

Summary
Reck, a GPI-anchored membrane protein, and Gpr124, an orphan GPCR, have been implicated in Wnt7a/Wnt7b signaling in the CNS vasculature. We show here that vascular endothelial cell (EC)-specific reduction in Reck impairs CNS angiogenesis and that EC-specific postnatal loss of Reck combined with loss of Norrin impairs blood-brain barrier (BBB) maintenance. The most N-terminal domain of Reck binds to the leucine-rich repeat (LRR) and immunoglobulin (Ig) domains of Gpr124, and weakening this interaction by targeted mutagenesis reduces Reck/Gpr124 stimulation of Wnt7a signaling in cell culture and impairs CNS angiogenesis. Finally, a soluble Gpr124(LRR-Ig) probe binds to cells expressing Frizzled, Wnt7a or Wnt7b, and Reck; and a soluble Reck(CC1–5) probe binds to cells expressing Frizzled, Wnt7a or Wnt7b, and Gpr124. These experiments indicate that Reck and Gpr124 are part of the cell-surface protein complex that transduces Wnt7a- and Wnt7b-specific signals in mammalian CNS ECs to promote angiogenesis and regulate the BBB.
Keywords: vascular endothelial cells, canonical Wnt signaling, Wnt reporter, mouse genetics
Graphical abstract
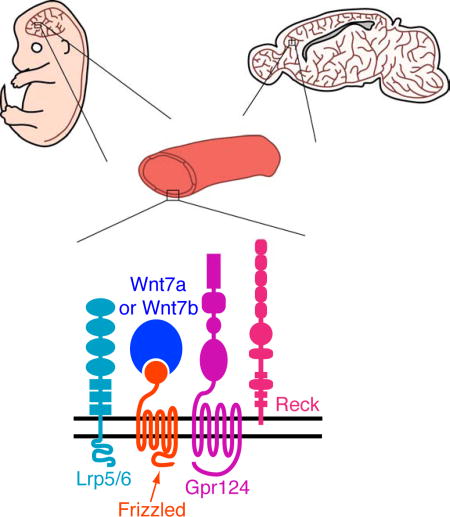
Introduction
Vascularization of the mammalian brain and spinal cord begins with the development of a perineural vascular plexus and the migration of endothelial cells from the plexus into the midgestation neural tube. An analogous process occurs during retinal development, with blood vessels first growing along the vitreal face of the retina to form the superficial plexus and then invading the retina to form the deep and intermediate capillary plexuses. A unifying feature of angiogenesis throughout the central nervous system (CNS) is its dependence on canonical Wnt signaling, with Wnt7a and Wnt7b serving as the dominant ligands in the brain and spinal cord, and Norrin serving as the dominant ligand in the retina (Xu et al., 2004; Liebner et al., 2008; Daneman et al., 2009; Stenman et al., 2008; Ye et al., 2009). These ligands are produced by neuroepithelial cells and their derivatives, and they act on Frizzled receptors and Lrp5/6 co-receptors on vascular endothelial cells (ECs). In humans, mutations in the genes coding for Norrin, Frizzled4 (Fz4), Lrp5, or Tspan12 (an integral membrane protein that enhances canonical Wnt signaling by the other three components) cause retinal hypovascularization (Junge et al., 2009; Nikopoulos et al., 2010; Ye et al., 2010). In zebrafish, it has been shown that tip cells are the predominant receivers and integrators of the Wnt signal for vascular sprouting and invasion (Vanhollebeke et al., 2015). In contrast to CNS angiogenesis, development of the peripheral vasculature is largely independent of canonical Wnt signaling.
The CNS vasculature is distinguished from the peripheral vasculature by a diffusion barrier between blood and tissue - the blood-brain barrier (BBB) and its retinal analogue, the blood-retina barrier (BRB) - that arise from molecular and cellular specializations in capillary ECs, including acquisition of tight junctions, suppression of transcytosis, absence of fenestrations, and expression of multiple extrusion pumps and small molecule transporters (Daneman and Prat, 2015). Barrier integrity requires interactions between ECs and pericytes in close association with astrocyte end feet, which together are referred to as the neurovascular unit. In addition to its role in CNS angiogenesis, canonical Wnt signaling in ECs plays a central role in the development and maintenance of the BBB and BRB. The requirement for ongoing Wnt signaling in mature CNS ECs has been shown by (1) the loss of barrier integrity that follows an acute reduction in canonical Wnt signaling, and (2) the restoration of barrier integrity that follows acute reactivation of canonical Wnt signaling (Liebner et al., 2008; Wang et al., 2012; Zhou et al., 2014). Although the normal role of CNS barrier plasticity is not presently known, it is of great interest to the medical community because a wide variety of neurologic and ophthalmologic diseases are associated with compromised BBB or BRB integrity (Zhao et al., 2015).
Recent work has implicated two additional proteins in Wnt7a/Wnt7b signaling in CNS ECs: Gpr124 and Reck (reversion inducing cysteine-rich protein with kazal motifs). In mice, Gpr124, a member of the adhesion GPCR superfamily, is required for angiogenesis in the cerebral cortex, medial and lateral ganglionic eminences (MGE and LGE), and spinal cord, and for expression of BBB markers in more mature vasculature (Kuhnert et al., 2010; Anderson et al., 2011; Cullen et al., 2011). In humans, GPR124 appears to play a role in tumor vascular biology, as GPR124 transcripts are dramatically up-regulated in vascular ECs in tumors relative to ECs in normal tissue (St Croix et al., 2000). Reck, a multi-domain GPI-anchored plasma membrane protein with matrix metalloproteinase (MMP) inhibitory activity, was initially identified based on the ability of human RECK cDNA clones to induce reversion of the transformed phenotype in NIH/3T3 cells (Noda et al., 1989). In mice, Reck plays an essential role in embryonic and placental vascular remodeling, and these activities have been ascribed to its function as an MMP inhibitor, with at least one site of action in pericytes (Oh et al., 2001; Chandana et al., 2010; de Almeida et al., 2015). In zebrafish, loss-of-function Gpr124 and Reck mutations have very similar phenotypes, with each gene playing an essential role in the development of dorsal root ganglia and intracerebral vascularization (Prendergast et al., 2012; Vanhollebeke et al., 2015; Ulrich et al., 2016). In cell culture, Gpr124 and Reck strongly enhance Frizzled- and Lrp5/6-dependent canonical Wnt signaling by Wnt7a and Wnt7b, but not by any of the other 17 mammalian Wnts or by Norrin, and in mice the Gpr124 knockout phenotype can be rescued by experimentally activating canonical Wnt signaling (Zhou and Nathans, 2014; Posokhova et al., 2015; Vanhollebeke et al., 2015).
In the present study we have addressed a series of open questions related to the role of Reck and Gpr124 in mammalian CNS angiogenesis and barrier maintenance. What is the functional and anatomic relationship between Reck and Gpr124 loss-of-function defects in the CNS vasculature? What are the functional relationships between Reck and Norrin pathways? Are there defined domains of Reck and Gpr124 that interact physically and functionally? Does signaling - and Wnt specificity - involve a multi-protein complex of ligand, receptor, and co-receptor, together with Gpr124 and Reck?
Results
Reck is Required for Canonical Wnt Signaling in CNS ECs and for Normal CNS Angiogenesis
To assess the role of Reck in CNS ECs, we studied ReckΔex2/Δex2 and Reckflex2/Δex1;Tie2-Cre vascular EC-specific knockout embryos (Figure 1). Reck exon 2 deletion produces a hypomorphic allele (Chandana et al., 2010), whereas Reck exon 1 deletion produces a null allele (Oh et al., 2001). Reckflex2 is a conditional allele in which exon 2 is flanked by loxP sites. The ReckΔex2/Δex2 and Reckflex2/Δex1;Tie2-Cre genotypes circumvent the embryonic day (E)10.5 lethality seen with the ReckΔex1/Δex1 genotype, which precludes an analysis of CNS vascularization (Oh et al., 2001; Chandana et al., 2010). In E13.5 embryos of both genotypes, hemorrhaging occurs in the forebrain and spinal cord (Figure 1B and 1C; Chandana et al., 2010; de Almeida et al., 2015), and ECs in the cortex, ganglionic eminences, and spinal cord form glomeruloid-like tufts instead of an interconnected vascular network (Figure 1A–C). Vascular density was significantly reduced in these regions (quantified in Figure 1E), and the hypovascular territories showed increased infiltration of non-endothelial GS Lectin+ cells that predominantly co-stain for macrophage markers F4/80 or Cd11b (referred to hereafter as GS Lectin+ macrophages; Figure S1). This phenotype closely matches the phenotype of Gpr124 null embryos (Kuhnert et al., 2010; Anderson et al., 2011; Cullen et al., 2011; Zhou and Nathans, 2014). In these and all embryos described below, the growth of the embryo and the appearance of non-CNS vasculature were normal, implying that vascular defects were confined to the CNS. A complete list of all embryonic genotypes examined and the severities of their vascular phenotypes can be found in Table S1.
Figure 1. Reck Activates Canonical Wnt Signaling to Promote CNS Angiogenesis.

(A–D) E13.5 embryos are shown (column 1) alongside coronal sections of brain (columns 2–4) and cross-sections of spinal cord (column 5). The white line in (A) depicts the plane for each brain section. Boxed regions in cortex (a) and MGE (b) in column 2 are shown at higher magnification in columns 3 and 4, respectively. (B) Reckflex2/Δex1;Tie2-Cre vascular knockout embryos show hemorrhaging in the forebrain (arrow) and distal spinal cord (white arrowhead). Vascular density is reduced in the cortex, MGE, and spinal cord, accompanied by infiltration of GS Lectin+ macrophages. (C) ReckΔex2/Δex2 embryos display vascular defects similar to their Tie2-Cre conditional knockout counterparts in (B). (D) Beta-catenin was artificially stabilized in ECs of ReckΔex2/Δex2;Ctnnb1flex3/+;Pdgfb-CreER embryos following IP injection of 1.5 mg of tamoxifen in gestational day (G)10.5 females. At E13.5, the resulting embryos exhibit little or no hemorrhaging in the forebrain and spinal cord, and show a near-complete rescue of vascularization in these regions.
(E) Quantification of the relative vascular density in cortex, MGE, and spinal cord for each genotype shown in (A–D).
(F) Quantification of GS Lectin+ macrophages in cortex, MGE, and spinal cord for each genotype in (A–D). In this and subsequent figures, bars represent mean ± SD. Statistical significance, determined by the unpaired t-test, is represented by * (P<0.05), **(P<0.01), *** (P<0.001), and **** (P<0.0001). Scale bars, 200 µm.
Given the emerging role of Reck as a specific co-activator of Wnt7a/Wnt7b signaling (Vanhollebeke et al., 2015), we investigated whether the CNS vascular defects in Reck mutant embryos are associated with a reduction in canonical Wnt signaling. Existing reporter lines for canonical Wnt signaling are based on transgenes expressing beta-galactosidase or GFP under the control of either the Axin2 promoter or a juxtaposition of multimerized TCF/LEF sites and a minimal promoter (Jho et al., 2002; Maretto et al., 2003; Ferrer-Vaquer et al., 2010). The utility of these reporter lines is limited by (1) their capacity for expression in many cell types, which can make it difficult to visualize changes in canonical Wnt signaling in a minor cell type, and (2) random variegation in transgene expression. To resolve these problems, we generated a new Wnt reporter line by knocking into the ubiquitously expressed Rosa26 locus the sequences coding for a histone H2B-GFP-6×myc fusion protein under the control of 8×TCF/LEF binding sites and a minimal promoter, with the added feature that reporter expression also requires removal of a loxP-stop-loxP (LSL) casette by Cre-mediated recombination (Figure S2A). This last feature represents a general strategy for cell-type-specific visualization of any reporter. Figure S2B–D shows that CNS ECs in phenotypically wild-type (WT) Reckflex2/+;Rosa26 Tcf/Lef-LSL-H2B-GFP;Tie2-Cre embryos show strong and specific nuclear GFP that is absent in neighboring, non-ECs. In contrast, Reckflex2/Δex1;Rosa26 Tcf/Lef-LSL-H2B-GFP;Tie2-Cre embryos show significantly reduced nuclear GFP in the vascular tufts in the cortex, ganglionic eminences, and spinal cord compared to the phenotypically WT control, implying that Reck activity is required in CNS ECs for canonical Wnt signaling (Figure S2B–H).
