence does not alter this regulation by the CRF system.
2. MATERIALS AND METHODS
2.1 Animals and slice preparation
We used 107 adult male Sprague-Dawley rats (average weight 346±10 g) from Charles River (Raleigh, NC), housed 2–3 per cage in a temperature- and humidity-controlled room on a 12-h light/dark cycle (lights on at 8:00 am) with food and water available ad libitum. We conducted all care procedures in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with the Institutional Animal Care and Use Committee (IACUC) policies of The Scripps Research Institute (TSRI).
2.2 Chronic Ethanol Treatment
We used the standard ethanol inhalation method of the TSRI Alcohol Research Center to induce ethanol dependence (Cruz et al., 2013; Kallupi et al., 2014; Roberto et al., 2010; Roberto et al., 2004a; Roberto et al., 2004b; Roberto and Siggins, 2006; Rogers et al., 1979). Briefly, ethanol-dependent rats (n=36) had their home cages placed inside ethanol vapor chambers, and were intermittently exposed (14 h on, 10 h off) to ethanol vapor for 5–7 weeks. We determined the blood alcohol levels (BALs) of the chronic ethanol-treated animals using weekly tail-blood samples. The target BAL range was 150–200 mg/dL and the mean BAL was 177±10 mg/dL. Naïve/control rats (n=71) were treated similarly, but exposed to air 24 h/day. On experiment days, chronic ethanol-exposed rats were maintained in the vapor chamber until preparation of the CeA slices in ethanol-free solutions. Thus, all electrophysiology experiments were performed in brain slices undergoing acute in vitro withdrawal (2–8 hours), as previously described (Kallupi et al., 2014; Roberto et al., 2010; Roberto et al., 2004a; Roberto et al., 2004b; Varodayan et al., 2017).
2.3 Slice preparation
We prepared CeA slices as previously described (Cruz et al., 2012; Herman et al., 2016; Roberto et al., 2003; Roberto et al., 2004a; Roberto et al., 2004b; Roberto and Siggins, 2006). Briefly, the animals were anesthetized with isoflurane (3%) prior to decapitation, and 300–400 μm coronal slices were sectioned on a Leica 1000S vibratome (Leica Microsystems, Buffalo Grove, IL).
2.4 Intracellular recording of evoked responses
CeA slices were incubated in an interface configuration for 15–20 min, and then completely submerged and continuously superfused (flow rate of 2–4 ml/min) with 95% O2/5% CO2 equilibrated artificial cerebrospinal fluid (aCSF) of the following composition in mM: NaCl, 130; KCl, 3.5; NaH2PO4, 1.25; MgSO4•7H2O, 1.5; CaCl2, 2.0; NaHCO3, 24; glucose, 10. We recorded from a total of 150 CeA neurons (from the medial subdivision of the CeA) with sharp micropipettes filled with 3M KCl using discontinuous current-clamp mode (Kallupi et al., 2014). Data were acquired with an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA) and stored for later analysis using pClamp software (Axon Instruments). We held most neurons near their resting membrane potential (RMP) and applied hyperpolarizing and depolarizing current steps (200 pA increments, 750 msec duration) to generate I–V curves. We evoked pharmacologically-isolated excitatory postsynaptic potentials (EPSPs) by stimulating locally within the CeA through a bipolar stimulating electrode and superfusing the slices with aCSF containing the GABA receptor blockers: 30 μM bicuculline (to block GABAA receptors) and 1 μM [1-(S)-3,4-dichlorophenyl)ethyl]amino-2-(S)-hydroxypropyl-p- benzylphosphonic acid (CGP 55845A) (to block GABAB receptors). At the end of the recording session we often added 30 μM 6,7-Dinitroquinoxaline-2,3-dione (DNQX) and 30 μM DL-2-Amino-5-phosphonopentanoic acid (DL-AP5) to the aCSF to confirm the glutamatergic nature of the EPSP. To determine the synaptic response parameters for each cell, we performed an input-output (I-O) protocol (Cruz et al., 2012; Kallupi et al., 2014; Roberto et al., 2010; Roberto et al., 2003; Roberto et al., 2004a) consisting of a range of five current stimulations (50–250 mA; 0.125 Hz), starting at the minimum current required to elicit an EPSP up to the strength required to elicit the maximum subthreshold amplitude. These stimulus strengths were maintained throughout the entire duration of the experiment.
We examined paired-pulse facilitation (PPF) in each neuron using paired stimuli at 50 and 100 ms inter-stimulus interval (Andreasen and Hablitz, 1994; Logrip M., 2017; Roberto et al., 2004b). PPF was calculated as the amplitude of the second EPSP over that of the first EPSP. The stimulus strength was adjusted such that the amplitude of the first EPSP was ~50% of the maximal amplitude determined by the I-O protocol. A drug-induced change in PPF reflects presynaptic effects such that an increase in PPF suggests a decrease in neurotransmitter (glutamate) release. All measures were taken before drug superfusion (control) and during drug superfusion (10–20 min).
2.5 Whole-cell patch-clamp recording of glutamate currents
CeA slices were sectioned and then incubated in oxygenated aCSF for 30 min at 37°C and then 30 min at room temperature. We recorded from 87 medial CeA neurons that were visualized with infrared differential interference contrast (IR-DIC) optics, a x60 water immersion objective (Olympus BX51WI, Tokyo, Japan) and a CCD camera (EXi Aqua, QImaging, Surrey, BC, Canada). Whole-cell voltage-clamp recordings were performed in gap-free acquisition mode with a sampling rate of 10 kHz and low-pass filtered at 10 kHz, using a Multiclamp 700B amplifier, Digidata 1440A and pClamp 10 software (Molecular Devices, Sunnyvale, CA). Pipettes (3–7 MΩ; King Precision, Claremont, CA) were filled with internal solution (in mM): 145 Kgluconate; 0.5 EGTA; 2 MgCl2; 10 HEPES; 2 Na-ATP; 0.2 Na-GTP. Spontaneous miniature excitatory postsynaptic currents (mEPSCs) were isolated with the GABA receptor blockers, 30 μM bicuculline and 1 μM CGP 55845A, as well as 0.5 μM tetrodotoxin (TTX). Neurons were clamped at −60 mV and the series resistance was monitored with a 10 mV pulse (experiments with a series resistance >15 MΩ or a >20% change were excluded).
2.6 Drugs
We purchased CGP 55845A and bicuculline from Sigma (St. Louis, MO, USA), DNQX and DL-AP5 from Tocris (Ellisville, MO), and tetrodotoxin (TTX) from Biotum (Hayward, CA). CRF, Astressin B (antagonist for both CRF1 and CRF2) and Astressin 2B (CRF2 antagonist) were synthesized and provided by Dr. Jean Rivier at the Salk Institute for Biological Studies. R121919 (CRF1 antagonist) was synthesized by Dr. Kenner Rice at the Drug Design and Synthesis Section, Chemical Biology Research Branch, National Institute on Drug Abuse, National Institutes of Health, Bethesda, MD. Bicuculline was first dissolved in dimethylsulfoxide (DMSO) before being added to aCSF superfusate (final concentration of 0.05–0.1% DMSO). Drugs were added to the aCSF from stock solutions to obtain known concentrations in the superfusate.
