

Abstract
Background
Angiogenesis has been a major target of novel drug development in hepatocellular carcinoma (HCC). It is hypothesized that the combination of two antiangiogenic agents, sorafenib and bevacizumab, will provide greater blockade of angiogenesis.
Objective
To determine the optimal dose, safety, and effectiveness of dual anti-angiogenic therapy with sorafenib and bevacizumab in patients with advanced HCC.
Patients and Methods
Patients with locally advanced or metastatic HCC not amenable for surgery or liver transplant were eligible. The phase I starting dose level was bevacizumab 1.25 mg/kg day 1 and 15 plus sorafenib 400 mg twice daily (BID) days 1–28. In the phase II portion, patients were randomized to receive bevacizumab and sorafenib at the maximum tolerated dose (MTD) or sorafenib 400 mg BID.
Results
17 patients were enrolled in the phase I component. Dose-limiting toxicities included grade 3 hand/foot skin reaction, fatigue, hypertension, alanine/aspartate aminotransferase increase, dehydration, hypophosphatemia, creatinine increase, hypoglycemia, nausea/vomiting, and grade 4 hyponatremia. 7 patients were enrolled onto the phase II component at the MTD: sorafenib 200 mg BID days 1–28 and bevacizumab 2.5 mg/kg every other week. 57% (4/7) had grade 3 AEs at least possibly related to treatment,. No responses were observed in the phase II portion. Estimated median time to progression and survival were 8.6 months (95% CI: 0.4–16.3) and 13.3 months (95% CI 4.4 – not estimable), respectively.
Conclusions
The MTD of the combination is sorafenib 200 mg twice daily on days 1–28 plus bevacizumab 2.5 mg/kg on days 1 and 15 of a 28-day cycle. In the phase II portion of the trial, concerns regarding excessive toxicity, low efficacy, and slow enrollment led to discontinuation of the trial. (Clinical Trials ID: NCT00867321.)
1. INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth leading cause of cancer and third leading cause of cancer-related deaths worldwide [1]. HCC accounts for approximately 30,000 new cancer diagnoses each year in the United States, a number that is steadily increasing due to virus-associated etiologies. Fewer than 20% of patients presenting with HCC are candidates for potentially curative treatment such as surgical resection, liver transplantation, or radiofrequency ablation [2]. Once HCC becomes advanced, there are few systemic therapy options for management.
HCC is a highly vascular malignancy, and the overexpression of vascular endothelial growth factor (VEGF) is felt to play a critical role in the development and progression of HCC [3, 4]. Sorafenib is an oral multi-kinase inhibitor, which targets the VEGF-receptor 2 (R2) intracellularly, as well as kinases downstream the VEGF signaling pathway including Raf [5]. A large randomized phase III trial demonstrated an overall survival (OS) of 10.7 months in the sorafenib arm compared to 7.9 months for placebo (p<0.001) [6], which led to regulatory approval for use in unresectable HCC at a dose of 400 mg twice daily.
Bevacizumab is a monoclonal antibody that targets VEGF by binding to the VEGF ligand thereby preventing its interaction with the VEGF-R2 and blocking VEGF-induced angiogenesis [7]. Results from a phase II trial of single agent bevacizumab for patients with unresectable HCC showed promising activity with median progression-free survival (PFS) time of 6.9 months (95% CI, 6.5 to 9.1 months) and a one-year OS rate of 53% [8].
The combination of bevacizumab and sorafenib could result in enhanced inhibition of VEGF-mediated angiogenesis by more broad inhibition of VEGF signaling and targeting downstream intracellular processes such as the RAF/RAS/MEK pathway. A previous phase I study in advanced solid tumors found the maximum-tolerated dose (MTD) of the combination was sorafenib 200 mg twice daily and bevacizumab 5 mg/kg every 2 weeks. Of the 37 assessable patients, seven had a partial response (PR), and PR or disease stabilization for ≥ 4 months was seen in 22 (59%) of 37 assessable patients [9].
A phase I/II clinical trial was conducted through the North Central Cancer Treatment Group (NCCTG) to evaluate the safety and efficacy of sorafenib and bevacizumab in HCC.
2. PATIENTS AND METHODS
2.1 Patient Selection
Patients ≥ 18 years of age with locally advanced or metastatic HCC not candidates for surgery or liver transplant were eligible for enrollment. Inclusion criteria for the trial included: no prior systemic chemotherapy or external beam radiation therapy for HCC, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, measurable disease, total bilirubin ≤ 1.5 × upper limit of normal (ULN), AST ≤ 5 × ULN, alkaline phosphatase ≤ 5 × ULN, and adequate bone marrow and renal function. Patients with Child Pugh A or B7 liver disease were allowed on the study. Patients must have had ≥ 3 months life expectancy to participate in the trial.