If Reck is responsible for activating canonical Wnt signaling at a level upstream of beta-catenin stabilization, then artificially stabilizing beta-catenin should rescue the Reck mutant vascular phenotype. Exon 3 of the beta-catenin gene (Ctnnb1) harbors key phosphorylation sites for glycogen synthase kinase-3 (GSK-3) that target beta-catenin for degradation. We therefore combined EC-specific expression of CreER (from a Pdgfb-CreER transgene) and the Ctnnb1flex3 allele, which contains loxP sites flanking exon 3, to produce an in-frame deletion of this region and artificially stabilize beta-catenin specifically in ECs. Gestational day (GD)10.5 females were injected intraperitoneally (IP) with 1.5 mg of tamoxifen, and their embryos were harvested three days later for analysis. The resulting E13.5 ReckΔex2/Δex2;Ctnnb1flex3/+;Pdgfb-CreER embryos display little to no hemorrhaging in the forebrain and spinal cord, and the vascular network in the cortex, ganglionic eminences, and spinal cord is substantially denser than in ReckΔex2/Δex2 embryos (compare Figure 1C and 1D; quantification in Figure 1E). We ascribe the incomplete phenotypic rescue to the relatively brief (three day) time window during which rapid CNS growth may make it difficult for angiogenesis in ReckΔex2/Δex2;Ctnnb1flex3/+;Pdgfb-CreER embryos to catch up to their WT counterparts. Control experiments in which Ctnnb1flex3/+;Pdgfb-CreER was used to stabilize beta-catenin in ECs on a phenotypically WT genetic background during the same E10.5–13.5 time window showed little or no effect on CNS angiogenesis (Zhou and Nathans, 2014). Despite the partial rescue of the CNS angiogenesis phenotype, ReckΔex2/Δex2;Ctnnb1flex3/+;Pdgfb-CreER embryos continue to show elevated numbers of GS Lectin+ macrophages in the CNS parenchyma (Figure 1D and 1F). Together, these data imply that Reck’s role in CNS angiogenesis is via activation of canonical Wnt signaling in ECs.
Reck is Required for the Development of the Blood-Brain Barrier
The development of the BBB is tightly coupled to CNS angiogenesis, and decreased canonical Wnt signaling perturbs both of these processes (Wang et al., 2012; Zhou et al., 2014). We therefore studied Reckflex2/Δex1;Tie2-Cre mice to determine whether Reck activity in ECs also plays a role in BBB development. Since BBB development is largely complete by E15.5 (Ben-Zvi et al., 2014) and Reckflex2/Δex1;Tie2-Cre embryos survive to birth, we assessed BBB integrity in neonatal pups by IP injection of a membrane impermeable sulfo-N-hydroxysuccinimide-long chain-biotin (sulfo-NHS-biotin) tracer. As expected from their appearance at E13.5, Reckflex2/Δex1;Tie2-Cre neonates exhibit impaired angiogenesis and severe hemorrhaging in the cortex (Figure 2). Interestingly, the hypothalamus and brainstem, where vascular density was close to normal and there was no hemorrhaging, showed extensive sulfo-NHS-biotin leakage, and lower level leakage was widely present in many subcortical regions. Consistent with a defect in BBB development, the fenestral diaphragm protein plasmalemma vesicle-associated protein (PLVAP), which serves as a marker for the differentiation state of ECs and correlates with a compromised BBB (Stan et al., 2004; Wang et al., 2012), was elevated throughout the Reckflex2/Δex1;Tie2-Cre CNS. These data point to a central role for endothelial Reck in BBB development.
Figure 2. Reck is Essential for the Development of the Blood-Brain Barrier.

(A,B) Reckflex2/+;Tie2-Cre and Reckflex2/Δex1;Tie2-Cre P0 mice were injected IP with 2–3 mg of sulfo-NHS-biotin 30–45 minutes before sacrifice. Dorsal view (a) and sagittal section (d) of P0 brains, with the boxed region in (d) shown at higher magnification in (b) and (c). Left, anterior; right, posterior. Reckflex2/+;Tie2-Cre control mice (A) have an intact BBB with no biotin leakage, except in the choroid plexus (arrows in (b) and (c)). Reckflex2/Δex1;Tie2-Cre mice (B) show severe hemorrhaging in the cortex along with biotin leakage in cortical, subcortical, and hindbrain regions. PLVAP is widely up-regulated in the CNS vasculature in Reckflex2/Δex1;Tie2-Cre mice. Scale bars, 50 µm.
Reck Signaling is Partially Redundant with Norrin Signaling in Promoting CNS Angiogenesis and BBB Maintenance
Norrin signaling, in addition to being largely responsible for retinal angiogenesis and the development of the BRB, has been shown to function in parallel with Gpr124 to regulate angiogenesis and barrier integrity in the brain (Zhou et al., 2014; Zhou and Nathans, 2014). To examine the functional and anatomic relationship between Reck- and Norrin-mediated Wnt signaling in CNS angiogenesis, we analyzed embryos that carry various allelic combinations of Reck and Ndp (Norrin) mutant alleles (Figure 3). (Since Ndp is an X-linked gene, Ndp null male mutant mice are designated NdpΔ/Y). Previous analyses of hindbrain vasculature of Gpr124 and Ndp mutant embryos were performed at E11.5 (Zhou and Nathans, 2014). To facilitate comparisons between that study and this one, we analyzed embryos at this time point. Consistent with the phenotypes observed at E13.5, E11.5 Reckflex2/Δex2;Tie2-Cre embryos show hemorrhaging in the forebrain and spinal cord (data not shown) with no obvious vascular defects in subcortical tissue, including the hindbrain (Figure 3B and 3F). Reckflex2/+;NdpΔ/Y;Tie2-Cre embryos appear phenotypically normal and show no vascular abnormalities in the developing CNS, similar to Reckflex2/+;Tie2-Cre embryos (Figure 3A, 3C, and 3F). However, a dramatic phenotype is observed in Reckflex2/Δex2;NdpΔ/Y; Tie2-Cre mutants: in addition to more pronounced hemorrhaging in the forebrain and spinal cord (data not shown), vascular density is markedly reduced in the hindbrain, and these hypovascular areas are infiltrated with a large numbers of GS Lectin+ macrophages (Figure 3D and 3F). Interestingly, vascular knockout of both Reck and Gpr124 produces a very similar hindbrain vascular phenotype (Figure 3E and 3F), implying that the actions of Reck and Gpr124 in Wnt7a/Wnt7b signaling in vivo are synergistic, as observed in cell culture experiments (Vanhollebeke et al., 2015), and that the absence of either one reduces but does not abolish Wnt7a/Wnt7b signaling. The genetic interaction between Reck and Ndp closely mirrors that observed between Gpr124 and Ndp (Zhou and Nathans, 2014), and together they support a model in which Reck/Gpr124- and Norrin-mediated signaling represent two parallel pathways that function redundantly in the hindbrain to promote angiogenesis.
Figure 3. Reck- and Norrin-Mediated Wnt Signaling Pathways Function Redundantly to Regulate CNS Angiogenesis and BBB Integrity.

(A–E) Coronal sections of E11.5 hindbrains (column 1). Boxed regions (a) and (b) are displayed at higher magnification in columns 2 and 3. In the hindbrain, Reckflex2/+;Tie2-Cre (A), Reckflex2/Δex2;Tie2-Cre (B), and Reckflex2/+;NdpΔ/Y;Tie2-Cre (C) animals show little or no vascular defects. Reckflex2/Δex2;NdpΔ/Y;Tie2-Cre (D) and Reckflex2/Δex2;Gpr124fl/Δ;Tie2-Cre (E) embryos show reduced vascular density with increased infiltration of GS Lectin+ macrophages.
(F) Quantification of relative vascular density (top) and GS Lectin+ macrophages (bottom) in the hindbrain for each genotype in (A–E).
(G–I) Coronal sections of P7 brains (column 1). Boxed regions are displayed at higher magnification (columns 2–4). Animals were injected IP with 50 µg of 4-HT at P3 and P4. Reckflex2/Δex1;Pdgfb-CreER (G) and Reckflex2/+;NdpΔ/Y;Pdgfb-CreER (H) animals show little or no biotin leakage into the brain parenchyma. Vessels are Claudin-5+;PLVAP−, except in the choroid plexus where they are Claudin-5−;PLVAP+ (upper left of each boxed region). Reckflex2/Δex1;NdpΔ/Y;Pdgfb-CreER (I) mice show extensive biotin leakage, most prominently in the cortex, with vessels in the brain parenchyma converted to Claudin-5−;PLVAP+.
(J) Quantification of the fraction of vessels that immunostain positive for Claudin-5 (top) and PLVAP (bottom) for each genotype in (G–I).
Scale bars, 200 µm.
Maintenance of the BBB depends on canonical Wnt signaling in mature ECs (Liebner et al., 2008; Wang et al., 2012; Zhou et al., 2014). We therefore investigated the functional relationship between Reck- and Norrin-mediated signaling on BBB maintenance postnatally. Mice carrying various combinations of Reck and Ndp alleles together with Pdgfb-CreER were injected IP with 50 µg of 4-hydroxytamoxifen (4-HT) at P3 and P4, followed by analysis of P7 brains for sulfo-NHS-biotin leakage. For all genotypes characterized, the vasculature was structurally intact and devoid of hemorrhaging. Reckflex2/Δex1;Pdgfb-CreER and Reckflex2/+;NdpΔ/Y;Pdgfb-CreER mice showed no evidence of vascular leakage in the brain parenchyma (Figure 3G and 3H). Consistent with the presence of an intact BBB, CNS ECs in these mice express the tight junction marker Claudin-5 with little or no expression of PLVAP (Claudin-5+;PLVAP−) (Figure 3G, 3H, and quantified in 3J). In contrast, postnatal loss of both Reck and Norrin (Reckflex2/Δex1;NdpΔ/Y;Pdgfb-CreER) produced extensive leakage throughout the brain, most prominently in the cortex (Figure 3I). In these mice, CNS ECs showed reduced Claudin-5 expression and widespread PLVAP upregulation (Claudin-5−;PLVAP+), consistent with the loss of BBB integrity (Figure 3I and quantified in 3J).
These changes closely resemble the BBB breakdown and Claudin-5−;PLVAP+ conversion produced in an analogous experiment featuring the combined postnatal loss of Gpr124 and Norrin (Zhou and Nathans, 2014). In both of these experiments, treatment with 4-HT in early postnatal life produced efficient Pdgfb-CreER-mediated recombination of the Reck or Gpr124 conditional alleles, as judged by the near-complete conversion of CNS ECs from PLVAP− to PLVAP+ (Figure 3I; Zhou and Nathans, 2014). To the extent that some ECs do not undergo recombination, then these experiments would be under-estimating the severity of the loss-of-function phenotypes. We also quantified cortical vascular density and observed a ~35% reduction for both Reckflex2/Δex1;Pdgfb-CreER and Reckflex2/Δex1;NdpΔ/Y;Pdgfb-CreER when compared to Reckflex2/+;NdpΔ/Y;Pdgfb-CreER, suggesting a role for Reck in post-natal vascular development and remodeling (p-value<0.01, data not shown). Together, these data imply functionally redundant roles for Reck/Gpr124 and Norrin signaling in maintaining BBB integrity.