2.7 Data analysis and statistics
To analyze data acquired from intracellular recordings, we quantified synaptic responses by calculating the EPSP amplitude with Clampfit 10 software. For whole cell recordings, the mEPSC frequency, amplitude (only mEPSCs >5 pA were included), and kinetics were analyzed using Mini Analysis software (Synaptosoft Inc., Fort Lee, NJ) and visually confirmed. Average mEPSC characteristics were determined over 3–5 min of recording trace and a minimum of 50 events. To control for cell-to-cell variation in all baseline electrophysiology properties, drug effects were normalized to their own baseline prior to group analyses. We used GraphPad Prism 6 software (GraphPad Software, La Jolla, CA) for all statistical analysis of results.
Pooled data for each experimental condition were analyzed by one-sample t-test for individual means comparisons to baseline to evaluate single drug (e.g. CRF) effects and paired t-test for comparison of successive drug effects (e.g. CRF antagonist vs. CRF antagonist + CRF) within the same animal treatment group. To assess differences resulting from ethanol exposure (i.e. naive vs. ethanol-dependent rats) and drug effects between these groups, we used a t-test or ANOVA as appropriate. Additionally, the Bonferroni post hoc test was used to assess significance between treatments as appropriate. We accepted statistical significance at the p<0.05 level. All data are presented as mean ± standard error of the mean (SEM). n is reported in the figure legends, and represents the cell number from a minimum of four animals (1 to 5 cells were recorded per animal).
3. RESULTS
3.1 Chronic ethanol exposure alters glutamatergic transmission at CeA synapses
To investigate whether chronic ethanol treatment altered baseline glutamatergic signaling in the rat CeA, we recorded both evoked (intracellular configuration with sharp pipettes) and spontaneous (whole-cell voltage-clamp configuration) glutamate transmission.
We recorded intracellularly from a total of 157 neurons in the medial subdivision of the CeA. The mean resting membrane potential (RMP) and input resistance for the 84 CeA neurons from naïve rats were −81±0.7 mV and 169±7 MΩ, and for the 73 CeA neurons from ethanol-dependent rats (cells recorded in ethanol-free conditions equivalent to early withdrawal) were −80±0.6 mV and 172±8 MΩ, respectively. We generated compound excitatory postsynaptic potential (EPSP) input-output (I-O) curves using 5 normalized stimulation intensities (Fig. 1A). A two-way ANOVA of stimulus intensity and ethanol treatment did not show a significant difference between the compound EPSP I-O curves in slices from naive and ethanol-dependent animals (F(1,155)=1.42, p=0.2, n=157 cells), indicating that chronic ethanol treatment did not alter evoked glutamate transmission in the CeA. There was a main effect of stimulus intensity (F(4,620)=483.4, p<0.001), with a Bonferroni post-hoc test indicating EPSP amplitudes were significantly different from each other (p<0.001 per comparison) among the five intensities. Finally, there was no interaction effect between the stimulus intensity and ethanol treatment (F(4,620)=0.96, p=0.4).
Figure 1. Chronic ethanol exposure alters CeA glutamatergic synapse function.

A) Top: Representative traces of evoked CeA EPSPs at the five normalized stimulus intensities from a naïve and an ethanol-dependent rat (Dep). Bottom: I-O relationship curves are similar for naïve and dependent rats (84 cells from 61 naïve rats, 73 cells from 31 dependent rats). B) Top: Representative traces of PPF of EPSPs at the 50 ms ISI from naïve and dependent rat CeA neurons. Bottom: Histograms representing group PPF ratio for naïve and dependent rats at 50 ms and 100 ms ISI (46 cells from 34 naïve rats, 30 cells from 17 dependent rats). C) Left: Representative mEPSC traces from CeA neurons of naive and ethanol-dependent rats in baseline conditions. Middle and Right: Group comparison showed significantly lower baseline mEPSC frequencies and amplitudes in dependent rats compared to naïve rats (42 cells from 19 naïve rats, 45 cells from 10 dependent rats). **p<0.01 by unpaired t-test.
We also examined paired-pulse facilitation (PPF), a phenomenon whereby a secondary synaptic response is influenced by a preceding primary stimulus of equal intensity (Andreasen and Hablitz, 1994; Manabe et al., 1993). Generally, changes in the PPF ratio (second EPSP/first EPSP) are inversely related to transmitter release, such that a reduction of the PPF ratio is associated with an increased probability of transmitter release (Roberto et al., 2004b). We examined the PPF at the half-maximal intensity determined from the I-O curve using inter-stimulus intervals (ISI) of 50 ms and 100 ms (Fig. 1B). In slices from naïve animals, the PPF was 1.45±0.08 and 1.23±0.04 for 50 ms and 100 ms ISI, respectively (n=46 cells). In slices from dependent animals, the PPF was 1.27±0.07 and 1.20±0.06 for 50 ms and 100 ms ISI (n=30 cells). An unpaired t-test between naïve and dependent animals found no differences between groups for both the 50 ms (t(74)=1.64, p=0.1) and 100 ms (t(74)=0.37, p=0.7) ISI.
Next, we determined the effects of chronic ethanol treatment on spontaneous glutamate transmission by studying miniature excitatory postsynaptic currents (mEPSCs). This vesicular form of glutamate release is recorded in the presence of TTX to pharmacologically block action potentials, and so is distinct from the evoked glutamatergic transmission (EPSP) that is produced by electrically stimulating the entire synaptic network (Atasoy et al., 2008; Kavalali, 2015). We found that ethanol-dependent rats had a significantly lower mEPSC frequency (t(85)=3.22, p<0.01, n=42 naïve cells, n=45 dependent cells) and amplitude (t(85)=2.64, p<0.01) than that of naïve rats, with no change in the rise or decay times (Fig. 1C). Specifically, in naïve rats the mean frequency was 0.90±0.13 Hz, amplitude was 26.1±1.0 pA, rise time was 1.23±0.06 ms and decay time was 0.90±0.11 ms, while in the ethanol-dependent rats, the mean frequency was 0.46±0.06 Hz, amplitude was 22.9±0.68 pA, rise time was 1.18±0.05 ms and decay time was 0.97±0.07 ms. As mEPSCs are action potential-independent, increased frequencies reflect higher glutamate release probabilities, whereas altered amplitudes/kinetics indicate changed glutamate receptor sensitivity (De Koninck and Mody, 1994; Otis et al., 1994). Therefore, chronic ethanol decreased both spontaneous glutamate release and postsynaptic glutamate receptor function at CeA synapses.
3.2 CRF decreases CeA evoked glutamatergic transmission similarly in naïve and ethanol-dependent rats
We first assessed the effects of 4 concentrations of CRF (25, 50, 100, and 200 nM) on evoked glutamate transmission in the CeA of naïve rats. The histograms in Figure 2A express the peak CRF effects as a percent of baseline evoked EPSP amplitudes at half-max stimulus intensity obtained from the I-O relationship during CRF superfusion. The lowest concentration of CRF (25 nM) caused a non-significant decrease in EPSP amplitude (90.0±5.2% of control; t(7)=1.91, p=0.1, n=8 cells), while 50, 100 and 200 nM CRF significantly decreased the EPSP amplitude to 89.3±3.4% (t(6)=3.19, p<0.05, n=7 cells), 89.6±2.7% (t(15)=3.91, p<0.01, n=16 cells) and 83.4±3.0% (t(6)=5.48, p<0.01, n=7 cells) of control, respectively. Overall, a one-way ANOVA demonstrated that there was no significant difference in the inhibitory effect of CRF among the different concentrations after 15 min of application (F(3,34)=0.75, p=0.5). In this experiment, we also found that CRF, at all concentrations applied, did not significantly alter the neuronal RMP or input resistance.
Figure 2. CRF decreases evoked EPSP amplitude in CeA neurons of naïve and ethanol-dependent rats.