Patients who had chemoembolization, radioembolization, radiofrequency ablation (RFA), or other local ablative therapies < 6 weeks prior to registration were excluded. Patients who had treatment of known or clinically suspected esophageal varices ≤6 months prior to registration were not eligible. Patients with bleeding diathesis or on chronic anticoagulation were ineligible. Patients with brain metastases, uncontrolled hypertension (defined as systolic blood pressure > 150 mmHg or diastolic blood pressure > 100 mmHg), class III or IV congestive heart failure, ventricular arrhythmia or arterial thrombotic event in the previous 6 months, non-healing wound, major surgery or trauma within 4 weeks, and any other illness which the investigator felt would make the patient inappropriate for study entry were excluded.
This study was approved by the Mayo Clinic Institutional Review Board (IRB) and by the IRBs of the individual memberships of the North Central Cancer Treatment Group (NCCTG) that participated in the study. Each participant signed an IRB-approved, protocol-specific informed consent in accordance with federal and institutional guidelines. Clinical Trials ID: NCT00867321.
2.2 Treatment
The goal of the phase I component was to determine the MTD for treating this patient population with sorafenib plus bevacizumab. The phase I portion of this trial was a standard 3 + 3 dose escalation/de-escalation design with the doses listed in Table 1. Sorafenib was administered twice daily (continuously per dose schedule) and bevacizumab was administered every 2 weeks on a 28-day cycle. Once the MTD was established, patients were randomly assigned using a Dynamic Allocation Procedure [10] to receive either the MTD of the dual agent combination or sorafenib monotherapy at 400 mg BID, days 1–28. Overall, patients were treated until they refused treatment or experienced progressive disease (PD) or unacceptable adverse events.
Table 1.
Treatment Administration, by Study Component
| Phase I Component | ||||
| Dose Level | Sorafenib (twice daily) | Bevacizumab (every 2 weeks) | ||
| +2 | 400 mg BID | 5 mg/kg | ||
| +1 | 400 mg BID | 2.5 mg/kg | ||
| 0* | 400 mg BID | 1.25 mg/kg | ||
| −1 | 400 mg BID, 5 consecutive days out of each 7 days | 1.25 mg/kg | ||
| (−2a) | 200 mg BID days 1–28 | 2.5 mg/kg | ||
| −2 | 200 mg BID days 1–28 | 1.25 mg/kg | ||
| (−3b) | 200 mg BID, days 1–5 out of each 7 days | 5 mg/kg | ||
| (−3a) | 200 mg BID, days 1–5 out of each 7 days | 2.5 mg/kg | ||
| −3 | 200 mg BID, days 1–5 out of each 7 days | 1.25 mg/kg | ||
| Phase II Component | ||||
| Agent | Dose Level | Route | Day | ReRx |
| Arm A | ||||
| Bevacizumab | 2.5 mg/kg | IV* | Day 1, 15(+/− 3 days) | Every 4 weeks |
| Sorafenib | 200 mg BID | PO | Days 1–28 | |
| Arm B | ||||
| Sorafenib | 400 mg BID | PO | Days 1–28 | Every 4 weeks |
Starting dose level
2.3 Patient Evaluation
Patients were evaluated every cycle for treatment adherence and adverse events. In the phase I portion, dose limiting toxicity (DLT) was defined as an adverse event (AE) ≥ grade 3 attributed (definitely, probably or possibly) to the study treatment based on NCI CTCAE v3.0 criteria in the first 28-day cycle. The maximum tolerated dose (MTD) was defined as the dose level below the lowest dose that induces DLT in at least one-third of patients. If dose level (−1) was not tolerable, but dose (−3) or (−2) was below or at MTD, testing of alternate dose levels (−2a, −3a, −3b) were to occur as outlined in the table. These alternate dose levels would only be tested if full-dose sorafenib could not be combined with any dose of bevacizumab and the combination would require a sorafenib dose reduction to 200 mg twice daily or lower (dose level −2 or −3).
The phase I portion of this study was monitored by the rules specified for identifying DLTs. In the phase II portion, the Mayo Clinic Cancer Center (MCCC) Data and Safety Monitoring Board (DSMB) reviewed accrual and safety data twice a year.
2.4 Disease Assessment
All eligible patients who signed the consent form and began treatment were evaluable for response. Radiologic studies were performed at baseline, after four weeks, and then every two cycles (8 weeks) of therapy to assess tumor response using the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 [11]. After completion of study treatment, patients were followed every 3 months until first documentation of progression and every 6 months after progression until death or for 2 years.
2.5 Statistical Considerations
The primary endpoint of the phase I component is the determination of the MTD (defined earlier), using the standard ‘3+3’ phase I study design. Patients were evaluable for MTD determination during the first cycle of treatment. Patients enrolled at the MTD were considered evaluable for the assessing efficacy in the phase II portion of the study. The primary goal of the phase II component of this study was to compare time to progression (TTP) in the experimental arm (bevacizumab/sorafenib) to the control arm (sorafenib alone), where the alternative hypothesis was that the estimated hazard ratio (control:experimental) was at least 1.8. We designed this study using a non-definitive (alpha=0.15, power=0.85), “screening” comparison of the dual-agent arm (bevacizumab/sorafenib) against monotherapy with sorafenib as described by Rubenstein et al [12]. After determining the appropriate dose levels in the phase I component, we intended to enroll up to 76 patients (70 with an additional 6 allowed to account for ineligible patients) using a 1:1 randomization scheme with patients stratified by gender, ECOG PS (0 vs 1), and Child-Pugh class (A vs B7). At the time all patients were followed for at least 6 months, the p-value associated with a 1-sided log-rank test between the two Kaplan-Meier curves [13] was calculated and compared to alpha=0.15. A p-value less than 0.15 was an indication that adding bevacizumab to sorafenib improved TTP.