Reck Genetically Interacts with Gpr124 and Wnt7a/Wnt7b
The severe E11.5 hindbrain angiogenesis defect produced by the combined EC-specific loss of Reck and Gpr124 suggested that a systematic exploration of the effects of reduced Reck and Gpr124 gene dosage would be of interest. Figure 4 shows such an analysis at a time point (E13.5) and in a format that are identical to those in Figure 1. Reckflex2/+;Gpr124fl/+;Tie2-Cre embryos appear phenotypically normal (Figure 4A), while Reckflex2/Δex2;Gpr124fl/+;Tie2-Cre and Gpr124fl/Δ;Tie2-Cre embryos exhibit vascular defects that are comparable to those of Reckflex2/Δex1;Tie2-Cre embryos (compare Figures 1B and 4B–C). Specifically, these embryos show: (1) moderate hemorrhaging in the forebrain and distal spinal cord; (2) reduced vascular density in the cortex, MGE, and spinal cord; and (3) increased abundance of GS Lectin+ macrophages in the MGE and spinal cord (Figure 4B, 4C, 4F, and 4G). This phenotype is exacerbated in Reckflex2/+;Gpr124fl/Δ;Tie2-Cre and Reckflex2/Δex2;Gpr124fl/Δ;Tie2-Cre mutants, the latter exhibiting the most severe phenotype with pronounced hemorrhaging in the forebrain and along the entire length of the spinal cord (Figure 4D and 4E). Additionally, the vascular density is further reduced in the cortex and MGE for both of these mutants, as well as in the spinal cord for Reckflex2/Δex2;Gpr124fl/Δ;Tie2-Cre mutants when compared to Reckflex2/Δex2;Gpr124fl/+;Tie2-Cre and Gpr124fl/Δ;Tie2-Cre mutants (Figure 4B–F). The progressive increase in severity of the angiogenic phenotype that is elicited by EC-specific decrements in Reck and/or Gpr124 gene dosage imply that there is a correspondingly graded cell biological response of developing ECs to different levels of Reck/Gpr124-mediated signaling.
Figure 4. Genetic Evidence for Reck-Gpr124 Interaction in CNS Angiogenesis.

(A–E) E13.5 embryos (columns 1 and 2), coronal sections of brain (columns 3–5), and cross-sections of spinal cord (column 6). Boxed regions of cortex (a) and MGE (b) in column 3 are shown at higher magnification in columns 4 and 5, respectively. Reckflex2/+;Gpr124fl/+;Tie2-Cre (A) embryos show normal vascular development, but ~2–3-fold greater numbers of GS Lectin+ macrophages compared to WT embryos (compare panel G to Fig. 1F). Reckflex2/Δex2;Gpr124fl/+;Tie2-Cre (B) and Gpr124fl/Δ;Tie2-Cre (C) embryos show hemorrhaging in the forebrain (arrow) and distal spinal cord (white arrowhead). Vascular density is reduced in the cortex, MGE, and spinal cord, and hypovascular regions show increased infiltration of GS Lectin+ macrophages. Reckflex2/+;Gpr124fl/Δ;Tie2-Cre (D) and Reckflex2/Δex2;Gpr124fl/Δ;Tie2-Cre (E) embryos show severe hemorrhaging in the forebrain (arrow) and along the length of the spinal cord (white arrowhead), as well as reduced vascular density in the cortex, MGE, and spinal cord.
(F) Quantification of relative vascular density in cortex, MGE, and spinal cord for each genotype in (A–E).
(G) Quantification of GS Lectin+ macrophages in cortex, MGE, and spinal cord for each genotype in (A–E).
Scale bars, 200 µm.
Although all of the CNS angiogenesis phenotypes described here are associated with accumulation of GS Lectin+ macrophages in the CNS parenchyma, the number of macrophages does not continue to increase with increasing severity of the angiogenic phenotype (i.e. reduced vascular density; Figure 4G). Instead, there is a drop in the number of macrophages in the cortex in Reckflex2/+;Gpr124fl/Δ;Tie2-Cre and Reckflex2/Δex2;Gpr124fl/Δ;Tie2-Cre embryos compared to Gpr124fl/Δ;Tie2-Cre embryos, and there is a similar drop in the number of macrophages in the spinal cord in Reckflex2/Δex2;Gpr124fl/Δ;Tie2-Cre embryos when compared to Reckflex2/+;Gpr124fl/Δ;Tie2-Cre embryos. These data suggest that a baseline vascular infrastructure is required for maximal macrophage infiltration into damaged CNS tissue.
Cell culture experiments show that Reck functions as a specific co-activator for Wnt7a/Wnt7b signaling (Vanhollebeke et al., 2015), but, as yet, there is no in vivo evidence for such an interaction. Previous work has shown that Wnt7aΔ Δ;Wnt7b+/Δ mutant mice have a near-normal neurovascular phenotype (Stenman et al., 2008; Daneman et al., 2009; Posokhova et al., 2015). Consistent with that work, Figure S3 (A, B, E and F) shows that the vascular densities in the cortex, MGE, and spinal cord of Wnt7aΔ/Δ;Wnt7b+/Δ and Reck+/Δex2;Wnt7a+/Δ;Wnt7b+/Δ embryos at E13.5 are comparable to that of WT embryos, the only difference being modest elevations in macrophage counts in the cortex in Reck+/Δex2;Wnt7a+/Δ;Wnt7b+/Δ embryos and in the cortex, MGE, and spinal cord in Wnt7aΔ/Δ;Wnt7b+/Δ embryos (Figure S3A, S3B, and S3F). We reasoned that if Reck and Wnt7a/Wnt7b function through the same signaling complex, then heterozygosity for ReckΔex2, which by itself confers a normal phenotype, on a genetically sensitized Wnt7aΔ/Δ; Wnt7b+/Δ background could bring out a vascular phenotype. In agreement with this idea, Figures S3C, S3E, and S3F show that Reck+/Δex2;Wnt7aΔ/Δ;Wnt7b+/Δ embryos have decreased CNS vascular density in the cortex and MGE together with increased numbers of GS Lectin+ macrophages in the MGE. Furthermore, homozygosity for ReckΔex2 on the Wnt7aΔ/Δ;Wnt7b+/Δ background produces a phenotype that is substantially more severe than that of ReckΔex2/Δex2 embryos (Figure S3D–F). Specifically, the cortex and spinal cord of ReckΔex2/Δex2;Wnt7aΔ/Δ;Wnt7b+/Δ mutants are almost completely avascular. We also observe a reduction in GS Lectin+ macrophages in the cortex and MGE of ReckΔex2/Δex2;Wnt7aΔ/Δ;Wnt7b+/Δ embryos compared to Reck+/Δex2;Wnt7aΔ/Δ;Wnt7b+/Δ embryos, consistent with the suggestion above that an intact vascular network is required for efficient infiltration of macrophages into the brain parenchyma (Figures S3C, S3D, and S3F). In sum, these data provide genetic evidence that Reck functionally cooperates with Gpr124 and Wnt7a/Wnt7b in CNS angiogenesis.
The N-terminal Domains of Reck and Gpr124 are Required for Synergistic Wnt7a/Wnt7b Activation in vitro
Given the evidence that Reck and Gpr124 function as specific co-activators of Wnt7a/Wnt7b signaling (Zhou and Nathans, 2014; Posokhova et al., 2015; Vanhollebeke et al., 2015), we sought to define the regions that are required for this activity. The ectodomain of Gpr124 consists of an N-terminal leucine-rich repeat domain (LRR), an immunoglobulin-like domain (Ig), a hormone receptor domain (HormR), and a putative GPCR-Autoproteolysis Inducing domain (GAIN) (Figure S4A). We generated deletion mutants for each of these domains and used cell-surface biotinylation of C-terminally epitope tagged proteins to show that the plasma membrane localization of each mutant in transfected cells was comparable to WT Gpr124 (Figure S4B). Using a Super TOP Flash (STF) luciferase reporter cell line for canonical Wnt signaling (Xu et al., 2004), we co-transfected Reck, Wnt7a, and each of the Gpr124 mutants and observed that deletion of the LRR and/or Ig domains largely eliminated Gpr124 activity, consistent with a previous report (Posokhova et al., 2015), whereas deletion of the HormR or GAIN domain had only a modest effect on activity (Figure S4C).
The function of Reck as an MMP inhibitor has been attributed to its Kazal-like domains (Chang et al., 2008). The functions of other domains, which include five tandem copies of a domain with a dicysteine motif (CC1–5), a Frizzled-like cysteine-rich domain (Fz-like CRD), and two EGF-like domains, remain largely uncharacterized. To identify the domains of Reck required for canonical Wnt signaling, we inserted a Myc-epitope immediately after the signal peptide and then generated various domain deletions (Figure S5A). Plasma membrane localization for each mutant was confirmed by both cell-surface biotinylation and live-cell immunostaining for Myc (Figure S5D and S5E). We note a pattern of cell-surface puncta in our live-cell immunostaining images, which implies that Reck localizes in membrane zones, perhaps in lipid rafts or caveolae (Figure S5E). Using STF reporter cells co-transfected with Gpr124, Wnt7a, and each of the Reck mutants, we observed that deletion of the five tandem CC domains (ΔCC1–5) completely abolished Reck activity (Figure S5B). Other truncation mutants (ΔFz-like CRD, ΔC-term1, ΔC-term2) substantially reduced but did not eliminate activity; it is unclear whether these reductions represent an indirect effect due to the shortened distance from the plasma membrane to more N-terminal domains. These data imply that the most N-terminal domains of both Gpr124 (LRR-Ig) and Reck (CC1–5) play a critical role in activating Wnt7a/Wnt7b signaling.
Based on these results, we focused on CC1–5 and constructed a series of deletion mutants to more precisely define the role of individual CC domains (Figure S5C–E). ΔCC2 and ΔCC5 were able to stimulate Wnt activity at levels comparable to WT Reck, while ΔCC3 had ~50% WT activity. The most dramatic defects in activity were observed for ΔCC1 and ΔCC4, with the latter showing a complete loss of function. We performed an initial analysis of Reck CC4 function by converting the presumptive surface residues to alanines in blocks of several amino acids per mutant; however, none of the 14 alanine-scanning mutants in CC4 showed a significant reduction in Wnt signaling when co-transfected with Wnt7a and Gpr124 (data not shown). One possible explanation for this result is that there are multiple non-adjacent surface regions that are functionally redundant for CC4 function. Alternatively, CC4 may be important only in relation to the overall architecture and/or spacing of other Reck domains.
Direct Binding Between Domains of Reck and Gpr124 that Activate Canonical Wnt Signaling
The biochemical mechanism by which Reck and Gpr124 synergistically enhance Wnt signaling has not been determined. Since both co-activators are membrane proteins, the simplest possibility is that they form part of the signaling complex with the receptor, co-receptor, and ligand. Thus far, the only reported experiment that directly addresses their physical association is a proximity ligation assay indicating that a measurable fraction of Reck and Gpr124 reside near one another when they are over-expressed in transiently co-transfected cells (Vanhollebeke et al., 2015). To test for a direct interaction between the domains of Reck and Gpr124 that are critical for canonical Wnt signaling, we generated alkaline phosphatase (AP) fusion proteins with the critical Wnt-activating domains of Reck (CC1–5) and Gpr124 (LRR-Ig). In these initial constructs, we included a cartilage oligomeric matrix protein (COMP) domain for pentamerization to increase the avidity of the probes (Figures 5A and S6A). These COMP-AP fusion proteins were tested for their ability to bind Gpr124, Reck, or various of their deletion derivatives on the surface of transfected COS-7 cells.
Figure 5. Direct Binding of the Domains of Reck and Gpr124 Responsible for Canonical Wnt Signaling.

(A) Diagram of COMP-AP probes (left) and Fc baits (right).
(B) AP binding assay using COS-7 cells transfected with Gpr124, Gpr124 deletion mutants, or Gpr125, a close homologue of Gpr124. Live cells were incubated with the Reck(CC1–5)-COMP-AP probe shown in (A). No Txn, no transfection.
(C) Cell-free binding assay using Gpr124(LRR-Ig) or Reck(CC1–5) (a negative control) Fc baits captured in Protein-G coated wells. Each bait was incubated with three COMP-AP probes: Reck(CC1), Reck(CC1–2), and Reck(CC1–5). After removing unbound probe, the bound probe was visualized with a colorimetric AP reaction.
(D) Amino acid sequence of mouse Reck CC1, with alanine scanning mutants indicated. Alanine substitution blocks 2, 7, 10, 14, and 16 were produced at near-WT levels and 9 and 15 at reduced levels (Figure S7A and S7B).