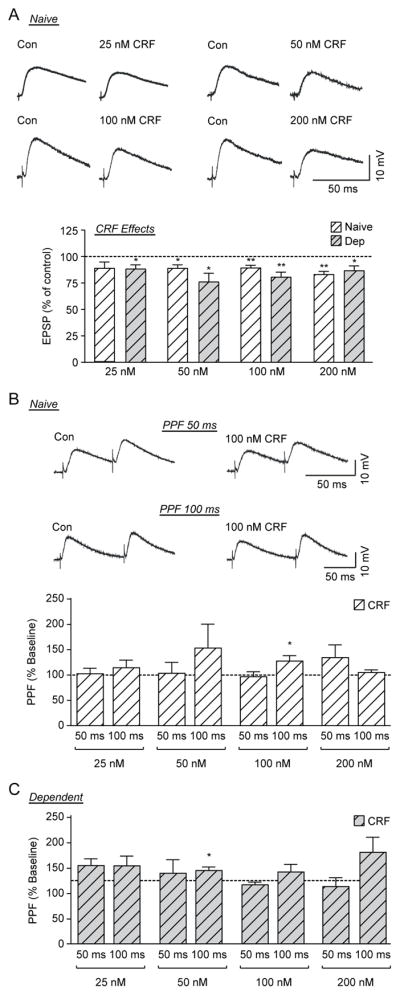
A) Top: Representative recordings of evoked EPSPs in CeA neurons from naïve rats before and during CRF application. Bottom: Histograms representing percent peak decrease in evoked (at half max stimulus intensity) EPSP amplitudes during superfusion of different concentrations (25, 50, 100 and 200 nM) of CRF on the CeA of naïve and ethanol-dependent rats (6–16 cells from at least 4 naïve or dependent rats per group). B) Top: Representative recordings of PPF at both 50 ms (top traces) and 100 ms (bottom traces) ISI in a CeA neuron from a naïve rat before and during superfusion of 100 nM CRF. Bottom: Histograms representing PPF ratio as a percentage of baseline after superfusion of 4 concentrations of CRF at 50 ms and 100 ms ISI in naïve rats. CRF significantly increased the PPF ratio of evoked EPSPs at the 100 ms ISI with 100 nM CRF C) Histograms representing PPF ratio as a percentage of baseline after superfusion of 4 concentrations of CRF at 50 ms and 100 ms ISI in ethanol-dependent rats. CRF significantly increased the PPF ratio of evoked EPSPs at the 100 ms ISI with 50 nM CRF. *p <0.05 and **p<0.01 by one-sample t-test.
We then tested whether chronic ethanol exposure altered the effects of CRF (25–200 nM) on CeA evoked glutamate transmission. All 4 concentrations of CRF significantly decreased the EPSP amplitude at the middle stimulus intensity in the CeA of ethanol-dependent rats, similar to naïve animals (Fig. 2A). CRF reduced EPSP amplitudes to 88.7±4.0% (t(6)=2.85, p<0.05, n=7 cells), 76.4±8.3% (t(5)=2.85, p<0.05, n=6 cells), 80.9±4.9% (t(8)=3.95, p<0.01, n=9 cells), and 87.1±4.5% (t(5)=2.86, p<0.05, n=6 cells) of control for 25, 50, 100, and 200 nM CRF respectively. A two-way ANOVA of CRF concentration and ethanol treatment showed no difference in the effect of CRF concentration (F(3,58)=0.36; p=0.8) or ethanol treatment (F(1,58)=1.00; p=0.3), with no interaction effect (F(3,58)=2.08, p=0.1), suggesting that ethanol dependence does not alter the CRF-induced decrease in glutamatergic transmission in the CeA.
We also examined the effects of CRF on PPF in CeA neurons of naïve and ethanol-dependent rats, and observed mixed responses (Fig. 2B and C). In the CeA of naïve rats, only 100 nM CRF at the 100 ms ISI produced a significant (t(12)=2.56, p<0.05, n=13 cells) change in PPF ratio, increasing it to 128.2±11.0% of baseline (Fig. 2B). Additionally, one-way ANOVAs at 50 ms (F(3,29)=0.92, p=0.4, n=33 cells) and 100 ms (F(3,30)=0.61, p=0.6, n=34 cells) ISI found no significant effects of CRF concentration on PPF ratios.
Similarly, in the CeA of ethanol-dependent rats, only 50 nM CRF at 100 ms ISI caused a significant (t(4)=2.86, p<0.05, n=5 cells) increase (116.2±5.6% of baseline) in PPF ratio (Fig. 2C). Also, one-way ANOVAs of CRF concentrations at the 50 ms (F(3,22)=1.77, p=0.2, n=26 cells) and 100 ms (F(3,19)=0.80, p=0.5, n=22 cells) ISI revealed no effects on PPF ratios. Finally, we used a two-way RM ANOVA of CRF treatment and ethanol exposure to examine whether the effects of CRF differed between naïve and ethanol-dependent animals. A two-way ANOVA of CRF concentration and ethanol treatment found no significant effects of CRF (F(3,51)=0.72, p=0.55, n=53 cells) or ethanol treatment (F(1,51)=0.20, p=0.7) with no interaction effect (F(3,51)=1.45, p=0.2) at the 50 ms ISI, and no significant effects of CRF (F(3,55)=0.61, p=0.6, n=57 cells) or ethanol treatment (F(1,55)=0.75, p=0.4), and no interaction effect (F(3,55)=0.46, p=0.7) at the 100 ms ISI. Therefore, CRF decreased evoked glutamate release at CeA synapses similarly in naïve and ethanol-dependent rats. Because 50, 100, and 200 nM CRF induced similar inhibitions of the evoked EPSPs, we used 100 nM CRF throughout the rest of the study.
3.3 CRF1 receptors mediate the CRF-induced decrease of evoked glutamatergic transmission
To assess whether endogenous CRF acts at CRF receptors to regulate evoked glutamate transmission, and to identify the specific CRF receptor(s) that mediates this effect, we used several pharmacological tools. We found that 200 nM Astressin B, a non-selective CRF1/2 receptor antagonist (Liu et al., 2004), significantly decreased evoked EPSP amplitudes (to 76.8±4.2% of control baseline; t(7)=5.57, p<0.001, n=8 cells) in the CeA of naïve rats (Fig. 3A). In 7 of these neurons, we co-applied 100 nM CRF in the continued presence of Astressin B and found that CRF did not further change the evoked EPSPs (72.9±6.6% of control baseline; t(6)=0.43, p=0.7 by paired t-test between AB and AB+CRF data) (Fig. 3A). These data indicate that the CRF1/2 system promotes excitatory transmission in the CeA by increasing glutamate release under basal conditions.
Figure 3. The CRF-induced decrease in evoked EPSP amplitude is mediated by CRF1.