Secondary endpoints across the components of the study included assessing adverse events, TTP, progression-free survival (PFS), and overall survival (OS). Summary statistics and categorical data methods were used to summarize the distributions of patient characteristics, treatment dosing, and adverse events. Adverse events were summarized as the maximum severity of each type of event, per patient. Kaplan-Meyer methodology was used to estimate the distributions of TTP, PFS, and OS. The data analysis for this paper was generated using SAS/STAT software, Version 9.3 of the SAS System for Linux. (© 2002–2008 SAS Institute Inc, Cary, NC, USA). All analyses were based on the study database frozen on May 5, 2014. Data collection and statistical analyses were conducted by the Alliance Statistics and Data Center.
3. RESULTS
3.1 PHASE I COMPONENT
3.11 Patient Accrual
From April 2009 to September 2010, a total of 17 patients were enrolled. Patient characteristics are shown in Table 2. The median age was 66 years (range 18 – 79). Two patients (12%) had prior radiation for bone metastasis, 1 patient (6%) had prior chemoembolization, 3 patients (18%) had vascular invasion, and 12 patients (71%) had ascites. Eight patients (47.1%) entered with intrahepatic (vs. extrahepatic) disease. Only 41% of patients had cirrhosis, which may be due to the relatively low incidence of viral hepatitis at the centers that participated in the trial.
Table 2.
Patient Characteristics, by Study Component
| Phase I | Phase II | |||
|---|---|---|---|---|
|
|
|
|||
| Sorafenib + Bevacizumab |
Sorafenib + Bevacizumab |
Sorafenib | Total | |
| Age | ||||
| Median (Range in years) | 66 (18–79) | 59 (54–80) | 55 (52–61) | 57 (52–80) |
| Gender | ||||
| Female | 3 (17.6%) | 1 (25.0%) | 1 (33.3%) | 2 (28.6%) |
| Male | 14 (82.4%) | 3 (75.0%) | 2 (66.7%) | 5 (71.4%) |
| Race | ||||
| White | 13 (76.5%) | 4 (100.0%) | 3 (100.0%) | 7 (100.0%) |
| Black | 2 (11.8%) | |||
| Asian | 2 (11.8%) | |||
| ECOG Performance Score | ||||
| 0 | 3 (75.0%) | 2 (66.7%) | 5 (71.4%) | |
| 1 | 1 (25.0%) | 1 (33.3%) | 2 (28.6%) | |
| History of Cirrhosis | ||||
| Yes | 7 (41.2%) | 2 (50.0%) | 2 (66.7%) | 4 (57.1%) |
| No | 10 (58.8%) | 2 (50.0%) | 1 (33.3%) | 3 (42.9%) |
| Current Disease Status | ||||
| Intrahepatic | 8 (47.1%) | 2 (50.0%) | 2 (66.7%) | 4 (57.1%) |
| Extrahepatic | 9 (52.9%) | 2 (50.0%) | 1 (33.3%) | 3 (42.9%) |
| Vascular Invasion | ||||
| Yes | 3 (20.0%) | |||
| No | 12 (80.0%) | 4 (100.0%) | 3 (100.0%) | 7 (100.0%) |
| Missing | 2 | |||
| Differentiation | ||||
| Well | 2 (11.8%) | |||
| Moderate | 11 (64.7%) | 3 (75.0%) | 2 (66.7%) | 5 (71.4%) |
| Poor | 3 (17.6%) | 1 (25.0%) | 1 (33.3%) | 2 (28.6%) |
| Undifferentiated, anaplastic | 1 (5.9%) | |||
| Tumor Status | ||||
| Recurrent | 1 (25.0%) | 1 (14.3%) | ||
| Unresected | 17 (100.0%) | 3 (75.0%) | 3 (100.0%) | 6 (85.7%) |
3.12 Determination of Phase II Starting Dose Level
At dose level 0, two patients experienced DLTs including hand/foot skin reaction, fatigue, and hypertension. At dose level −1, one of three patients experienced a DLT (grade 3 events of alanine and aspartate aminotransferase increase, dehydration, and hypophosphatemia, and grade 4 hyponatremia). Two of the 3 subsequent patients enrolled at dose level −1 experienced DLTs: one patient experienced grade 3 creatinine increase and hypoglycemia, as well as grade 4 acidosis and syncope, and subsequently died (grade 5 sudden death) less than a month into treatment; another patient experienced grade 3 nausea and vomiting. At dose level −2 one patient had a treatment violation and was replaced. There were no DLT’s experienced by the 3 evaluable patients on dose level −2. The next cohort of patients was enrolled to dose level −2a, and there were no DLTs in this cohort. Because dose level −1 was already deemed too toxic, further dose escalation was halted, and the MTD and recommended phase II dose was determined to be sorafenib 200 mg twice daily and bevacizumab 2.5 mg/kg every other week.