(E) Cell-free binding assay with the indicated alanine scanning mutants (top) and images of the AP reactions at the final time point (bottom). N.C., negative control.
(F) Cell-free binding assay with the indicated single alanine substitutions (top) and images of the AP reactions at the final time point (bottom). See Figure S7A and S7B for protein yield.
(G) Alignment and conservation of Reck CC1 across vertebrates, generated by Clustal Omega. Alignment symbols denote: (*) fully conserved residues; (:) residues with strongly similar properties; and (.) residues with weakly similar properties.
Scale bars, 200 µm.
The Reck(CC1–5)-COMP-AP probe bound robustly to COS-7 cells transfected with Gpr124 but not its close homologue Gpr125, which does not activate Wnt7a/Wnt7b signaling or synergize with Reck (Figure 5B; Zhou and Nathans, 2014; Vanhollebeke et al., 2015). Deletion of the Wnt-activating domains of Gpr124 (ΔLRR, ΔIg, and ΔLRR+Ig) abolished Reck(CC1–5)-COMP-AP binding, while deletion of the GAIN domain had little or no effect (Figure 5B). Deletion of the Gpr124 HormR domain, which reduces canonical signaling by ~50% in STF cells (Figure S4C), reduced Reck(CC1–5)-COMP-AP binding below the limit of detection (Figure 5B). It is possible that in Gpr124(ΔHormR), the LRR and Ig domains are positioned in a way that hinders their interaction with Reck(CC1–5)-COMP-AP. [In reciprocal experiments involving incubation of Gpr124(LRR-Ig)-COMP-AP to COS-7 cells transfected with Reck and its deletion derivatives, the greatest decrements in binding were with Reck ΔCC1–5, ΔCC2–5, ΔCC1, and ΔCC2 (Figures S6A and S6C), but we note that these binding experiments have a relatively low signal-to-noise ratio.] These data point to the Reck(CC1–5) domain as important for both Gpr124(LRRIg) binding and Wnt7a/Wnt7b signaling.
To investigate protein-protein binding in a cell-free system, Gpr124(LRR-Ig)-Fc and Reck(CC1–5)-Fc fusion proteins (“baits”) were captured on Protein-G-coated wells and incubated with various COMP-AP probes. Consistent with the COS-7 binding data, Reck(CC1–5)-COMP-AP bound robustly to Gpr124(LRR-Ig)-Fc but not to a negative control bait (Figure 5C). Reciprocally, Gpr124(LRR-Ig)-COMP-AP bound to Reck(CC1–5)-Fc, albeit weakly, but not to a negative control bait (Figure S6B). Importantly, Reck(CC1–2)-COMP-AP and Reck(CC1)-COMP-AP bound robustly to Gpr124(LRR-Ig)-Fc, showing that Reck(CC1) is not only necessary but also sufficient for direct binding (Figure 5C).
To identify the region of Reck(CC1) that contacts Gpr124, we constructed 16 alanine scanning block substitutions in Reck(CC1)-COMP-AP, together covering nearly all CC1 residues other than glycine and cysteine (Figure 5D). By immunoblotting and AP enzyme activity, seven of the 16 CC1 mutants expressed at readily detectable levels (Ala Scan-2, -7, -9, -10, -14, -15, and -16; Figure S6D and S6E), and these were used for binding experiments after equalizing their concentrations. Only two of the seven CC1 mutants failed to bind to Gpr124(LRR-Ig)-Fc: Ala Scan-9 and -10 (Figure 5E). Single alanine substitution of the five residues encompassed by these two adjacent block substitutions showed that three of the five – R69A, P71A, and Y73A – greatly decreased binding (Figure 5F). These three residues are highly conserved across vertebrates, whereas adjacent residues that are dispensable for binding are not (Figure 5G). Taken together, these experiments pinpoint a highly conserved region – most likely a binding surface – on Reck CC1 that is required for Gpr124(LRR-Ig) binding.
The Reck CC1 Binding Site for Gpr124 is Important for Wnt Signaling and CNS Angiogenesis
To assess the importance of the Reck CC1 binding site for canonical Wnt signaling, we generated full-length Reck derivatives that contained the Ala Scan-9, -10, and -9+10 substitutions. The three mutants exhibited a level of plasma membrane accumulation similar to WT, as determined by surface biotinylation (Figure S6F). When cotransfected with Gpr124 and Wnt7a in STF cells, all three mutants showed ~50% of the activity level of WT Reck (Figure 6A), and this reduction was consistent over a wide range of Gpr124 and Reck DNA concentrations (Figure 6B and 6C).
Figure 6. Reck-Gpr124 Complex Formation is Important for Wnt Activation and CNS Angiogenesis.

(A) STF cells were transfected with: Wnt7a, Gpr124, and Reck WT or Ala Scan mutant plasmids. Luciferase assays were performed as described in ‘STAR Methods’.
(B) As in (A), but showing the dose response curve for varying concentrations of Gpr124 DNA in the presence of constant WT Reck (blue) or Ala Scan 9+10 (red).
(C) As in (A), but showing the dose response curve for varying concentrations of WT Reck (blue) or Ala Scan 9+10 (red) in the presence of constant WT Gpr124.
(D) Diagram of the mouse Reck WT and CRISPR-edited alleles. The region corresponding to Ala Scan mutants 9 and 10 in Reck(CC1) is encoded by exon 3.
(E) Reck protein abundance at E13.5 is unaltered in ReckCr/Cr embryos compared to a WT control, as determined by immunoblotting. The disappearance of the full-size Reck band in the ReckΔex2/Δex2 embryos confirms the specificity of the anti-Reck antibody. (*) denotes a non-specific band in the actin immunoblot. MW standards are shown at left.
(F–G) E13.5 coronal sections of brain (columns 1–3) and cross-sections of spinal cord (column 4). Boxed regions in LGE (a) and cortex (b) are shown at higher magnification in columns 2 and 3, respectively. ReckCr/Δex2;Gpr124+/Δ mutant embryos (G) show reduced vascular density and glomeruloid tufts in the LGE, accompanied by infiltration of GS Lectin+ macrophages. Vascular density in the spinal cord is also reduced.
(H) Quantification of relative vascular density (top) and the number of GS Lectin+ macrophages (bottom) in LGE, cortex, and spinal cord for the genotypes shown in (F–G).
Values represent means ± SD. Scale bars, 200 µm.
If the Reck-Gpr124 interaction is important for Wnt7a/Wnt7b signaling, then its disruption in vivo should impair CNS angiogenesis. We therefore used CRISPR/Cas9 to generate mice carrying a Reck allele with the five Ala Scan-9+10 substitutions, hereafter referred to as ReckCr (Figure 6D). At E13.5, immunoblotting shows that the abundance and electrophoretic mobility of the Reck protein was the same in WT and ReckCr/Cr embryos (Figure 6E), implying that any phenotypic consequences of the ReckCr mutation likely reflect a functional defect rather than a folding or stability defect.
Both ReckCr/+ and ReckCr/Cr mice are viable, with the former showing no CNS vascular abnormalities and the latter showing very mild angiogenesis defects in the lateral ganglionic eminence (LGE) that are incompletely penetrant (data not shown). Different brain regions differ in their sensitivity to the loss of Wnt7a/Wnt7b signaling components. Listed in order from most to least sensitive, the regions are LGE, MGE, cortex, and hindbrain. Based on the genetic interactions between Reck and Gpr124 shown in Figure 4, we asked whether the ReckCr allele could be distinguished phenotypically from a WT Reck allele on a genetic background sensitized by heterozygous loss of Gpr124. Figure 6F–H shows that at E13.5 Reck+/Δex2;Gpr124+/Δ embryos exhibit a normal vascular network, whereas ReckCr/Δex2;Gpr124+/Δ embryos show vascular defects in the LGE, with glomeruloid-like tufts and increased infiltration of GS Lectin+ macrophages. The ReckCr/Δex2;Gpr124+/Δ spinal cord shows a modest reduction in vascular density; the very small reduction in vascular density in the cortex is not statistically significant. These data are consistent with the STF data showing that the Ala Scan-9+10 substitutions that correspond to the ReckCr allele partially disrupt Reck function, and they support a model in which Reck-Gpr124 binding is important for Wnt7a/Wnt7b signaling during CNS angiogenesis.
Frizzled- and Wnt7a/Wnt7b-dependent assembly of Reck and Gpr124 into a cell-surface complex
We next asked: does the Reck-Gpr124 complex interact directly with the Wnt/Frizzled complex to distinguish Wnt7a/Wnt7b from other Wnts and from Norrin? To address this question, we developed an experimental platform in which cell-surface binding by the essential N-terminal domains of Reck or Gpr124 could be used to assess protein-protein interactions with various co-expressed and co-assembled ligand/receptor components.
In an initial set of experiments, we explored the binding properties of Reck(CC1–5)-AP and AP-Gpr124(LRR-Ig) – i.e. AP fusion proteins that lack the COMP pentamerization motif (Figure 7A). In contrast with their higher avidity COMP-AP counterparts (Figures 5 and S6), Reck(CC1–5)-AP did not detectably bind to COS-7 or HEK293T cells expressing Gpr124, and AP-Gpr124(LRR-Ig) did not detectably bind to COS-7 or HEK293T cells expressing Reck. Additionally, Reck(CC1–5)-AP and AP-Gpr124(LRR-Ig) did not detectably bind to cells expressing Wnt7a, Fz4, or Wnt7a+Fz4; Reck(CC1–5)-AP did not detectably bind to cells expressing Gpr124+Fz4; and AP-Gpr124(LRR-Ig) did not detectably bind to cells expressing Reck+Fz4. However, expression of Gpr124+Wnt7a+Fz4 or Gpr124+Wnt7a+Fz4+Lrp5 dramatically enhanced the binding of Reck(CC1–5)-AP, and expression of Reck+Wnt7a+Fz4 or Reck+Wnt7a+Fz4+Lrp5 dramatically enhanced the binding of AP-Gpr124(LRR-Ig) (Figure 7A). [Interestingly, Reck(CC1–5)-AP bound at a low level to cells expressing Gpr124+Wnt7a, and AP-Gpr124(LRR-Ig) bound at a moderate level to cells expressing Reck+Wnt7a. We attribute the binding signals observed in the absence of a co-transfected Frizzled or co-transfected Lrp5/Lrp6 to the low level expression of many Frizzed family members and Lrp5/Lrp6 by HEK293 cells, as determined by RNAseq (unpublished data).] Importantly, in all cases in which there was a positive binding signal, the co-expression of Wnt7a was required, suggesting that high affinity complex formation is ligand-dependent (Figure 7A, S7A, and S7B).
Figure 7. Reck-Gpr124 Assembles into a Multi-Protein Complex with Wnt7a/Wnt7b and Frizzled.

(A) AP binding assay using HEK293T cells transfected with Gpr124, Wnt7a, Fz4, and/or Lrp5 (left) or Reck, Wnt7a, Fz4, and/or Lrp5 (right). Live cells were incubated with Reck(CC1–5)-AP (left) or AP-Gpr124(LRR-Ig) (right).
(B) AP binding assay using HEK293T cells transfected with Gpr124 and Fz4, along with each of the 19 Wnts and Norrin. Live cells were incubated with Reck(CC1–5)-AP (left) or AP-Gpr124(LRR-Ig) (right).
(C) AP binding assay using HEK293T cells transfected with Gpr124 and Wnt7a (top) or Wnt7b (bottom), toegther with each of the 10 Fz receptors or a no transfection control. Live cells were incubated with Reck(CC1–5)-AP.
(D) Model of the multi-protein signaling complex, consisting of Reck, Gpr124, Wnt7a or Wnt7b, Frizzled, and Lrp5 or Lrp6. The straight double-headed arrow indicates a direct interaction between Reck(CC1) and Gpr124(LRR-Ig). The curved double-headed arrow indicates a direct interaction between Reck/Gpr124 and Fz/(Wnt7a or Wnt7b) sub-complexes. Modified from Zhou and Nathans (2014).
Scale bars, 200 µm.