A) Top: Representative recordings of evoked EPSPs in a CeA neuron from a naïve rat recorded in control conditions, during Astressin B (AB) application, and during AB+CRF application. Bottom: Plot representing the percent peak decrease in evoked EPSP amplitudes for the middle three stimulus intensities during superfusion of AB and AB+CRF (7–8 cells from 7–8 naïve rats). CRF receptor antagonism decreases the evoked EPSP amplitude in naïve rats and blocks any further CRF-induced decrease. B) Top: Representative evoked EPSPs in a CeA neuron from a naïve rat recorded in control conditions, during R121919 (R12) application, and during R12+CRF application. Bottom: Plot representing the percent peak decrease in evoked EPSP amplitudes for the middle three stimulus intensities during superfusion of R12 and R12+CRF (9–20 cells from 9–17 naïve rats). CRF1 antagonism decreases the evoked EPSP amplitude in naïve rats and blocks any further CRF-induced decrease. C) Top: Representative evoked EPSPs in a CeA neuron from a naïve rat in control conditions, during Astressin 2B (A2B) application, and during A2B+CRF application. Bottom: Plot representing the percent peak decrease in evoked EPSP amplitudes for the middle three stimulus intensities during superfusion of A2B and A2B+CRF (10 cells from 9 naïve rats). CRF2 antagonism does not alter the evoked EPSP amplitude from baseline at the middle stimulus intensity in naïve rats, while co-application of CRF with A2B decreases the evoked EPSP. D) Histograms representing the percent peak decrease in evoked EPSP amplitudes for the middle stimulus intensity during superfusion of R12 and A2B show no difference between naïve and ethanol-dependent rats (Dep). E) Left: Representative PPF of EPSPs at 50 ms (top) and 100 ms (bottom) ISI in CeA neurons from an ethanol-dependent rat before and during superfusion of R12. Right: Histograms representing PPF ratio as a percentage of baseline after superfusion of R12 and A2B at 50 ms and 100 ms ISI in naïve and ethanol-dependent rats. R12 significantly increased the PPF ratio of evoked EPSPs for naïve rats at the 100 ms ISI. *p<0.05 from baseline by one-sample t-test.
We next tested R121919, a selective antagonist for CRF1, to probe for a potential role of this receptor in CeA evoked glutamate transmission. R121919 (1 μM) (Herman et al., 2016) significantly decreased evoked EPSPs (86.4±2.7% of control at half maximal stimulus intensity; t(19)=4.96, p<0.001, n=20 cells; Fig. 3B and D). To investigate whether CRF decreased EPSPs in the CeA by activating CRF1, we applied CRF in the presence of R121919 in 9 cells. We found that CRF no longer decreased the EPSP amplitude (R121919: 88.0±3.1% of control; R121919+CRF: 85.7±4.4% of control; t(8)=0.57, p=0.6; Fig. 3B), indicating that the inhibitory effect of CRF occurs mainly through CRF1.
We also tested whether selective CRF2 blockade affects CRF-induced decreases in EPSPs. Application of the selective CRF2 antagonist Astressin 2B (200 nM) (Silberman and Winder, 2013) did not significantly affect evoked EPSPs (92.3±3.7% of control; t(9)=2.08, p=0.1, n=10 cells) and the addition of CRF decreased EPSPs (to 84.7±4.7% of control; t(9)=3.25, p<0.05; Fig. 3C and D). However, a paired t-test between the effects of Astressin 2B alone and Astressin 2B+CRF showed no significant difference in their effects on EPSPs (t(9)=1.65, p=0.1), indicating that CRF2 may also partially contribute to CRF’s inhibitory effects.
We next examined the effects of R121919 and Astressin 2B on PPF in naïve rats. R121919 increased the PPF ratio to 115.3±9.2% of baseline for the 50 ms ISI and to 122.3±8.9% of baseline for the 100 ms ISI (Fig. 3E). This increase in PPF ratio was significant at the 100 ms ISI (t(13)=2.50, p<0.05, n=14 cells) indicating a decrease of evoked glutamate release, but not at the 50 ms ISI (t(14)=1.66, p=0.1, n=15 cells). Astressin 2B did not alter PPF ratios (Fig. 3E).
To determine whether chronic ethanol exposure altered the CRF system’s regulation of evoked glutamatergic transmission in CeA neurons, we tested 1 μM R121919 and 200 nM Astressin 2B. The CRF1 antagonist significantly (t(8)=3.12, p<0.05) decreased the EPSP amplitude to 90.8±3.0% of control (at half maximal stimulus intensity; n=9 cells; Fig. 3D), suggesting that endogenous CRF/CRF1 signaling regulates CeA activity in ethanol-dependent rats. This effect of R121919 was not significantly different between naïve and ethanol-dependent rats (t(27)=0.96, p=0.3, n=29 cells). Also similar to naïve rats, R121919 increased PPF ratios in the dependent rats to 149.7±32.1% of baseline for the 50 ms ISI and 125.2±16.5% of baseline for the 100 ms ISI, however, neither of these effects were significantly different from baseline (t(8)=1.55, p=0.2, n=9 cells and t(8)=1.53, p=0.2, n=9 cells for 50 and 100 ms ISI respectively; Fig. 3E). However, there was no significant difference between the effect of R121919 on PPF ratios between naïve and dependent rats for either the 50 ms (t(22)=1.26, p=0.1, n=24 cells) or the 100 ms (t(21)=0.17, p=0.9, n=23 cells) ISI. The CRF2 antagonist did not significantly alter (t(6)=0.87, p=0.4, n=7 cells) the EPSP amplitude in the ethanol-dependent rats (94.14±6.7% of baseline), and an unpaired t-test found no differences in the effects of Astressin 2B between naïve and dependent animals (t(15)=0.27, p=0.8, n=17 cells) (Fig 3D). Similar to CeA neurons of naive rats, Astressin 2B did not alter PPF ratios in 7 neurons of ethanol-dependent rats (Fig. 3E). Therefore, the CRF/CRF1 system continues to modulate evoked glutamatergic transmission in the CeA after ethanol dependence.
3.4 CRF increases action potential-independent glutamate release similarly in the CeA of naïve and ethanol-dependent rats
To further pursue the site of CRF’s action at CeA glutamatergic synapses, we examined mEPSCs in neurons of naive and ethanol-dependent rats. We found that CRF (100 nM) increased the mEPSC frequency in CeA cells from naïve rats (155.7±16.9%; t(8)=3.29, p<0.05, n=9 cells; Fig. 4A); however, in 2/9 cells CRF had no effect or slightly decreased (<5% change from baseline) mEPSC frequencies, indicating that CRF does not enhance the glutamatergic input to all CeA neurons (similar to (Herman et al., 2016)). There were no CRF-induced changes in mEPSC amplitude or kinetics (Fig. 4A). Therefore, our data suggest that CRF acts presynaptically to increase vesicular (action potential-independent) glutamate release in the CeA of naïve rats. Similarly, CRF significantly increased the mEPSC frequency in cells from ethanol-dependent rats (141.2±17.1%; t(9)=2.41, p<0.05, n=10 cells), though it had no effect or a slight decrease on mEPSC frequencies in 4/10 cells and did not alter the overall mEPSC amplitude or kinetics (Fig. 4A). Therefore, CRF enhanced action potential-independent glutamate release at a subset of CeA synapses similarly in naïve and ethanol-dependent rats, without significantly affecting the region’s glutamate receptor composition or expression.
Figure 4. CRF1 activation promotes action potential-independent glutamate release.

A) Left: Representative mEPSC traces from CeA neurons of naive and ethanol-dependent (Dep) rats in baseline conditions and during 100 nM CRF superfusion. Right: CRF significantly increased mEPSC frequencies in the CeA of naïve and ethanol-dependent rats (9 cells from 4 naïve rats, 10 cells from 5 dependent rats). B) Left: Representative mEPSCs from CeA neurons of naive rats in baseline conditions and during 200 nM Astressin B (AB) superfusion. Right: AB significantly reduced the mEPSC frequency, but had no effect on amplitudes or kinetics (10 cells from 6 naïve rats). C) Co-application of CRF and AB did not significantly alter the mEPSC frequency compared to AB alone (7 cells from 5 naïve rats). D) Left: Representative mEPSCs from CeA neurons of naive rats in baseline conditions and during 1 μM R121919 (R12) superfusion. Right: R12 significantly reduced the mEPSC frequency, but had no effect on amplitudes or kinetics (17 cells from 10 naïve rats). E) CRF+R12 did not significantly alter the mEPSC frequency compared to R12 alone (7 cells from 4 naïve rats). F) Left: Representative mEPSCs from CeA neurons of naive rats in baseline conditions and during 1 μM Astressin 2B (A2B) superfusion. Right: A2B significantly increased the mEPSC frequency, but had no effect on amplitudes or kinetics (7 cells from 5 naïve rats). G) CRF+A2B significantly increased the mEPSC frequency compared to A2B alone (6 cells from 5 naïve rats). *p<0.05 by one-sample or paired t-test, as appropriate.