Overall, 17 patients received a total of 76 cycles of sorafenib (median 3 cycles, range: 0–19) and a total of 66 cycles of bevacizumab (median 3 cycles, range: 1–19). All patients have discontinued study treatment for reasons including disease progression (12), adverse events (4), and refusal (1).
3.13 Adverse Event Summary, Across Cohorts
All 17 patients are evaluable for toxicity. The frequency and severity of frade 3 and above adverse events at least possibly related to treatment appear in Table 3. Overall, 14 patients (82.4%) patients experienced grade 3+ adverse events at least possibly related to treatment, and 3 patients (17.6%) patients experienced grade 4+ adverse events at least possibly related to treatment. There was one grade 5 sudden death possibly related to treatment. Five patients have experienced grade 4 adverse events: Thrombosis (not related), hyponatremia (possibly related), acidosis (possibly related), hyperkalemia (possibly related), muscle weakness (unlikely related), hyperglycemia (unlikely related), syncope (possibly related), and dehydration (unlikely related).
Table 3.
Maximum Adverse Event1, by Study Component
| Body System | Toxicity | Arm | Grade 3 | Grade 4 | Grade 5 | |||
|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | |||
| PHASE I COHORT | ||||||||
| Constitutional Symptoms | Fatigue | Sorafenib + Bevacizumab | 4 | 23.5 | 0 | 0.0 | 0 | 0.0 |
| Weight loss | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Dermatology/Skin | Hand-and-foot syndrome/reaction | Sorafenib + Bevacizumab | 3 | 17.6 | 0 | 0.0 | 0 | 0.0 |
| Hematology | Platelet count decreased | Sorafenib + Bevacizumab | 2 | 11.8 | 0 | 0.0 | 0 | 0.0 |
| Lymphocyte count decreased | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Leukocyte count decreased | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Hepatic | Alanine aminotransferase increased | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 |
| Aspartate aminotransferase increased | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Bilirubin increased | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Metabolic/Laboratory | Acidosis | Sorafenib + Bevacizumab | 0 | 0.0 | 1 | 5.9 | 0 | 0.0 |
| Serum glucose decreased | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Serum phosphate decreased | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Serum potassium increased | Sorafenib + Bevacizumab | 0 | 0.0 | 1 | 5.9 | 0 | 0.0 | |
| Serum sodium decreased | Sorafenib + Bevacizumab | 0 | 0.0 | 1 | 5.9 | 0 | 0.0 | |
| Neurology | Syncope | Sorafenib + Bevacizumab | 0 | 0.0 | 1 | 5.9 | 0 | 0.0 |
| Renal /Genitourinary | Creatinine increased | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 |
| Death | Sudden death | Sorafenib + Bevacizumab | 0 | 0.0 | 0 | 0.0 | 1 | 5.9 |
| Cardiovascular | Arrhythmia | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 |
| Hypertension | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Electrocardiogram QTc interval prolonged | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Gastrointestinal | Anorexia | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 |
| Dehydration | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Esophageal ulcer | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Nausea | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| Vomiting | Sorafenib + Bevacizumab | 1 | 5.9 | 0 | 0.0 | 0 | 0.0 | |
| PHASE II COHORT | ||||||||
| Dermatology/Skin | Hand-and-foot syndrome/reaction | Sorafenib + Bevacizumab | 1 | 25.0 | 0 | 0.0 | 0 | 0.0 |
| Rash desquamating | Sorafenib | 1 | 33.3 | 0 | 0.0 | 0 | 0.0 | |
| Hematology | Lymphocyte count decreased | Sorafenib | 1 | 33.3 | 0 | 0.0 | 0 | 0.0 |
| Hepatic | Alanine aminotransferase increased | Sorafenib + Bevacizumab | 1 | 25.0 | 0 | 0.0 | 0 | 0.0 |
| Aspartate aminotransferase increased | Sorafenib + Bevacizumab | 0 | 0.0 | 1 | 25.0 | 0 | 0.0 | |
| Bilirubin increased | Sorafenib | 1 | 33.3 | 0 | 0.0 | 0 | 0.0 | |
| Serum albumin decreased | Sorafenib | 1 | 33.3 | 0 | 0.0 | 0 | 0.0 | |
| Metabolic/Laboratory | Serum phosphate decreased | Sorafenib + Bevacizumab | 1 | 25.0 | 0 | 0.0 | 0 | 0.0 |
| Serum potassium decreased | Sorafenib + Bevacizumab | 1 | 25.0 | 0 | 0.0 | 0 | 0.0 | |
| Gastrointestinal | Dehydration | Sorafenib + Bevacizumab | 1 | 25.0 | 0 | 0.0 | 0 | 0.0 |
| Infection/Febrile Neutropenia | Urinary tract infection(gr 0/1/2 ANC) | Sorafenib + Bevacizumab | 1 | 25.0 | 0 | 0.0 | 0 | 0.0 |
| Lymphatics | Localized edema | Sorafenib | 1 | 33.3 | 0 | 0.0 | 0 | 0.0 |
| Musculoskeletal | Muscle weakness | Sorafenib + Bevacizumab | 1 | 25.0 | 0 | 0.0 | 0 | 0.0 |
AEs at least possibly related to study treatment. Events are by patient across all cycles of treatment.