Having earlier identified the critical residues on Reck CC1 that facilitate binding to Gpr124 (Figure 5D–5F), we next tested whether the AP-Gpr124(LRR-Ig) probe could assemble into a complex in the presence of Wnt7a and Fz4 along with either WT Reck or the Reck-Ala9+10 mutant (Figure S7C). Consistent with the in vitro Reck CC1-COMP-AP binding experiments in Figure 5D–5F, mutating the critical CC1 amino acids to alanine abolishes AP-Gpr124(LRR-Ig) binding to the cell surface (Figure S7C). These data further support the importance of Reck CC1 in binding to Gpr124(LRR-Ig).
The simplest explanation for the binding data in Figure 7A, S7A, and S7B is that the AP probes co-assemble into a multi-protein signaling complex at the cell surface. However, it is conceivable that binding of Reck(CC1–5)-AP reflects high-level accumulation of Gpr124 at the cell surface and that binding of AP-Gpr124(LRR-Ig) reflects high-level accumulation of Reck at the cell surface – in both cases as a consequence of the combined production of multiple signaling proteins. To test this idea, we assessed total and cell-surface levels of Fz4-1D4 (1D4 is a C-terminal epitope tag), Wnt7a-1D4, Gpr124-3×HA, and Reck with various combinations of co-expressed proteins, by cell surface biotinylation, NeutrAvidin pull-down, and Western blotting [Figure S7D; the bioactivity for Fz4-1D4 and Wnt7a-1D4 was comparable to that of their WT counterparts by STF reporter activity (data not shown)]. For most combinations of co-transfected proteins, Fz4, Gpr124, and Reck exhibited little or no change in cell surface levels (Figure S7D). The only exception to this pattern is seen for Gpr124 co-transfected with Fz4, which leads to a ~2–4-fold increase in cell surface Gpr124 (Figure S7D). However, we note that co-transfection of Gpr124 and Fz4 does not lead to detectable Reck(CC1–5)-AP binding. Wnt7a-1D4 accumulation at the plasma membrane could not be defined as it was below the limit of detection of this assay (Figure S7D). These data show that cell-surface accumulation of individual binding proteins (Gpr124 and Reck) cannot account for the dramatically enhanced binding signals observed in Figures 7A, S7A, and S7B.
To determine whether the Reck(CC1–5)-AP and AP-Gpr124(LRR-Ig) binding reactions exhibit Wnt specificity, we co-transfected HEK293T cells with Gpr124+Fz4 or Reck+Fz4 together with each of the 19 Wnt ligands or Norrin. Strikingly, Reck(CC1–5)-AP only bound when cells expressed Gpr124+Fz4 together with Wnt7a or Wnt7b, and AP-Gpr124(LRR-Ig) only bound when cells expressed Reck+Fz4 together with Wnt7a or Wnt7b (Figure 7B). These binding specificities precisely mirror the signaling specificity of Reck and Gpr124 previously defined in STF cells (Zhou and Nathans, 2014; Vanhollebeke et al., 2015), and they further support the conclusion that binding reflects the assembly of a multi-protein ligand/receptor/co-activator complex.
To assess Frizzled specificity, we expressed Wnt7a+Gpr124 or Wnt7b+Gpr124 together with each of the 10 Frizzled receptors and probed the cells with Reck(CC1–5)-AP (Figure 7C). Seven of ten Frizzleds showed clear binding signals, with only Fz3, Fz6, and Fz10 showing no detectable binding. The strongest signals were observed with Fz5 and Fz8. [We did not perform the analogous experiment using AP-Gpr124(LRR-Ig) because, as shown in Figure 7A and noted above, this probe produces a moderate binding signal even in the absence of Frizzled transfection.] Consistent with these data, previous work using the STF reporter assay showed that (1) Wnt7a and Wnt7b elicited the strongest responses when co-transfected with Fz5 or Fz8, and (2) Fz3 and Fz6 gave little or no response with any Wnt ligand and do not synergize with Reck and Gpr124 in the presence of Wnt7a (Yu et al., 2012; Vanhollebeke et al., 2015). RNAseq of FACS-purified P7 mouse brain ECs shows expression of Fz6 (~110 FPKM), Fz4 (35 FPKM), Fz1 (6 FPKM), and Fz8 (~4 FPKM) (Zhang et al., 2014). Integrating this gene expression data with the Reck/Gpr124 signaling and binding data suggests that Wnt7a/Wnt7b signaling in brain ECs is likely mediated by the combined actions of Fz4, Fz1, and Fz8.
Discussion
The experiments reported here reveal an essential role for Reck and Gpr124 as integral parts of the cell-surface protein complex that transduces Wnt7a- and Wnt7b-specific signals in mammalian CNS ECs to promote angiogenesis and BBB formation and maintenance. In particular, we show that (1) in the embryo, EC-specific reduction in Reck is accompanied by a reduction in canonical Wnt pathway activation in CNS ECs and by angiogenesis defects that can be largely rescued by artificially stabilizing beta-catenin; (2) the phenotypes produced by EC-specific reduction in Reck closely resemble the phenotypes produced by loss of Gpr124 in their appearance, timing, and anatomic selectivity; (3) EC-specific reduction in Reck synergizes with loss of Norrin in CNS angiogenesis and BBB maintenance in a manner closely resembling the synergy produced by the combined loss of Gpr124 and Norrin; (4) EC-specific reduction in Reck synergizes with reductions in Gpr124 and in Wnt7a/Wnt7b in CNS angiogenesis; (5) the CC1 domain of Reck interacts directly with the LRR-Ig domains of Gpr124, and weakening this interaction by targeted mutagenesis of CC1 reduces Reck/Gpr124 stimulation of Wnt7a-mediated canonical Wnt signaling and impairs CNS angiogenesis; (6) Gpr124(LRR-Ig)-AP binds to cells expressing Frizzled, Wnt7a or Wnt7b, and Reck; and (7) Reck(CC1–5)-AP binds to cells expressing Frizzled, Wnt7a or Wnt7b, and Gpr124. Taken together, these data are most consistent with a model in which Wnt7a- and Wnt7b-mediated canonical signaling in ECs occurs in the context of a multi-protein complex consisting of Wnt, Frizzled, Lrp5 or Lrp6, Gpr124, and Reck (Figure 7D).
Ligand-Receptor Specificity in Canonical Wnt Signaling
In the twenty years since the discovery that Frizzleds constitute the principal receptors for Wnts and the roughly contemporaneous appreciation that mammalian genomes code for 10 Frizzleds and 19 Wnts, the extent of Wnt-Frizzled specificity and the biological roles that such specificity might play have largely remained open questions. The potential for a high degree of ligand-receptor specificity in canonical Wnt signaling is seen in the high selectivity of the non-Wnt ligand Norrin for its receptor Frizzled4 (Xu et al., 2004; Smallwood et al., 2007; Chang et al., 2015). Commensurate with this high degree of specificity, the crystal structure of the Norrin dimer in complex with a pair of Frizzled4 CRDs shows ~3,500 Å2 of buried surface area for the dimeric interaction (Chang et al., 2015; Shen et al., 2015).
In contrast to the specificity of Norrin-Frizzled4 interactions, signaling assays with the 190 pairwise combinations of 19 Wnts and 10 Frizzleds show relatively promiscuous interactions (Yu et al., 2012). Although only a few Wnts are available in biochemically well-behaved form for direct binding assays, the several that have been tested bind to multiple Frizzleds (Hsieh et al., 1999; Dijksterhuis et al., 2015). Consistent with this relatively promiscuous binding, the crystal structure of Xenopus Wnt8 in complex with mouse Fz8 CRD shows: (1) ~600 Å2 of buried surface area associated with a hydrophobic groove on the CRD and the lipid that is covalently-linked to Wnt8, a conserved feature among mammalian Wnts, and (2) an additional ~1,400 Å2 of surface area buried by protein-protein contacts that largely consist of residues from each protein that are conserved across family members (Janda et al., 2012).
Overlap in Wnt-Frizzled specificity could plausibly explain the greater severity of several double Wnt or double Frizzled KO phenotypes compared to the constituent single KO phenotypes [e.g., Wnt1 and Wnt4 (Mulroy et al., 2002); Wnt3a and Wnt8a (Cunningham et al., 2015); Fz1, Fz2, and Fz7 (Yu et al., 2010, 2012)]. Under this scenario, WT function would be redundantly mediated by two Wnts signaling through the same Frizzled receptor(s) or by two Frizzleds stimulated by the same Wnt(s), and, in each case, loss of both redundant genes would be required to produce a severe phenotype. Overlap in specificity is also possible for pairs of Wnt genes or pairs of Frizzled genes with non-interacting loss-of-function phenotypes, since the absence of a genetic interaction could simply reflect non-overlap in the spatiotemporal pattern of expression rather than intrinsic differences in ligand-receptor specificity. As described here, the existence of additional membrane components (Reck and Gpr124) that are part of the Wnt/Frizzled/Lrp signaling complex represents an evolutionary strategy that increases Wnt-Frizzled specificity without requiring a change in the Wnt-Frizzled binding interface.
Gpr124 and Reck Regulate Canonical Wnt Signaling at the Ligand/Receptor Level
The ability of Gpr124 and Reck to enhance signaling by Wnt7a and Wnt7b, with little or no effect on signaling by other Wnts, argues for a direct interaction between Reck/Gpr124 and the Wnt ligand. This model is strongly supported by the AP-Gpr124(LRR-Ig) and Reck(CC1–5)-AP cell-surface binding experiments in Figure 7. Less direct models are possible, including models in which a distinctive Wnt7a/Wnt7b-induced conformational change in Frizzled and/or Lrp5/6 is recognized by Reck/Gpr124. However, models in which Reck/Gpr124 regulates the stability or trafficking of unliganded Frizzled or Lrp5/6 seem less likely because they do not account for the observed Wnt7a/Wnt7b specificity.
A technical challenge in defining the biochemical mechanism of Reck and Gpr124 function is the relatively low affinities of their interactions. This lower affinity likely reflects (1) the proximity of the interacting plasma membranes, which provides a restricted search space and insures favorable binding kinetics, and (2) the presence of multiple copies of the two proteins, which provides high avidity. For laterally interacting proteins on the plasma membrane of the same cell, we can rationalize the low affinity of protein-protein interactions in an analogous way: since the constraint of a two dimensional diffusion space favors binding, low affinities suffice for efficient assembly of the multi-protein complex. Indeed, high affinity interactions between components would be disfavored if the system operates with rapid exchange of subunits or a steady state mixture of free and assembled subunits. In the case of canonical Wnt signaling in CNS ECs, rapid exchange of subunits may be an efficient way for Frizzled4 and Lrp5/6 to mediate signaling via Norrin, with Tspan12 as a co-activator, and via Wnt7a/Wnt7b, with Reck/Gpr124 as coactivators.
Reck, a Multifunctional Regulator
The present work extends Reck’s biochemical and functional repertoire by revealing a role for its N-terminal CC domains. In addition to the role of CC1 in binding to Gpr124 and stimulating Wnt7a/Wnt7b signaling, CC4 also appears to play an important role based on the large decrease in Wnt7a/Wnt7b signaling that accompanies its deletion. The five CC domains, each of which has six cysteines, have no readily detectable homologues in the current protein database.
Previous work on Reck activity in vascular development has largely focused on MMP inhibition, which derives from Reck’s three tandem repeats of a Kazal-type serine protease inhibitor domain (Takahashi et al., 1998; Oh et al., 2001; Chang et al., 2008). Reck is essential for angiogenesis and vascular remodeling in multiple non-CNS tissues, including in the yolk sac and abdomen (Chandana et al., 2010), and Reck’s role in these contexts has been ascribed to MMP inhibition on the surface of ECs and mural cells (de Almeida et al., 2015). The importance of Reck-mediated MMP inhibition is supported by the observation that the vascular defects and mid-gestational lethality caused by ubiquitous knockout of Reck can be partially suppressed by knocking out MMP2 (Oh et al., 2001).