3.5 CRF1 activation promotes action potential-independent glutamate release
We next assessed the effects of the CRF1/2 antagonist Astressin B (200 nM) and found that it significantly decreased the mEPSC frequency (to 82.4±6.1% of control; t(9)=2.89, p<0.05, n=10 cells), but had no effect on the amplitude or kinetics (Fig. 4B). In 7 of these neurons, we co-applied CRF in the presence of Astressin B and found that there was no significant difference in mEPSC frequency between the antagonist alone and Astressin B+CRF (Astressin B: 80.5±8.4% of control; Astressin B+CRF: 88.9±10.5% of control; t(6)=1.61, p=0.16; Fig. 4C). There were also no drug-induced changes in the mEPSC amplitudes and kinetics. Overall, these data indicate that the CRF system tonically promotes excitatory transmission in the CeA by increasing glutamate release.
Similar to the effects of Astressin B on vesicular glutamate release, the CRF1 antagonist R121919 (1 μM) significantly decreased the mEPSC frequency (to 76.2±4.8% of control; t(16)=4.94, p<0.05, n=17 cells), with no effect on the amplitude or kinetics (Fig. 4D). In 7 of these neurons, we applied CRF in the presence of R121919 and there was no further change in mEPSC frequency (R121919: 85.1±4.7% of control; R121919+CRF: 80.0±5.6% of control; t(6)=1.09, p=0.32; Fig. 4E). However, CRF decreased the mEPSC amplitude (t(6)=3.29, p<0.05) and decay time (t(6)=3.65, p<0.05) in this R121919+CRF experiment (data not shown), suggesting that CRF may have additional postsynaptic effects that are not mediated by CRF1. Overall, the R121919 data indicate that CRF1 mediates the tonic effects of endogenous CRF to promote CeA glutamate release, as well as the exogenous CRF-induced facilitation of glutamate release we observed in this region.
In a third set of experiments, we used the CRF2 antagonist Astressin 2B. Notably, Astressin 2B alone significantly increased the mEPSC frequency (to 133.3±9.8% of control; t(6)=3.39, p<0.05, n=7 cells), with no effect on the amplitude or kinetics (Fig. 4F), indicating that the basal activation of CRF2 in the CeA limits glutamate release. Additionally, co-application of Astressin 2B and CRF in 6 of these cells led to a further significant increase in mEPSC frequency (Astressin 2B: 136.5±11.0% of control; Astressin 2B+CRF: 173.0±20.6%; t(5)=2.82, p<0.05; Fig. 4G), with no change in the mEPSC amplitude or kinetics. Thus, CRF appears to activate both CRF1 and CRF2 to regulate vesicular glutamate transmission in the CeA, but its facilitation of glutamate release occurs predominantly via CRF1.
4. DISCUSSION
The amygdalar CRF/CRF1 system plays a critical role in ethanol dependence (Gilpin et al., 2015; Koob, 1999; Koob and Volkow, 2010) and chronic ethanol treatment upregulates the expression of CRF and CRF1 in the CeA (Merlo Pich et al., 1995; Roberto et al., 2010). Systemic CRF1 antagonism prevented the escalated ethanol drinking of dependent rats (Roberto et al., 2010), and intra-CeA administration of a CRF1 antagonist reduced both the ethanol consumption (Funk et al., 2006; Varodayan et al., 2017) and anxiety-like behavior (Rassnick et al., 1993) of these rats. Additionally, CRF acting at CRF1 increased CeA GABAergic transmission in naïve rats, with a greater effect observed in ethanol-dependent animals (Roberto et al., 2010). Despite the key implications of this neuropeptide in the CeA with regard to alcohol-related behaviors, the present study is the first to our knowledge to determine whether ethanol dependence produces neuroadaptation within the CRF system to impact its neuromodulation of CeA glutamatergic synapse function.
Here we showed that CRF increases vesicular glutamate release at CeA synapses in both naive and ethanol-dependent animals, and that tonic CRF1 activity enhances vesicular glutamate release, whereas tonic CRF2 activity inhibits it. This CRF-induced vesicular glutamate release was prevented by CRF1/2 and CRF1 antagonists, but not by the CRF2 antagonist. CRF decreases evoked glutamatergic responses, and this decrease was smaller in the presence of the CRF2 antagonist, but blocked by CRF1 antagonism. Therefore, despite the fact that CRF produces opposite effects on evoked and spontaneous glutamatergic transmission in the CeA of both naïve and ethanol-dependent rats, it acts predominantly via CRF1 at these synapses.
We and others have previously shown that CRF modulates glutamate transmission in the CeA (Gallagher et al., 2008; Herman et al., 2016; Liu et al., 2004; Orozco-Cabal et al., 2006; Silberman and Winder, 2013). Consistent with our previous work in Wistar rats (Herman et al., 2016), we found here that CRF decreases evoked glutamatergic transmission in CeA neurons of naïve Sprague Dawley rats. This effect most likely results from decreases in evoked glutamate release, though we cannot rule out the possibility of CRF having postsynaptic effects across the synaptic network. Moreover, previous work by Liu et al. (Liu et al., 2004) reported a similar CRF-induced inhibition of basolateral amygdala glutamatergic afferents into the CeA (Liu et al., 2004), suggesting that CRF may modulate these projections in the present study. In contrast to these evoked glutamatergic responses, CRF increased the frequency of mEPSCs in the majority of CeA neurons both in the present study and our previous work using Wistar rats (Herman et al., 2016), indicating that it increases spontaneous action potential-independent glutamate release onto a subset of CeA neurons similarly in both naïve and ethanol-dependent rats. Notably, the two forms of glutamate transmission examined here (evoked and spontaneous action potential-independent) are differentially regulated by CRF. Evoked neurotransmission results from the stimulation of the entire synaptic network to elicit widespread classical neural communication, whereas spontaneous action potential-independent neurotransmission reflects isolated synaptic communication that can maintain homeostasis and mediate plasticity at mature synapses (Kavalali, 2015). Thus, the differences we observed in CRF’s modulation of spontaneous and evoked glutamatergic release are likely the result of distinct presynaptic neurotransmitter release mechanisms (e.g. vesicle fusion machinery, spatial segregation of the vesicles and/or vesicle populations) (Atasoy et al., 2008; Kavalali, 2015).