3.14 Outcome Measures
Sixteen patients had at least one post-baseline assessment of disease. During treatment, there were two (12.5%) confirmed partial responses, 9 (56.3%) patients had stable disease, and 5 (31.3%) patients experienced progressive disease (Table 4). A 58-year-old-male experienced a PR after one cycle of treatment (Dose Level −2), lasting 6.1 months prior to the appearance of extrahepatic lesions. A 73-year-old male experienced a PR after nine cycles of treatment (Dose Level −2a), lasting 10 months until disease progression. Fourteen of the 16 patients progressed (5 - intrahepatic, 3-extrahepatic, and 3 - both intrahepatic and extrahepatic). One patient is alive with 18.2 months of follow-up. The estimated median time to progression (Table 4, Figures 2a–b) and survival were 3 months (95% CI: 1.7–7.2) and 9.2 months (95% CI: 3.7–11.9), respectively.
Table 4.
Patient Outcome, by Study Component
| Outcome | Estimate (% or 95% CI) |
|||
|---|---|---|---|---|
|
| ||||
| Phase I Cohort | Phase II Cohort | |||
|
|
|
|||
| Sorafenib + Bevacizumab |
Sorafenib + Bevacizumab |
Sorafenib | All Patients | |
| Best Objective Response | ||||
| Evaluablea | 16 | 4 | 2 | 6 |
| Not Evaluable | 1 | 0 | 1 | 1 |
| CR | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| PR | 2 (12.5%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| SD | 9 (56.3%) | 2 (50.0%) | 2 (100.0%) | 4 (66.7%) |
| PD | 5 (31.3%) | 0 (0.0%) | 0 (0.0%) | 2 (33.3%) |
| Confirmed CR/PR | 1 (6.3%) | 2 (50.0%) | 0 (0.0%) | 0 (0.0%) |
| Time to Progression | ||||
| Events | 14 | 4 | 2 | 6 |
| Medianb (95% CI) | 3.0 (1.7–7.2) | 2.1 (0.4–8.6) | 16.3 (11.4–16.3) | 8.6 (0.4–16.3) |
| % Progression-Free | ||||
| 2 mos | 69.7% (41.7–86.1) | 50.0% (5.8–84.5) | 100.0% | 71.4% (25.8–92.0) |
| 4 mos | 44.3% (20.2–66.1) | 25.0% (0.9–66.5) | 100.0% | 57.1% (17.2–83.7) |
| Survival | ||||
| Events | 16 | 4 | 1 | 5 |
| Medianb (95% CI) | 9.2 (3.7–11.9) | 10.4 (4.4–13.3) | NE (13.9-NE) | 13.3 (4.4-NE) |
| % Alive | ||||
| 6 mos | 64.7% (37.7–82.3) | 75.0% (12.8–96.1) | 100.0% | 85.7% (33.4–97.9) |
| 1 year | 23.5% (7.3–44.9) | 50.0% (5.8–84.5) | 100.0% | 71.4% (25.8–92.0) |
Number of patients having at least 1 post-baseline assessment
Kaplan-Meier methodology
Figure 2.
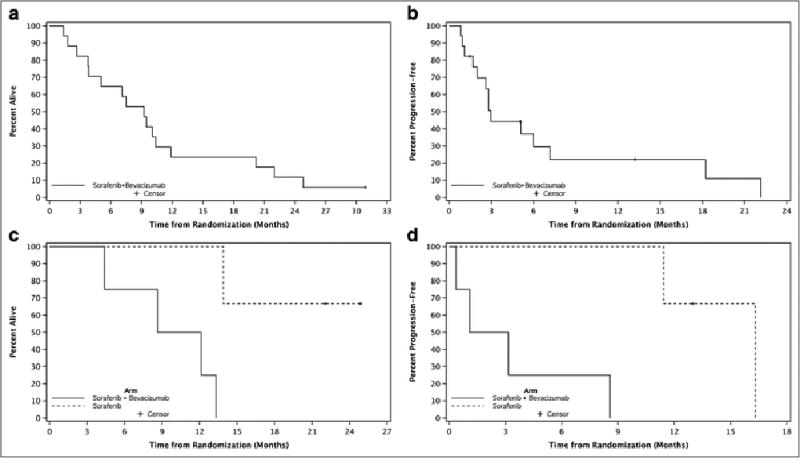
a–d: Time to Progression & Survival, by Study Component
3.2 PHASE II COMPONENT
3.21 Patient Accrual
From November 2010 to October 2011, a total of 7 (4 bevacizumab/sorafenib, 3 sorafenib) patients were accrued at a rate of less than 1 patient per month. The study was subsequently closed after a review of toxicity and lower than expected time to progression for patients on the combination arm. At study entry (Table 2), patients aged from 54 to 80 (median 58.5) in the bevacizumab/sorafenib arm and from 52 to 61 (median 55) in sorafenib arm. All patients were Caucasian; 5 were male. Four patients presented with intrahepatic (vs. extrahepatic) disease. Neither prior chemoembolization nor vascular invasion was reported in patients. Overall, 3 (28.6%) patients had single metastatic sites: liver metastasis (2, bevacizumab/sorafenib arm) and abdominal metastases (1, sorafenib arm).