In addition to its role in vascular biology, Reck inhibits a disintegrin and metalloproteinase (ADAM)-10 (and possibly other ADAM family members) in cortical and spinal cord progenitors, which increases Notch signaling by suppressing ADAM-catalyzed cleavage of the Notch ligand Delta-like 1 (Dll1) (Muraguchi et al., 2007). In the developing spinal cord, glycerophosphodiester phosphodiesterase 2 (GDE2) releases Reck from the plasma membrane by cleaving its GPI-anchor, thereby reducing Reck concentration locally (Park et al., 2013). The resulting dis-inhibition of ADAM-10 enhances cleavage of Dll1 and thereby down-regulates Notch signaling, which promotes differentiation of spinal motor neurons (Muraguchi et al., 2007).
Gel filtration and electron microscopy of negatively stained samples indicates that Reck is a dimer (Omura et al., 2009). As current evidence indicates that the Lrp5 and Lrp6 co-receptors are also dimers (Liu et al., 2003; Chen et al., 2014), and Norrin is a disulfide linked dimer (Chang et al., 2015; Shen et al., 2015), it appears plausible that the native signaling complexes comprising Norrin/Frizzled4/(Lrp5 or Lrp6)/Tspan12 and (Wnt7a or Wnt7b)/Frizzled/(Lrp5 or Lrp6)/Gpr124/Reck consist of two copies of each component. For the latter complex, this would imply a molecular mass of ~900 kDa.
Interplay of Norrin and Wnt7a/Wnt7b Signaling in Human Disease
Compromised BBB integrity is associated with a wide variety of neurologic disorders, including traumatic brain injury, stroke, multiple sclerosis, infection, and neurodegenerative diseases such as Alzheimer’s disease (Zhao et al., 2015). Recent studies using mouse models of glioblastoma and ischemic stroke found a greater loss of CNS microvascular integrity in mice with EC-specific loss of Gpr124 compared to heterozygous controls, suggesting that, in these and other disorders characterized by loss of CNS vascular integrity, reduced Wnt7a/Wnt7b signaling could predispose to worse clinical outcomes and enhanced Wnt7a/Wnt7b signaling could be of therapeutic value (Chang et al., 2017). Direct evidence that canonical Wnt signaling may be relevant for human BBB integrity comes from the observation that ~10% of Norrie disease patients suffer from a chronic seizure disorder, which could plausibly reflect leakage of excitatory serum contents, such as glutamate, into the CNS (Smith et al., 2012; Okumura et al., 2015).
A distinctive feature of the control of CNS angiogenesis and BBB integrity by canonical Wnt signaling is the anatomic regionalization of Norrin and Wnt7a/Wnt7b/Gpr124/Reck action. In the embryonic hindbrain, both systems are active and they show nearly complete redundancy in promoting angiogenesis. In the embryonic cerebral cortex and ganglionic eminences, Wnt7a/Wnt7b/Gpr124/Reck signaling is predominant and Norrin signaling is minimal. In contrast, in the retina, Norrin signaling is predominant and Wnt7a/Wnt7b/Gpr124/Reck signaling is minimal, with the result that loss of Norrin signaling leads to a severe defect in retinal angiogenesis. It is interesting to consider that if angiogenesis in the retina more closely resembled angiogenesis in the hindbrain, with redundant Norrin and Wnt7a/Wnt7b/Gpr124/Reck signaling, then Norrie Disease, the most severe inherited disorder of retinal vascularization, would likely not exist.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Jeremy Nathans (jnathans@jhmi.edu).
Experimental Model and Subject Details
Gene Targeting
To construct the Cre-dependent reporter for canonical Wnt signaling, the following elements were inserted (in the order listed) into a standard Rosa26 targeting vector: an frt-phosphoglycerate kinase (PGK)-neomycin (Neo)-frt (FNF) cassette, which includes a strong poly-adenylation signal; six tandem repeats of a consensus LEF/TCF binding site CCTTTGATC and a minimal Heat shock protein 68 promoter (Ferrer-Vaquer et al., 2010); a LSL cassette; an open reading frame coding for a histone H2B-GFP-6×Myc fusion protein; and a bovine growth hormone 3’UTR. The R26-Tcf/Lef-LSL-H2B-GFP targeting construct with a flanking Diphteria toxin-A coding sequence was electroporated into R1 ES cells, which were then subjected to G418 selection. Clones harboring the targeted allele were identified by Southern blot hybridization, karyotyped, and injected into blastocysts. Germline transmission to progeny of founder males was determined by PCR.
The ReckCr mouse was generated using CRISPR/Cas9 gene editing. A sgRNA (gattattgccctgaaacaat) targeting exon 3 of Reck was selected and synthesized, as described by Pelletier et al. (2015). Briefly, a dsDNA template containing the sequences for a T7 promoter, the sgRNA, and a tracrRNA scaffold was generated by tandem PCR amplification and purification on a Qiagen column (Qiagen 28106). In vitro transcription of this dsDNA template was performed using the MEGAshortscript T7 Kit (Invitrogen AM1354), and products were purified using the MEGAclear Transcription Clean-Up Kit (Invitrogen AM1908). The Cas9 mRNA was transcribed from a modified pX330 plasmid (Addgene 42230), which contains a T7 promoter (pX330+T7), using the mMESSAGE mMACHINE T7 Ultra Transcription Kit (Invitrogen AM1345). The transcript was purified by LiCl precipitation. The quality of all purified transcripts was confirmed using the Agilent 2100 Bioanalyzer. The ssODN HDR template (tttctcctcaaaaagtgaatcccgactgaaacatctgttggctgctgctgctgctgcttgccctgaaacaatggtgagtcttacggagctgtggaag) was synthesized by IDT. The sgRNA, Cas9 mRNA, and HDR template were injected into C57BL/6 × SJL F2 embryos and correctly targeted founders were backcrossed to C57BL/6 mice for a total of four generations.
Mice
The following mouse alleles were used: Reckflex2 (Acc. No. CDB0488K: http://www.cdb.riken.jp/arg/mutant%20mice%20list.html; Chandana et al., 2010), ReckΔex1 (Oh et al., 2001), Ctnnb1flex3 (Harada et al., 1999), NdpΔ (JAX 012287; Ye et al., 2009), Gpr124fl (JAX 016881; Cullen et al., 2011), Wnt7afl (Dunlap et al., 2011), Wnt7bfl (JAX 008467; Rajagopal et al., 2008), Tie2-Cre (JAX 008863), and Pdgfb-CreER (Claxton et al., 2008). The following null alleles were generated by breeding mice carrying the germline Sox2-Cre transgene with floxed (fl) mice: ReckΔex2, Gpr124Δ, Wnt7aΔ, and Wnt7bΔ. All mice were housed and handled according to the approved Institutional Animal Care and Use Committee (IACUC) protocol MO16M369 of the Johns Hopkins Medical Institutions.
Method Details
Antibodies and Other Reagents
The following antibodies were used for tissue immunohistochemistry: rat anti-mouse PECAM/CD31 (BD Biosciences 553370); rabbit anti-Glut1 (Thermo Fisher Scientific RB-9052-P1); rat anti-mouse PLVAP/MECA-32 (BD Biosciences 553849); mouse anti-Claudin5, Alexa Fluor 488 conjugate (Thermo Fisher Scientific 352588); Texas Red streptavidin (Vector Laboratories SA-5006); rabbit anti-GFP, Alexa Fluor 488 conjugate (Thermo Fisher Scientific A21311); rabbit anti-6×Myc (JH6204); Brilliant Violet 421 rat anti-mouse/human Cd11b (BioLegend 101235); and rat anti-F4/80, Alexa Fluor 488 conjugate (Bio-Rad MCA497A488T). Alexa Fluor-labeled secondary antibodies and GS Lectin (Isolectin GS-IB4) were from Thermo Fisher Scientific.
The following antibodies were used for immunoblot analysis: rabbit anti-6×Myc (JH6204); rabbit anti-HA (Cell Signaling 3724); mouse anti-actin (EMD Millipore MAB1501); rabbit anti-Reck (Cell Signaling 3433); mouse anti-1D4 (MacKenzie et al., 1984); and rat anti-alpha tubulin (Invitrogen MA1-80017). Fluorescent secondary antibodies for immunoblotting were from Li-Cor.
Tissue Processing and Immunohistochemistry
Tissue were prepared and processed for immunohistochemical analysis as described by Wang et al. (2012) and Zhou et al. (2014). Briefly, embryos and early postnatal brains were harvested and immersion fixed overnight at 4°C in 1% PFA, followed by 100% MeOH dehydration overnight at 4°C. Embryos that were specifically prepared for F4/80 or Cd11b staining were harvested and dehydrated without fixation in 100% acetone overnight at 4°C. All tissues were re-hydrated the following day in 1× PBS at 4°C for at least 3 hours before embedding in 3% agarose. Tissue sections of 150–180 µm thickness were cut using a vibratome (Leica).
Sections were incubated overnight with primary antibodies (1:400) or Texas Red streptavidin (1:100) diluted in 1× PBSTC (1× PBS + 0.5% Triton X-100 + 0.1mM CaCl2) + 10% normal goat serum (NGS). Sections were washed at least 3 times with 1× PBSTC over the course of 6 hours, and subsequently incubated overnight with secondary antibodies (1:400) diluted in 1× PBSTC + 10% NGS. If a primary antibody raised in rat was used, secondary antibodies were additionally incubated with 1% normal mouse serum (NMS) as a blocking agent. The next day, sections were washed at least 3 times with 1× PBSTC over the course of 6 hours, and flat-mounted using Fluoromount G (EM Sciences 17984-25). Sections were imaged using a Zeiss LSM700 confocal microscope, and processed with ImageJ, Adobe Photoshop, and Adobe Illustrator software. Incubation and washing steps were performed at 4°C.
4-HT and Tamoxifen Preparation and Administration
4-HT and Tamoxifen were prepared as described by Zhou and Nathans (2014). In short, solid 4-HT (Sigma-Aldrich H7904) or Tamoxifen (Sigma-Aldrich T5648) was dissolved in an ethanol:sunflower seed oil (Sigma-Aldrich S5007) mixture (1:5 volume) to a final concentration of 4 mg/ml or 50 mg/ml, respectively, and stored in aliquots at −80°C. Thawed aliquots of 4-HT and Tamoxifen were diluted to a final concentration of 1 mg/ml and 10 mg/ml, respectively. All injections were performed intraperitoneally. Pups were injected with 50 µl of 4-HT at P3 and again at P4. Gestational day (G)10.5 females were injected with 150 µl of Tamoxifen.
Sulfo-NHS-Biotin Preparation and Administration
Sulfo-NHS-Biotin (Thermo Scientific 21335) was dissolved in 1× PBS to a final concentration of 20 mg/ml. Pups were injected with 100–150 µl Sulfo-NHS-Biotin IP 30–45 minutes before sacrifice.
Plasmids
The mouse Reck cDNA sequence (GE Dharmacon; clone BC138065) was cloned into the pRK5 mammalian expression vector. The Myc epitope (EQKLISEEDL) was inserted at an endogenous XmaI site downstream of the Reck signal sequence to generate Myc-Reck. Myc-tagged Reck was used as a backbone to generate ΔCC1–5 (Δ amino acids (Δaa)30–339), ΔFz-like CRD (Δaa348–468), ΔC-term1 (Δaa477–830), and ΔC-term2 (Δaa477–906). An 11 amino acid spacer (GGTRGSGAPGG) replaced each of these deleted segments. Additional deletions in Myc-Reck were constructed without the spacer: ΔCC1 (Δaa30–103), ΔCC2 (Δaa104–150), ΔCC3 (Δaa151–215), ΔCC4 (Δaa216–291), ΔCC5 (Δaa292–342), ΔCC2–5 (Δaa104–342), ΔCC3–5 (Δaa151–342), and ΔCC4–5 (Δaa216–342). The COMP domain in the COMP-AP vector encompassed the pentameric domain of COMP (Malashkevich et al., 1996).
The Gpr124 plasmid was a kind gift from Dr. Calvin Kuo (Stanford) and was cloned into a pRK5 expression vector. A 3×HA epitope tag (SGSSGYPYDVPDYAGGYPYDVPDYAGGYPYDVPDYATRG) was inserted between amino acids 1322 and 1323 to generate Gpr124-3×HA. The epitope tagged Gpr124 was used as a backbone to generate the following deletions: ΔLRR (Δaa37–245), ΔIg (Δaa253–348), ΔLRR+Ig (Δaa37–348), ΔHormR (Δaa352–427), and ΔGAIN (Δaa428–751).