To better understand the mechanisms underlying the opposing actions of CRF at CeA glutamatergic synapses, we performed a series of pharmacology studies using its receptor antagonists. CRF and its G protein-coupled receptors CRF1 and CRF2 are widely expressed in the extended amygdala (Chalmers et al., 1996; Justice et al., 2008; Van Pett et al., 2000). In the CeA, CRF1 activation increases anxiety-like responses (Muller et al., 2003; Smith et al., 1998; Zorrilla et al., 2002a; Zorrilla et al., 2002b), whereas CRF2 activity has mixed behavioral effects that appear to be circuit- and synapse-specific (Radulovic et al., 1999). We identified dual effects of the CRF system on basal action potential-independent glutamatergic transmission; activation of CRF1 promotes glutamate release under basal conditions, while activation of CRF2 limits it. As the CRF1/2 antagonist and the CRF1-specific antagonist produce similar decreases in mEPSC frequency, endogenous CRF acts primarily at CRF1 to increase glutamate release. CRF1 also mediates the increased action potential-independent glutamatergic release that we observed upon exogenous CRF application.
Regarding evoked glutamatergic responses, exogenous CRF acts primarily through CRF1, although CRF2 may also be involved. Notably, the CRF1 antagonist and CRF each inhibited evoked glutamate responses, which may seem counter-intuitive. We speculate that the heterogeneous expression of CRF1 and CRF2 at both pre- and postsynaptic sites (Gallagher et al., 2008; Liu et al., 2004; Orozco-Cabal et al., 2006), as well as the CRF system’s selective modulation of specific CeA glutamatergic inputs as determined by the distribution of CRF1 and CRF2 (Gallagher et al., 2008; Liu et al., 2004), results in a predominant upstream network inhibition of evoked glutamate release that acts on distinct sites on our pool of stimulated presynaptic terminals. Accordingly, the Gallagher group (Gallagher et al., 2008; Liu et al., 2004; Orozco-Cabal et al., 2006) reported that CRF and urocortin I (a CRF-related peptide that has a higher affinity for CRF2 than CRF1) have opposing effects on CeA glutamate transmission, potentially due to their different binding affinities for each CRF receptor subtype and the presynaptic CRF1 vs. pre- and postsynaptic CRF2 distributions of their main targets (but see also (Henckens et al., 2016a; Henckens et al., 2016b)). Thus, similar to how the CRF system’s neuromodulation of CeA glutamate release varies based on its pre- and/or postsynaptic receptor expression, it likely also varies in terms of the origin of glutamatergic afferents and CeA target cell types.
Glutamatergic dysfunction is a prime molecular mechanism for many of the long-term behavioral effects resulting from chronic ethanol consumption (Lovinger and Roberto, 2013; Roberto et al., 2006; Roberto et al., 2004b), and the CRF system within the CeA is positioned to play a critical role in the emergence of the negative affect associated with dependence (Koob and Zorrilla, 2010). We have previously observed increased CRF and CRF1 expression in the CeA of ethanol-dependent rats (Roberto et al., 2010) and an increased CRF/CRF1 influence over CeA GABAergic transmission in ethanol-dependent rats, suggesting that these synapses may play an important role in the transition to alcohol dependence. However, a recent study reported decreased amygdalar CRF2 density in ethanol-naïve rats selectively bred for their alcohol preference (P rats) compared to their non-preferring counterparts (NP rats) (Yong et al., 2014). Although we have studied the effects of acute ethanol on CeA glutamatergic signaling in naïve and ethanol-dependent rats (Roberto et al., 2006; Roberto et al., 2004b), to our knowledge, this is the first study exploring whether the effects of CRF on CeA glutamatergic transmission are altered by chronic ethanol exposure. Surprisingly, we found no differences in the CRF-induced glutamatergic responses in the CeA of naïve versus ethanol-dependent rats, perhaps due to compensatory mechanisms at these synapses. Therefore, our present work highlights the critical functional role of the CeA glutamatergic system in the maintenance of homeostasis during chronic ethanol exposure. Interestingly, CRF has been reported to potentiate CeA glutamate transmission after 2 weeks, but not 24 hours, of withdrawal from repeated cocaine exposure (Pollandt et al., 2006), highlighting the important contribution of both stress history and current stress levels in shaping the CRF system’s neuroadaptation at CeA glutamatergic synapses (Henckens et al., 2016a; Henckens et al., 2016b).
CONCLUSION
The CeA CRF system has been hypothesized to drive the escalation of ethanol intake and the kindling of anxiety-like behaviors in dependent individuals (Gilpin et al., 2015; Koob and Volkow, 2010). We have previously shown that alcohol dependence is associated with increased CRF/CRF1 influence over GABA transmission in the CeA, leading to greater local inhibition (Roberto et al., 2010). Here we found that CRF primarily acts at presynaptic CRF1 to produce opposite effects on CeA evoked and spontaneous glutamatergic release in both naïve and ethanol-dependent animals. In addition, CRF also appears to have modest effects at CRF2, highlighting the complexity of the CRF system’s neuromodulation of CeA glutamatergic synapses. This diversity in receptor type, localization and action allows greater flexibility for the CRF system to respond to different levels of stress, to both promote stress responses and to ensure stress recovery and the maintenance of homeostasis (Henckens et al., 2016a; Henckens et al., 2016b). As we observed no alteration in the CRF system’s ability to regulate CeA glutamatergic synapses in ethanol-dependent animals, our data identify the CRF system as a sustained regulator of CeA glutamatergic synapses throughout the development of alcohol dependence.
CRF acts primarily via CRF1 at central amygdala glutamatergic synapses.
CRF decreases evoked glutamate transmission at central amygdala synapses.
CRF increases spontaneous glutamate release at central amygdala synapses.
CRF affects glutamate transmission similarly in naïve and ethanol-dependent rats.
Acknowledgments
This is manuscript number 29506 from The Scripps Research Institute. This work was supported by the National Institute on Alcohol Abuse and Alcoholism/National Institutes of Health [AA017447, AA015566, AA06420, AA021491, AA013498] and the Pearson Center for Alcoholism and Addiction Research. We thank Maury Cole for his assistance with the ethanol vapor chambers. We thank J. Rivier of the Salk Institute for Biological Studies and K. Rice of the Drug Design and Synthesis Section of the National Institute on Drug Abuse for their generous gifts of CRF, Astressin B and Astressin 2B (all J.R.) and R121919 (K.R.).