3.22 Outcome Measures
Six of 7 patients (4 bevacizumab/sorafenib, 2 sorafenib alone) have had at least one post-baseline assessment of disease. One patient was unable to comply with the study and refused further treatment prior to obtaining a post-baseline assessment. No responses were observed (Table 4). Five of the 7 patients have had disease progression (2 - intrahepatic, 3 - extrahepatic). Two patients remain alive with a median of 23.5 months of follow-up (range 22–24.9). The estimated median time to progression and survival were 8.6 months (95% CI: 0.4 –16.3) and 13.3 months (95% CI 4.4 - NE) (Table 4, Figures 2c–d). At 1 year, survival was 71.4% (95% CI: 25.8– 92%). The small sample size precludes meaningful conclusions with respect to study endpoints for the phase II component.
3.23 Treatment Summary
Patients on the bevacizumab/sorafenib arm received 11 (median 2, range 1–6) cycles of treatment; patients receiving sorafenib monotherapy received 21 (median 8, range 1–12) cycles of treatment. Two cycles of treatment (1 bevacizumab/sorafenib, 1 sorafenib alone) were delayed for the following reasons: dermatitis/rash/skin condition, and for sorafenib shipping. Sorafenib dose reductions at the initiation of a cycle occurred 6 times in 3 patients (1 bevacizumab/sorafenib, 2 sorafenib alone); no reductions were reported for bevacizumab. Seven patients completed study treatment. Reasons for discontinuing treatment include: disease progression (4), adverse events (1), refusal (1), and for inability to comply with the treatment schedule (1).
3.24 Adverse Event Summary
Overall, 4 of 7 (57%) patients (2 bevacizumab/sorafenib, 2 sorafenib alone) experienced grade 3+ adverse events at least possibly related to treatment, and 1 patient receiving bevacizumab/sorafenib reported a grade 4+ adverse events at least possibly related to treatment (elevated aspartate aminotransferase). The frequency and severity of grade 3 and above adverse events at least possibly related to treatment appear in Table 3.
4. DISCUSSION
The MTD of bevacizumab combined with sorafenib in this patient population was sorafenib 200 mg twice daily and bevacizumab 2.5 mg/kg every other week. The dose of bevacizumab was lower than a prior phase I trial of this combination among patients with solid tumors reported by Azad et al [9] where the MTD was found to be sorafenib 200 mg twice daily plus bevacizumab 5 mg/kg every 2 weeks. Other patient populations have been able to tolerate the latter doses. For instance, the clinical trial N054C, which evaluated the combination of bevacizumab and sorafenib in patients with refractory metastatic colorectal cancer (mCRC), used the MTD from the aforementioned trial reported by Azad [14]. In the N054C trial, 68% of patients had a grade ≥3 toxicity at least possibly related to treatment, compared to 82.4% of patients in the phase I portion of this study. The reasons for the difference in dosing in our study are unclear, but possibly due to the poorer tolerability of cancer treatments in the HCC patient population. Liver dysfunction that coexists in patients with HCC may lead to different pharmacokinetics and drug deposition potentially increasing toxicity compared to cancer patients with normal liver function [15], However, since less than half of patients on the study had underlying cirrhosis, the poor tolerability of the regimen is not entirely explained by liver dysfunction.
For unclear reasons, attempts to the broaden the targets affecting the antiangiogenic pathway have not led to improved outcomes for patients with HCC [16]. Multiple randomized phase III trials involving kinase inhibitors have failed to demonstrate a survival benefit beyond sorafenib alone. Sunitinib, which targets VEGFR and PDGFR, was compared to sorafenib in the first-line treatment setting of HCC. Compared to sorafenib, patients in the sunitinib arm had inferior overall survival along with more frequent and severe toxicity [17]. Brivanib (targeting fibroblastic growth factor receptor [FGFR] and VEGFR) was not superior to sorafenib as first-line therapy for HCC patients, and those on the brivanib arm had poorer tolerability leading to a higher discontinuation rate (33% vs 43%) [18]. Linifanib, targeting VEGFR and PDGFR, was also found to be less effective compared to sorafenib in first-line setting for treatment of HCC [19].