Inserts for AP and Fc constructs were PCR amplified from Reck and Gpr124 pRK5 expression vectors. Alanine scanning mutagenesis was performed by tandem PCR. All constructs were confirmed by sequencing.
Expression plasmids for the mouse Wnts and Frizzleds are described in Yu et al. (2012).
Luciferase Assays
Dual luciferase assays were performed as described by Xu et al. (2004). Briefly, STF cells were plated on 96-well plates at a confluency of 30–40%. The following day, fresh DMEM/F-12 (Thermo Fisher Scientific 12500) supplemented with 10% fetal bovine serum (FBS) was replaced in each of the wells. Three hours later, cells were transfected in triplicate with expression plasmids (180–240 ng of DNA per 3 wells) using FuGENE HD Transfection Reagent (Promega E2311). The DNA master mix (unless otherwise specified) included: 1.5 ng of the internal control Renilla luciferase plasmid (pRL-TK), and 60 ng each of the pRK5 expression plasmids for Gpr124, Reck, Wnt7a, mutants, and control vector. 48 hours post-transfection, cells were harvested in 1× Passive Lysis Buffer (Promega E194A) for 20 minutes at room temperature. Lysates were used to measure Firefly and Renilla luciferase activity using the Dual-Luciferase Reporter Assay System (Promega E1910) and a Turner BioSystems Luminometer (TD-20/20). Relative luciferase activity was calculated by normalizing Firefly/Renilla values. GraphPad Prism 7 software was used to generate plots and perform statistical analysis. The mean ± standard deviations are shown.
Alkaline Phosphatase Binding Assays
Conditioned DMEM/F-12 media containing AP and Fc fusion proteins were collected from HEK293T cells that were transfected with pRK5 expression plasmids using FuGENE HD Transfection Reagent. For Fc fusion proteins, the conditioned media was removed 24 hours after transfection, and cells were washed at least 3 times with 1× PBS prior to incubating them in serum-free DMEM/F-12. All conditioned media were collected 72 hours post-transfection and spun-down to remove detached cells. To quantify yield of AP fusion proteins, an aliquot of the conditioned medium was incubated with BluePhos phosphatase substrate solution (5-bromo-4-chloro-3-indolyl phosphate/tetrazolium; Kirkegaard and Perry Laboratories 50-88-00).
For cell-based COMP-AP binding assays (Figures 5 and S6), COS-7 cells were transfected for 48 hours and subsequently incubated with the COMP-AP probe in serum-containing conditioned medium at 4°C for 2 hours. Cells were washed 6 times with cold PBS prior to fixing with cold 4% paraformaldehyde (PFA) in PBS. Fixed cells were placed in a 70°C water bath for 1 hour to heat denature endogenous AP. Bound AP was visualized by incubation with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (NBT/BCIP) substrate (Roche 11383213001) at room temperature.
For cell-based AP binding assays to assess multi-protein complexes (Figure 7), HEK293T cells were used because their high transfection efficiency facilitates co-transfection of multiple plasmids. Cells were plated on 0.2% gelatin coated coverslips, and 48 hours post-transfection the coverslips were incubated with serum-containing conditioned medium at 4°C for 2 hours. Cells were washed 3 times with cold serum-free DMEM/F-12 media prior to fixation, heat denaturation, and AP visualization as described above.
The following probes were tested for cell-based binding assays: Reck(CC1–5)-AP, Gpr124(LRR-Ig)-AP (data not shown), AP-Gpr124(LRR-Ig), Reck(CC1–5)-COMP-AP, and Gpr124(LRR-Ig)-COMP-AP. Gpr124(LRR-Ig)-AP bound minimally to COS-7 cells expressing Reck (data not shown); a stronger signal was obtained with Gpr124(LRR-Ig)-COMP-AP. AP-Gpr124(LRR-Ig), which contains a 3×Myc linker between AP and Gpr124(LRR-Ig), did not bind to COS-7 cells expressing Reck, but bound specifically to HEK293T cells expressing Frizzled/Wnt/Reck multi-protein complexes.
For cell-free AP binding assays, Fc fusion proteins in serum-free conditioned medium were captured on 96-well Protein-G plates (Thermo Fisher Scientific 15131) at 4°C overnight. Wells were washed 3× with PBS, and incubated with conditioned medium containing COMP-AP fusion proteins at 25°C for 1–2 hours. Wells were subsequently washed 6× with PBS, and incubated with BluePhos phosphatase substrate solution. AP activity was detected by measuring absorbance at 620 nm using a SpectraMax M3 Microplate Reader (Molecular Devices). For experiments involving a comparison among different COMP-AP probes, probe concentrations were equalized based on AP activity.
Non-permeabilized Live-Cell Immunostaining, Cell Surface Biotinylation Assay, and Immunoblot Analysis
For non-permeabilized live-cell immunostaining, COS-7 cells were transfected with pRK5 expression plasmids using FuGENE HD Transfection Reagent. The media was removed 48 hours post-transfection, and cells were washed 3 times with PBS. Cells were incubated with diluted primary antibodies (1:500) in serum-containing media at 4°C for 1 hour, and then washed 5–6 times with cold PBS, prior to fixation with 4% PFA/PBS. Coverslips were mounted using Fluoromount G and imaged using a Zeiss LSM700 confocal microscope. Images were processed with ImageJ, Adobe Photoshop, and Adobe Illustrator software.
Cell surface biotinylation was performed as described by Pavel et al. (2014). In short, COS-7 cells were transfected with pRK5 expression plasmids with FuGENE HD Transfection Reagent. The media was removed 48 hours post-transfection, and cells were washed 3 times with PBS. Cells were incubated in PBS containing Sulfo-NHS-Biotin (250 µg/ml) at 4°C for 30 minutes. Excess biotin was quenched by adding Tris-HCl pH 7.4 to a final concentration of 50 mM at 4°C for 5 minutes. After removing the Tris buffer, cells were detached, washed 3 times in 1× Tris buffered saline (TBS), and lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, and 0.5% deoxycholate) containing protease inhibitor (Roche 11836170001). Cell lysates were incubated at 4°C for 30 minutes and subsequently centrifuged at 10,000×g at 4°C for 20 minutes to remove cellular debris. Cell-surface proteins were captured by incubating cleared lysates with NeutrAvidin Agarose Resin (Thermo Fisher Scientific 29200) overnight at 4°C. Resin was washed 5 to 6 times with RIPA buffer, and captured proteins, along with input controls, were resolved by SDS-PAGE and transferred to PVDF membranes (EMD Millipore IPFL00010) for immunoblotting. Immunoblots were incubated with primary antibodies (1:10,000 mouse anti-HA and rabbit anti-6×Myc; 1:2000 rabbit anti-Reck) diluted in Odyssey Blocking Buffer (LiCor 927–40000) overnight at 4°C. Membranes were washed with 1× PBS-T (1× PBS + 0.1% Tween 20), and incubated with LiCor secondary antibodies (1:10,000) diluted in Odyssey Blocking Buffer for 1 hour at room temperature. Membranes were washed at least 3 times with 1× PBS-T and developed using the Odyssey Fc Imaging System (LiCor).
For whole tissue lysates, E13.5 embryos were harvested and homogenized using a plastic pestle in RIPA buffer supplemented with protease inhibitors. Tissue lysates were incubated at 4°C for 1 hour, cleared by centrifugation, and prepared for immunoblot analysis, as described above.
Quantification and Statistical Analysis
For quantifying relative vascular density, 150–180 µm thick coronal sections of embryonic brain or cross sections of embryonic spinal cord were stained with GS Lectin, anti-PECAM, and anti-Glut1. Confocal images were scanned at 10–12 µm intervals along the Z-axis, of which 8–10 images were Z-stacked. The following areas of the Z-stacked images were analyzed: cortex (1000 × 600 µm), MGE (500 × 500 µm), LGE (300 × 300 µm), hindbrain (300 × 500 µm), and spinal cord (350 × 500 µm). All of the blood vessels within the designated volume were manually traced using a Wacom tablet and Adobe Illustrator software. The lengths of the traced vessels were quantified by calculating pixel coverage as a fraction of the total area using ImageJ. For quantifying GS Lectin+ macrophages, individual GS Lectin+ cells that did not co-localize with a vascular marker (i.e. anti-PECAM and anti-Glut1) were manually counted in each of the designated volumes. Quantification of the fraction of macrophages that immunostained positive for F4/80, Cd11b, and/or GS Lectin, was conducted as follows: non-ECs (cells that did not co-localize with anti-Glut1) were manually counted for each of the different markers and divided by the total number of non-ECs that immunostained positive for F4/80 and/or GS Lectin or Cd11b and/or GS Lectin.
For quantifying GFP+ nuclei in the Rosa26 Tcf/Lef-lsl-H2B-GFP Wnt reporter mouse, 150–180 µm thick coronal sections of embryonic brain or cross sections of embryonic spinal cord were stained with anti-GFP and anti-PECAM. Quantification followed the procedure outlined above, with the following areas cropped: cortex (1000 × 600 µm) and spinal cord (150 × 150 µm). GFP+ nuclei that co-localized with PECAM+ cells were manually counted in each of the designated volumes. Relative vascular density within that volume was also measured (as described above). To calculate the number of GFP+ EC nuclei normalized to vascular density, the absolute number of GFP+ EC nuclei was divided by the length of the vasculature within the designated volume.
For quantifying the fraction of vessels that immunostained positive for Claudin-5 or PLVAP, vessels in the designated volume of 1000 × 1000 × 100 µm were manually traced and pixel coverage of the traced image was determined as a fraction of the total designated volume (as described above) for each marker. The vascular density for Claudin-5+ and PLVAP+ vessels was then divided by the total vascular density in the designated volume.
GraphPad Prism 7 software was used to generate plots and to perform statistical analysis. The mean ± standard deviations are shown. Statistical significance was determined by the unpaired t-test, and is represented by * (P<0.05), **(P<0.01), *** (P<0.001), and **** (P<0.0001).
The analysis of alignment and conservation of Reck CC1 across vertebrates was generated using Clustal Omega (McWilliam et al., 2013).
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Makoto Noda (Kyoto University) for the Reckflex2 mice, Dr. Chiaki Takahashi (Kanazawa University) and Dr. Shan Sockanathan (Johns Hopkins University) for the ReckΔex1 mice, Dr. Thomas Spencer (University of Missouri) for the Wnt7afl mice, and Dr. Maketo Taketo (Kyoto University) for the Ctnnb1flex3 mice, the Johns Hopkins University School of Medicine Transgenic Core Laboratory for blastocyst and egg injections, and Mark Sabbagh for helpful comments on the manuscript. This study was supported by the Howard Hughes Medical Institute, the National Eye Institute (R01 EY018637), and the Arnold and Mabel Beckman Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
C.C. and J.N. designed experiments, analyzed data, and wrote the manuscript. P.M.S. constructed many of the plasmids and the Wnt reporter mice. C.C. conducted the experiments.