Abbreviations
- AB
Astressin B
- A2B
Astressin 2B
- aCSF
artificial cerebrospinal fluid
- AP-5
DL-2-amino-5-phosphonovalerate
- BAL
blood alcohol level
- CeA
central amygdala
- CRF
corticotropin-releasing factor
- CRF1
corticotropin-releasing factor type 1 receptor
- CRF2
corticotropin-releasing factor type 2 receptor
- DMSO
dimethylsulfoxide
- DNQX
6,7-dinitroquinoxaline-2,3-dione
- EPSP
excitatory postsynaptic potential
- GABA
γ-aminobutyric acid
- I-O
input-output protocol
- ISI
inter-stimulus interval
- mEPSC
miniature excitatory postsynaptic current
- mSP
Marchigian Sardinian Preferring rat
- PPF
paired-pulse facilitation
- RMP
resting membrane potential
- SEM
standard error
- TSRI
The Scripps Research Institute
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreasen M, Hablitz JJ. Paired-pulse facilitation in the dentate gyrus: a patch-clamp study in rat hippocampus in vitro. J Neurophysiol. 1994;72:326–336. doi: 10.1152/jn.1994.72.1.326. [DOI] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci. 2008;28:10151–10166. doi: 10.1523/JNEUROSCI.2432-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Chu K, Dayas CV, Funk D, Knapp DJ, Koob GF, Le DA, O'Dell LE, Overstreet DH, Roberts AJ, Sinha R, Valdez GR, Weiss F. Stress enhancement of craving during sobriety: a risk for relapse. Alcohol Clin Exp Res. 2005;29:185–195. doi: 10.1097/01.alc.0000153544.83656.3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain ST, Owens MJ, Nemeroff CB. Subcellular distribution of corticotropin-releasing-factor-like immunoreactivity in rat central nervous system. Neuroendocrinology. 1991;54:36–41. doi: 10.1159/000125848. [DOI] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, Grigoriadis DE, Behan DP, De Souza EB. Corticotrophin-releasing factor receptors: from molecular biology to drug design. Trends Pharmacol Sci. 1996;17:166–172. doi: 10.1016/0165-6147(96)81594-x. [DOI] [PubMed] [Google Scholar]
- Charlton ME, Sweetnam PM, Fitzgerald LW, Terwilliger RZ, Nestler EJ, Duman RS. Chronic ethanol administration regulates the expression of GABAA receptor alpha 1 and alpha 5 subunits in the ventral tegmental area and hippocampus. J Neurochem. 1997;68:121–127. doi: 10.1046/j.1471-4159.1997.68010121.x. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Economidou D, Cippitelli A, Cucculelli M, Ubaldi M, Soverchia L, Lourdusamy A, Massi M. Genetically selected Marchigian Sardinian alcohol-preferring (msP) rats: an animal model to study the neurobiology of alcoholism. Addict Biol. 2006;11:339–355. doi: 10.1111/j.1369-1600.2006.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz MT, Herman MA, Cote DM, Ryabinin AE, Roberto M. Ghrelin Increases GABAergic Transmission and Interacts with Ethanol Actions in the Rat Central Nucleus of the Amygdala. Neuropsychopharmacology. 2013 Jan;38(2):364–75. doi: 10.1038/npp.2012.190. Epub 2012 Sep 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz MT, Herman MA, Kallupi M, Roberto M. Nociceptin/orphanin FQ blockade of corticotropin-releasing factor-induced gamma-aminobutyric acid release in central amygdala is enhanced after chronic ethanol exposure. Biol Psychiatry. 2012;71:666–676. doi: 10.1016/j.biopsych.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koninck Y, Mody I. Noise analysis of miniature IPSCs in adult rat brain slices: properties and modulation of synaptic GABAA receptor channels. J Neurophysiol. 1994;71:1318–1335. doi: 10.1152/jn.1994.71.4.1318. [DOI] [PubMed] [Google Scholar]
- Eckardt MJ, File SE, Gessa GL, Grant KA, Guerri C, Hoffman PL, Kalant H, Koob GF, Li TK, Tabakoff B. Effects of moderate alcohol consumption on the central nervous system. Alcohol Clin Exp Res. 1998;22:998–1040. doi: 10.1111/j.1530-0277.1998.tb03695.x. [DOI] [PubMed] [Google Scholar]
- Fischman AJ, Moldow RL. Extrahypothalamic distribution of CRF-like immunoreactivity in the rat brain. Peptides. 1982;3:149–153. doi: 10.1016/0196-9781(82)90044-4. [DOI] [PubMed] [Google Scholar]
- Funk CK, O'Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JP, Orozco-Cabal LF, Liu J, Shinnick-Gallagher P. Synaptic physiology of central CRH system. Eur J Pharmacol. 2008;583:215–225. doi: 10.1016/j.ejphar.2007.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Herman MA, Roberto M. The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol Psychiatry. 2015;77:859–869. doi: 10.1016/j.biopsych.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs SC, Menzaghi F, Merlo Pich E, Britton KT, Koob GF. The role of CRF in behavioral aspects of stress. Ann N Y Acad Sci. 1995;771:92–104. doi: 10.1111/j.1749-6632.1995.tb44673.x. [DOI] [PubMed] [Google Scholar]
- Henckens MJ, Deussing JM, Chen A. Region-specific roles of the corticotropin-releasing factor-urocortin system in stress. Nat Rev Neurosci. 2016a;17:636–651. doi: 10.1038/nrn.2016.94. [DOI] [PubMed] [Google Scholar]
- Henckens MJ, Printz Y, Shamgar U, Dine J, Lebow M, Drori Y, Kuehne C, Kolarz A, Eder M, Deussing JM, Justice NJ, Yizhar O, Chen A. CRF receptor type 2 neurons in the posterior bed nucleus of the stria terminalis critically contribute to stress recovery. Mol Psychiatry. 2016b Aug 23; doi: 10.1038/mp.2016.133. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Herman MA, Varodayan FP, Oleata CS, Luu G, Kirson D, Heilig M, Ciccocioppo R, Roberto M. Glutamatergic transmission in the central nucleus of the amygdala is selectively altered in Marchigian Sardinian alcohol-preferring rats: Alcohol and CRF effects. Neuropharmacology. 2016;102:21–31. doi: 10.1016/j.neuropharm.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice NJ, Yuan ZF, Sawchenko PE, Vale W. Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. J Comp Neurol. 2008;511:479–496. doi: 10.1002/cne.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallupi M, Varodayan FP, Oleata CS, Correia D, Luu G, Roberto M. Nociceptin/orphanin FQ decreases glutamate transmission and blocks ethanol-induced effects in the central amygdala of naive and ethanol-dependent rats. Neuropsychopharmacology. 2014;39:1081–1092. doi: 10.1038/npp.2013.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET. The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci. 2015;16:5–16. doi: 10.1038/nrn3875. [DOI] [PubMed] [Google Scholar]
- Koob GF. Circuits, drugs, and drug addiction. Adv Pharmacol. 1998;42:978–982. doi: 10.1016/s1054-3589(08)60910-2. [DOI] [PubMed] [Google Scholar]
- Koob GF. Corticotropin-releasing factor, norepinephrine, and stress. Biol Psychiatry. 1999;46:1167–1180. doi: 10.1016/s0006-3223(99)00164-x. [DOI] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Zorrilla EP. Neurobiological mechanisms of addiction: focus on corticotropin-releasing factor. Curr Opin Investig Drugs. 2010;11:63–71. [PMC free article] [PubMed] [Google Scholar]
- Krettek JE, Price JL. A description of the amygdaloid complex in the rat and cat with observations on intra-amygdaloid axonal connections. J Comp Neurol. 1978;178:255–280. doi: 10.1002/cne.901780205. [DOI] [PubMed] [Google Scholar]