Trials examining antiangiogenesis agents in the second-line setting have yielded mixed results. The use of brivanib failed to improve overall survival when compared to placebo in the second-line setting for HCC patients [20]. Ramucirumab, a monoclonal antibody against VEGF-R2, led to a 9.2 month overall survival versus 7.6 months for placebo as second-line therapy, but this did not reach statistical significance (p = 0.14).[21] Upon further analysis, it did appear that HCC patients with an alpha-fetoprotein (AFP) level > 400 ng/mL and Child-Pugh class 5 or 6 did have a significant survival benefit with ramurcirumab (HR, 0.61; 95% CI, 0.43–0.87; P = .01 and HR, 0.64; 95% CI, 0.42–0.98; P = .04 respectively. Finally, regorafenib, a multikinase inhibitor targeting VEGF-R2 and mechanisms of VEGF therapy resistance including TIE2, did show an overall survival benefit compared to placebo among patients with advanced HCC who were intolerant of or progressed on sorafenib. Patients in the regorafenib arm had on overall survival of 10.6 months compared to 7.8 months in the placebo arm (HR 0.62, 95% CI: 0.50–0.78; p<0.001).[22]
Investigators are now evaluating drugs with alternative targets involved in HCC development and progression. Tivantinib is a novel inhibitor of the MET receptor. In a randomized [23]phase II randomized trial in the second-line setting, tivantinib had an improved TTP compared to placebo, 1.6 versus 1.4 months (HR 0.64, p = 0.04) [24]. A subgroup of patients whose tumor tissue demonstrated high MET expression levels had a longer TTP (2.7 months), and based on that subgroup analysis, a randomized phase III trial of tivantinib versus placebo in second-line setting for MET overexpression tumors is underway (ClinicalTrials.gov identifier: NCT01755767). A phase I/II study in patients in unresectable HCC with or without prior therapy investigating the mTOR inhibitor everolimus led to a median PFS of 3.8 months and an OS of 8.4 months. Currently a phase III randomized trial of everolimus versus placebo second-line after failure of sorafenib is ongoing (ClinicalTrials.gov identifier: NCT01035229).
In conclusion, the combination of sorafenib plus bevacizumab was difficult to administer in patients with unresectable HCC, leading to an MTD using sub-standard doses of both sorafenib and bevacizumab. Concerns over excess toxicity and decreased efficacy as well as slow enrollment led to early discontinuation of the randomized phase II portion of the trial. Novel agents that broaden the spectrum of angiogenic targets have not led to improvements in survival above sorafenib alone leading to a new focus on systemic therapies targeting alternative pathways such as RET and MET. The development of well-tolerated, effective therapies in the setting of liver dysfunction remains a challenge in this patient population.
Figure 1.
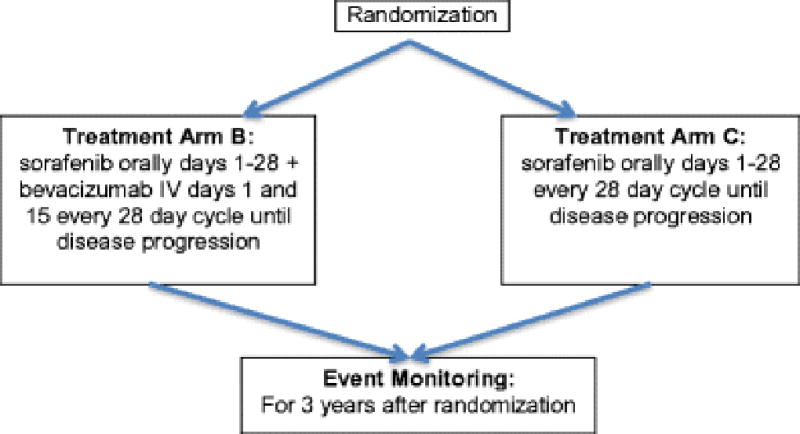
Consort diagram for the Phase II portion
KEY POINTS.
The maximum tolerable dose of the combination of sorafenib and bevacizumab in HCC patients (200 mg twice daily and 2.5 mg/kg every 2 weeks respectively) is lower than the standard doses of these medications.
Due to toxicity concerns and poor accrual, the phase II portion of the trial was unable to be completed.
Despite the highly vascular nature of HCC tumors, the combination of bevacizumab and sorafenib in HCC patients had disappointing results.
Acknowledgments
Funding: Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U10CA180821 and U10CA180882 to the Alliance for Clinical Trials in Oncology (Alliance) and the following funding to the legacy North Central Cancer Treatment Group (NCCTG): NCI Grant CA25224 and Bayer Pharmaceuticals and Bayer's REACH Program.
The following institutions participated in this study:
Geisinger Clinic & Medical Center CCOP, Danville, PA 17822 (Christian S. Adonizio, M.D.) - Grant #CA-35448
Grand Rapids Clinical Oncology Program, Grand Rapids, MI 49503 (Gilbert D.A. Padula, M.D.)
Mayo Clinic Arizona, Scottsdale, AZ 85259-5404 (William W. Wong, M.D.)
Mayo Clinic Rochester, Rochester, MN 55905 (Steven R. Alberts, M.D.)
Metro-Minnesota Community Clinical Oncology Program, St. Louis Park, MN 55416 (David M. Anderson) – Grant #CA-35267
Michigan Cancer Research Consortium, Ann Arbor, MI 48106 (Philip J. Stella, M.D.) - Grant #CA-63848
Montana Cancer Consortium, Billings, MT 59101 (Benjamin T. Marchello, M.D.)