References
- Anderson KD, Pan L, Yang XM, Hughes VC, Walls JR, Dominguez MG, Simmons MV, Burfeind P, Xue YZ, Wei Y, et al. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc. Natl. Acad. Sci. USA. 2011;108:2807–2812. doi: 10.1073/pnas.1019761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, Gu C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509:507–511. doi: 10.1038/nature13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandana EP, Maeda Y, Ueda A, Kiyonari H, Oshima N, Yamamoto M, Kondo S, Oh J, Takahashi R, Yoshida Y, et al. Involvement of the Reck tumor suppressor protein in maternal and embryonic vascular remodeling in mice. BMC Dev. Biol. 2010;10:84. doi: 10.1186/1471-213X-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CK, Hung WC, Chang HC. The Kazal motifs of RECK protein inhibit MMP-9 secretion and activity and reduce metastasis of lung cancer cells in vitro and in vivo. J. Cell Mol. Med. 2008;12:2781–2789. doi: 10.1111/j.1582-4934.2008.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Mancuso M, Maier C, Liang X, Yuki K, Yang L, Kwong JW, Wang J, Rao V, Vallon M, et al. Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat. Med. 2017;23:450–460. doi: 10.1038/nm.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TH, Hsieh FL, Zebisch M, Harlos K, Elegheert J, Jones EY. Structure and functional properties of Norrin mimic Wnt for signalling with Frizzled4, Lrp5/6, and proteoglycan. Elife. 2015;4:06554. doi: 10.7554/eLife.06554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Yan H, Ren DN, Yin Y, Li Z, He Q, Wo D, Ho MS, Chen Y, Liu Z, et al. LRP6 dimerization through its LDLR domain is required for robust canonical Wnt pathway activation. Cell. Signal. 2014;26:1068–1074. doi: 10.1016/j.cellsig.2013.12.020. [DOI] [PubMed] [Google Scholar]
- Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala-Dilke K, Fruttiger M. Efficient, inducible Cre-recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. doi: 10.1002/dvg.20367. [DOI] [PubMed] [Google Scholar]
- Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, Yang MY, Li XJ, Chaudhary A, Xu LH, Hilton MB, et al. GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc. Natl. Acad. Sci. USA. 2011;108:5759–5764. doi: 10.1073/pnas.1017192108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ, Kumar S, Yamaguchi TP, Duester G. Wnt8a and Wnt3a cooperate in the axial stem cell niche to promote mammalian body axis extension. Dev. Dyn. 2015;244:797–807. doi: 10.1002/dvdy.24275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA. 2009;106:6422–6422. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015;7:a020412. doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida GM, Yamamoto M, Morioka Y, Ogawa S, Matsuzaki T, Noda M. Critical roles for murine Reck in the regulation of vascular patterning and stabilization. Sci. Rep. 2015;5:17860. doi: 10.1038/srep17860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijksterhuis JP, Baljinnyam B, Stanger K, Sercan HO, Ji Y, Andres O, Rubin JS, Hannoush RN, Schulte G. Systematic mapping of WNT-FZD protein interactions reveals functional selectivity by distinct WNT-FZD pairs. J. Biol. Chem. 2015;290:6789–6798. doi: 10.1074/jbc.M114.612648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap KA, Filant J, Hayashi K, Rucker EB, 3rd, Song G, Deng JM, Behringer RR, DeMayo FJ, Lydon J, Jeong JW, et al. Postnatal deletion of Wnt7a inhibits uterine gland morphogenesis and compromises adult fertility in mice. Biol. Reprod. 2011;85:386–396. doi: 10.1095/biolreprod.111.091769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer-Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, Hadjantonakis AK. A sensitive and bright single-cell resolution live imaging reporter of Wnt/β-catenin signaling in the mouse. BMC Dev. Biol. 2010;10:121. doi: 10.1186/1471-213X-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh JC, Rattner A, Smallwood PM, Nathans J. Biochemical characterization of Wnt-frizzled interactions using a soluble, biologically active vertebrate Wnt protein. Proc. Natl. Acad. Sci. USA. 1999;96:3546–3551. doi: 10.1073/pnas.96.7.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural Basis of Wnt Recognition by Frizzled. Science. 2012;337:59–64. doi: 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, Costa M, Rice DS, Ye WL. TSPAN12 Regulates Retinal Vascular Development by Promoting Norrin- but Not Wnt-Induced FZD4/beta-Catenin Signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- Kuhnert F, Mancuso MR, Shamloo A, Wang HT, Choksi V, Florek M, Su H, Fruttiger M, Young WL, Heilshorn SC, et al. Essential Regulation of CNS Angiogenesis by the Orphan G Protein-Coupled Receptor GPR124. Science. 2010;330:985–989. doi: 10.1126/science.1196554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Bafico A, Harris VK, Aaronson SA. A novel mechanism for Wnt activation of canonical signaling through the LRP6 receptor. Mol. Cell. Biol. 2003;23:5825–5835. doi: 10.1128/MCB.23.16.5825-5835.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie D, Arendt A, Hargrave P, McDowell JH, Molday RS. Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry. 1984;23:6544–6549. doi: 10.1021/bi00321a041. [DOI] [PubMed] [Google Scholar]
- Malashkevich VN, Kammerer RA, Efimov VP, Schulthess T, Engel J. The crystal structure of a five-stranded coiled coil in COMP: a prototype ion channel? Science. 1996;274:761–765. doi: 10.1126/science.274.5288.761. [DOI] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulroy T, McMahon JA, Burakoff SJ, McMahon AP, Sen J. Wnt-1 and Wnt-4 regulate thymic cellularity. Eur. J. Immunol. 2002;32:967–971. doi: 10.1002/1521-4141(200204)32:4<967::AID-IMMU967>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Muraguchi T, Takegami Y, Ohtsuka T, Kitajima S, Chandana EP, Omura A, Miki T, Takahashi R, Matsumoto N, Ludwig A, et al. RECK modulates Notch signaling during cortical neurogenesis by regulating ADAM10 activity. Nat. Neurosci. 2007;10:838–845. doi: 10.1038/nn1922. [DOI] [PubMed] [Google Scholar]
- Nikopoulos K, Venselaar H, Collin RWJ, Riveiro-Alvarez R, Boonstra FN, Hooymans JMM, Mukhopadhyay A, Shears D, van Bers M, de Wijs IJ, et al. Overview of the Mutation Spectrum in Familial Exudative Vitreoretinopathy and Norrie Disease with Identification of 21 Novel Variants in FZD4, LRP5, and NDP. Hum. Mutat. 2010;31:656–666. doi: 10.1002/humu.21250. [DOI] [PubMed] [Google Scholar]
- Noda M, Kitayama H, Matsuzaki T, Sugimoto Y, Okayama H, Bassin RH, Ikawa Y. Detection of genes with a potential for suppressing the transformed phenotype associated with activated ras genes. Proc. Natl. Acad. Sci. USA. 1989;86:162–126. doi: 10.1073/pnas.86.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- Okumura A, Arai E, Kitamura Y, Abe S, Ikeno M, Fujimaki T, Yamamoto T, Shimizu T. Epilepsy phenotypes in siblings with Norrie disease. Brain Dev. 2015;37:978–982. doi: 10.1016/j.braindev.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Omura A, Matsuzaki T, Mio K, Ogura T, Yamamoto M, Fujita A, Okawa K, Kitayama H, Takahashi C, Sato C, et al. RECK forms cowbell-shaped dimers and inhibits matrix metalloproteinase-catalyzed cleavage of fibronectin. J. Biol. Chem. 2009;284:3461–3469. doi: 10.1074/jbc.M806212200. [DOI] [PubMed] [Google Scholar]
- Park S, Lee C, Sabharwal P, Zhang M, Meyers CL, Sockanathan S. GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science. 2013;339:324–328. doi: 10.1126/science.1231921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavel MA, Lam C, Kashyap P, Salehi-Najafabadi Z, Singh G, Yu Y. Analysis of the cell surface expression of cytokine receptors using the surface protein biotinylation method. Methods Mol. Biol. 2014;1172:185–192. doi: 10.1007/978-1-4939-0928-5_16. [DOI] [PubMed] [Google Scholar]
- Pelletier S, Gingras S, Green DR. Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity. 2015;42:18–27. doi: 10.1016/j.immuni.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posokhova E, Shukla A, Seaman S, Volate S, Hilton MB, Wu B, Morris H, Swing DA, Zhou M, Zudaire E, et al. GPR124 functions as a WNT7-specific coactivator of canonical β-catenin signaling. Cell Rep. 2015;10:123–130. doi: 10.1016/j.celrep.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast A, Linbo TH, Swarts T, Ungos JM, McGraw HF, Krispin S, Weinstein BM, Raible DW. The metalloproteinase inhibitor Reck is essential for zebrafish DRG development. Development. 2012;139:1141–1152. doi: 10.1242/dev.072439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal J, Carroll TJ, Guseh JS, Bores SA, Blank LJ, Anderson WJ, Yu J, Zhou Q, McMahon AP, Melton DA. Wnt7b stimulates embryonic lung growth by coordinately increasing the replication of epithelium and mesenchyme. Development. 2008;135:1625–1634. doi: 10.1242/dev.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner A, Yu H, Williams J, Smallwood PM, Nathans J. Endothelin-2 signaling in the neural retina promotes the endothelial tip cell state and inhibits angiogenesis. Proc. Natl. Acad. Sci. USA. 2013;110:E3830–3839. doi: 10.1073/pnas.1315509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen G, Ke J, Wang Z, Cheng Z, Gu X, Wei Y, Melcher K, Xu HE, Xu W. Structural basis of the Norrin-Frizzled 4 interaction. Cell Res. 2015;25:1078–1081. doi: 10.1038/cr.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood PM, Williams J, Xu Q, Leahy DJ, Nathans J. Mutational analysis of Norrin-Frizzled4 recognition. J. Biol. Chem. 2007;282:4057–4068. doi: 10.1074/jbc.M609618200. [DOI] [PubMed] [Google Scholar]
- Smith SE, Mullen TE, Graham D, Sims KB, Rehm HL. Norrie disease: extraocular clinical manifestations in 56 patients. Am. J. Med. Genet. A. 2012;158A:1909–1917. doi: 10.1002/ajmg.a.35469. [DOI] [PubMed] [Google Scholar]
- Stan RV, Tkachenko E, Niesman IR. PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol. Biol. Cell. 2004;15:3615–3630. doi: 10.1091/mbc.E03-08-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B, et al. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202. doi: 10.1126/science.289.5482.1197. [DOI] [PubMed] [Google Scholar]
- Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt Signaling Regulates Organ-Specific Assembly and Differentiation of CNS Vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- Takahashi C, Sheng Z, Horan TP, Kitayama H, Maki M, Hitomi K, Kitaura Y, Takai S, Sasahara RM, Horimoto A, et al. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc. Natl. Acad. Sci. USA. 1998;95:13221–13226. doi: 10.1073/pnas.95.22.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich F, Carretero-Ortega J, Menéndez J, Narvaez C, Sun B, Lancaster E, Pershad V, Trzaska S, Véliz E, Kamei M, et al. Reck enables cerebrovascular development by promoting canonical Wnt signaling. Development. 2016;143:147–159. doi: 10.1242/dev.123059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhollebeke B, Stone OA, Bostaille N, Cho C, Zhou Y, Maquet E, Gauquier A, Cabochette P, Fukuhara S, Mochizuki N, et al. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. Elife. 2015;4:e06489. doi: 10.7554/eLife.06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YS, Rattner A, Zhou YL, Williams J, Smallwood PM, Nathans J. Norrin/Frizzled4 Signaling in Retinal Vascular Development and Blood Brain Barrier Plasticity. Cell. 2012;151:1332–1344. doi: 10.1016/j.cell.2012.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, et al. Vascular development in the retina and inner ear: Control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- Ye X, Wang YS, Cahill H, Yu MZ, Badea TC, Smallwood PM, Peachey NS, Nathans J. Norrin, Frizzled-4, and Lrp5 Signaling in Endothelial Cells Controls a Genetic Program for Retinal Vascularization. Cell. 2009;139:285–298. doi: 10.1016/j.cell.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Wang YS, Nathans J. The Norrin/Frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol. Med. 2010;16:417–425. doi: 10.1016/j.molmed.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Smallwood PM, Wang Y, Vidaltamayo R, Reed R, Nathans J. Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development. 2010;137:3707–3717. doi: 10.1242/dev.052001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Ye X, Guo N, Nathans J. Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: evidence for a network of interacting genes. Development. 2012;139:4383–4394. doi: 10.1242/dev.083352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and Dysfunction of the Blood-Brain Barrier. Cell. 2015;163:1064–1078. doi: 10.1016/j.cell.2015.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Nathans J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical Wnt signaling. Dev. Cell. 2014;31:248–256. doi: 10.1016/j.devcel.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Tischfield M, Williams J, Smallwood PM, Rattner A, Taketo MM, Nathans J. Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Invest. 2014;124:3825–3846. doi: 10.1172/JCI76431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.