- Liu J, Yu B, Neugebauer V, Grigoriadis DE, Rivier J, Vale WW, Shinnick-Gallagher P, Gallagher JP. Corticotropin-releasing factor and Urocortin I modulate excitatory glutamatergic synaptic transmission. J Neurosci. 2004;24:4020–4029. doi: 10.1523/JNEUROSCI.5531-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yu B, Orozco-Cabal L, Grigoriadis DE, Rivier J, Vale WW, Shinnick-Gallagher P, Gallagher JP. Chronic cocaine administration switches corticotropin-releasing factor2 receptor-mediated depression to facilitation of glutamatergic transmission in the lateral septum. J Neurosci. 2005;25:577–583. doi: 10.1523/JNEUROSCI.4196-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logrip M, OCS, Roberto M. Sex differences in responses of the BLA-CeA circuit to alcohol, corticosterone and their interaction. Neuropharmacology Neuropharmacology. 2017 Mar 1;114:123–134. doi: 10.1016/j.neuropharm.2016.11.021. Epub 2016 Nov 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Roberto M. Synaptic Effects Induced by Alcohol. In: Sommer WH, Spanagel R, editors. Current Topics in Behavioral Neurosciences. Vol. 13. Springer-Verlag; Berlin-Heidelberg: 2013. pp. 31–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Wyllie D, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P, Kormann MS, Droste SK, Kuhn R, Reul JM, Holsboer F, Wurst W. Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci. 2003;6:1100–1107. doi: 10.1038/nn1123. [DOI] [PubMed] [Google Scholar]
- Natividad LA, Buczynski MW, Herman MA, Kirson D, Oleata CS, Irimia C, Polis I, Ciccocioppo R, Roberto M, Parsons LH. Constitutive Increases in Amygdalar Corticotropin-Releasing Factor and Fatty Acid Amide Hydrolase Drive an Anxious Phenotype. Biol Psychiatry Biol Psychiatry. 2017 Jan 13; doi: 10.1016/j.biopsych.2017.01.005. 2017. pii: S0006-3223(17)30037-9. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco-Cabal L, Pollandt S, Liu J, Shinnick-Gallagher P, Gallagher JP. Regulation of synaptic transmission by CRF receptors. Rev Neurosci. 2006;17:279–307. doi: 10.1515/revneuro.2006.17.3.279. [DOI] [PubMed] [Google Scholar]
- Otis TS, De Koninck Y, Mody I. Lasting potentiation of inhibition is associated with an increased number of gamma-aminobutyric acid type A receptors activated during miniature inhibitory postsynaptic currents. Proc Natl Acad Sci U S A. 1994;91:7698–7702. doi: 10.1073/pnas.91.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge JG, Forcelli PA, Luo R, Cashdan JM, Schulkin J, Valentino RJ, Vicini S. Stress increases GABAergic neurotransmission in CRF neurons of the central amygdala and bed nucleus stria terminalis. Neuropharmacology. 2016;107:239–250. doi: 10.1016/j.neuropharm.2016.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pich EM, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Stefanacci L, Farb CR, Go GG, LeDoux JE, Amaral DG. Intrinsic connections of the rat amygdaloid complex: projections originating in the lateral nucleus. J Comp Neurol. 1995;356:288–310. doi: 10.1002/cne.903560211. [DOI] [PubMed] [Google Scholar]
- Pollandt S, Liu J, Orozco-Cabal L, Grigoriadis DE, Vale WW, Gallagher JP, Shinnick-Gallagher P. Cocaine withdrawal enhances long-term potentiation induced by corticotropin-releasing factor at central amygdala glutamatergic synapses via CRF, NMDA receptors and PKA. Eur J Neurosci. 2006;24:1733–1743. doi: 10.1111/j.1460-9568.2006.05049.x. [DOI] [PubMed] [Google Scholar]
- Radulovic J, Ruhmann A, Liepold T, Spiess J. Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress: differential roles of CRF receptors 1 and 2. J Neurosci. 1999;19:5016–5025. doi: 10.1523/JNEUROSCI.19-12-05016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Fernhout BJ, Shinnick-Gallagher P. Differential actions of corticotropin releasing factor on basolateral and central amygdaloid neurones, in vitro. J Pharmacol Exp Ther. 1992;263:846–858. [PubMed] [Google Scholar]
- Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res. 1993;605:25–32. doi: 10.1016/0006-8993(93)91352-s. [DOI] [PubMed] [Google Scholar]
- Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology. 2006;31:988–996. doi: 10.1038/sj.npp.1300840. [DOI] [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, Cottone P, Madamba SG, Stouffer DG, Zorrilla EP, Koob GF, Siggins GR, Parsons LH. Corticotropin Releasing Factor-Induced Amygdala Gamma-Aminobutyric Acid Release Plays a Key Role in Alcohol Dependence. Biol Psychiatry. 2010:831–839. doi: 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Gilpin NW, Siggins GR. The Central Amygdala and Alcohol: Role of gamma-Aminobutyric Acid, Glutamate, and Neuropeptides. Cold Spring Harb Perspect Med. 2012 doi: 10.1101/cshperspect.a012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci U S A. 2003;100:2053–2058. doi: 10.1073/pnas.0437926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci. 2004a;24:10159–10166. doi: 10.1523/JNEUROSCI.3004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci. 2004b;24:1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Siggins GR. Nociceptin/orphanin FQ presynaptically decreases GABAergic transmission and blocks the ethanol-induced increase of GABA release in central amygdala. Proc Natl Acad Sci U S A. 2006;103:9715–9720. doi: 10.1073/pnas.0601899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Cole M, Koob GF. Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol Clin Exp Res. 1996;20:1289–1298. doi: 10.1111/j.1530-0277.1996.tb01125.x. [DOI] [PubMed] [Google Scholar]
- Rogers J, Wiener SG, Bloom FE. Long-term ethanol administration methods for rats: advantages of inhalation over intubation or liquid diets. Behav Neural Biol. 1979;27:466–486. doi: 10.1016/s0163-1047(79)92061-2. [DOI] [PubMed] [Google Scholar]
- Savander V, Go CG, LeDoux JE, Pitkanen A. Intrinsic connections of the rat amygdaloid complex: projections originating in the basal nucleus. J Comp Neurol. 1995;361:345–368. doi: 10.1002/cne.903610211. [DOI] [PubMed] [Google Scholar]
- Silberman Y, Winder DG. Corticotropin releasing factor and catecholamines enhance glutamatergic neurotransmission in the lateral subdivision of the central amygdala. Neuropharmacology. 2013;70:316–323. doi: 10.1016/j.neuropharm.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Liouterman L, Van Bockstaele EJ. Evidence for regional heterogeneity in corticotropin-releasing factor interactions in the dorsal raphe nucleus. J Comp Neurol. 2001a;435:450–463. doi: 10.1002/cne.1043. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Rudoy C, Saunders A, Liu XB, Van Bockstaele EJ. Corticotropin-releasing factor is preferentially colocalized with excitatory rather than inhibitory amino acids in axon terminals in the peri-locus coeruleus region. Neuroscience. 2001b;106:375–384. doi: 10.1016/s0306-4522(01)00279-2. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Varodayan FP, de Guglielmo G, Logrip ML, George O, Roberto M. Alcohol dependence disrupts amygdalar L-type voltage-gated calcium channel mechanisms. J Neurosci J Neurosci. 2017 Mar 31; doi: 10.1523/JNEUROSCI.3721-16.2017. 2017. pii: 3721-16. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong W, Spence JP, Eskay R, Fitz SD, Damadzic R, Lai D, Foroud T, Carr LG, Shekhar A, Chester JA, Heilig M, Liang T. Alcohol-preferring rats show decreased corticotropin-releasing hormone-2 receptor expression and differences in HPA activation compared to alcohol-nonpreferring rats. Alcohol Clin Exp Res. 2014;38:1275–1283. doi: 10.1111/acer.12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Shinnick-Gallagher P. Corticotropin-releasing factor increases dihydropyridine- and neurotoxin-resistant calcium currents in neurons of the central amygdala. J Pharmacol Exp Ther. 1998;284:170–179. [PubMed] [Google Scholar]
- Zorrilla EP, Schulteis G, Ormsby A, Klaassen A, Ling N, McCarthy JR, Koob GF, De Souza EB. Urocortin shares the memory modulating effects of corticotropin-releasing factor (CRF): mediation by CRF1 receptors. Brain Res. 2002a;952:200–210. doi: 10.1016/s0006-8993(02)03345-0. [DOI] [PubMed] [Google Scholar]
- Zorrilla EP, Valdez GR, Nozulak J, Koob GF, Markou A. Effects of antalarmin, a CRF type 1 receptor antagonist, on anxiety-like behavior and motor activation in the rat. Brain Res. 2002b;952:188–199. doi: 10.1016/s0006-8993(02)03189-x. [DOI] [PubMed] [Google Scholar]