Rapid City Regional Oncology Group, Rapid City, SD 59709 (Mark T. Schroeder, M.D.)
St. Vincent Regional Cancer Center CCOP, Green Bay, WI 54303 (Anthony J. Jaslowski, M.D.)
Wichita Community Clinical Oncology Program, Wichita, KS 67214-3882 (Shaker R. Dakhil, M.D.) - Grant #CA-35431
We thank BobbiAnn Jebens for administrative and technical support with the preparation and submission of this manuscript.
Footnotes
Compliance with Ethical Standards
The authors declare no conflict of interest.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–76. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Lin S, Hoffmann K, Schemmer P. Treatment of Hepatocellular Carcinoma: A Systematic Review. Liver Cancer. 2012;1(3–4):144–158. doi: 10.1159/000343828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poon RT, et al. Quantitative correlation of serum levels and tumor expression of vascular endothelial growth factor in patients with hepatocellular carcinoma. Cancer Res. 2003;63(12):3121–6. [PubMed] [Google Scholar]
- 4.Ng IO, et al. Microvessel density, vascular endothelial growth factor and its receptors Flt-1 and Flk-1/KDR in hepatocellular carcinoma. Am J Clin Pathol. 2001;116(6):838–45. doi: 10.1309/FXNL-QTN1-94FH-AB3A. [DOI] [PubMed] [Google Scholar]
- 5.Wilhelm SM, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Research. 2004;64(19):7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 6.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 7.Grothey A, Allegra C. Antiangiogenesis therapy in the treatment of metastatic colorectal cancer. Ther Adv Med Oncol. 2012;4(6):301–19. doi: 10.1177/1758834012454464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siegel AB, et al. Phase II trial evaluating the clinical and biologic effects of bevacizumab in unresectable hepatocellular carcinoma. J Clin Oncol. 2008;26(18):2992–8. doi: 10.1200/JCO.2007.15.9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azad NS, et al. Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. Journal of Clinical Oncology. 2008;26(22):3709–14. doi: 10.1200/JCO.2007.10.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pocock SJ, Simon R. Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics. 1975;31(1):103–15. [PubMed] [Google Scholar]
- 11.Therasse P, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 12.Rubinstein LV, et al. Design issues of randomized phase II trials and a proposal for phase II screening trials. J Clin Oncol. 2005;23(28):7199–206. doi: 10.1200/JCO.2005.01.149. [DOI] [PubMed] [Google Scholar]
- 13.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. Journal of the American Statistical Association. 1958;53(282):457–481. [Google Scholar]
- 14.Grothey A, et al. 2010 ASCO Annual Meeting. Chicago, IL: Dual VEGF inhibition with sorafenib and bevacizumab (BEV) as salvage therapy in metastatic colorectal cancer (mCRC): Results of the phase II North Central Cancer Treatment Group study N054C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venook AP. Key research issues in the management of hepatocellular carcinoma. Cancer Chemother Pharmacol. 2004;54(Suppl 1):S87–90. doi: 10.1007/s00280-004-0893-z. [DOI] [PubMed] [Google Scholar]
- 16.Welker MW, Trojan J. Antiangiogenic treatment in hepatocellular carcinoma: the balance of efficacy and safety. Cancer Manag Res. 2013;5:337–347. doi: 10.2147/CMAR.S35029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng AL, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol. 2013;31(32):4067–75. doi: 10.1200/JCO.2012.45.8372. [DOI] [PubMed] [Google Scholar]
- 18.Johnson PJ, et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: results from the randomized phase III BRISK-FL study. J Clin Oncol. 2013;31(28):3517–24. doi: 10.1200/JCO.2012.48.4410. [DOI] [PubMed] [Google Scholar]
- 19.Cainap C, et al. Phase III trial of linifanib versus sorafenib in patients with advanced hepatocellular carcinoma (HCC); 2013 Gastrointestinal Cancers Symposium; 2013. [Google Scholar]
- 20.Llovet JM, et al. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: results from the randomized phase III BRISK-PS study. J Clin Oncol. 2013;31(28):3509–16. doi: 10.1200/JCO.2012.47.3009. [DOI] [PubMed] [Google Scholar]
- 21.Zhu AX, et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015;16(7):859–70. doi: 10.1016/S1470-2045(15)00050-9. [DOI] [PubMed] [Google Scholar]
- 22.Bruix J, et al. Efficacy and safety of regorafenib versus placebo in patients with hepatocellular carcinoma (HCC) progressing on sorafenib: Results of the international, randomized phase 3 RESORCE trial. 2016 [Google Scholar]
- 23.Zhu AX, et al. Ramucirumab as Second-Line Treatment in Patients With Advanced Hepatocellular Carcinoma: Analysis of REACH Trial Results by Child-Pugh Score. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2016.4115. [DOI] [PubMed] [Google Scholar]
- 24.Santoro A, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14(1):55–63. doi: 10.1016/S1470-2045(12)70490-4. [DOI] [PubMed] [Google Scholar]