

Abstract
Objective
To report a rare association of central pontine myelinolysis (CPM) with hyperosmolar hyperglycaemic state (HHS).
Clinical Presentation and Intervention
A diabetic female presented with HHS and prolonged severe hypernatraemia. The metabolic derangement was adequately treated with proper correction of both hyperglycaemia and hypernatraemia. Lack of improvement in the presenting confusional state and the development of a fresh neurological deterioration led to the suspicion of CPM that was confirmed with magnetic resonance imaging. She fully recovered after 4 weeks with no specific medical treatment.
Conclusion
This case report showed that osmotic demyelination was linked to hypernatraemia and that CPM could result from severe hypernatraemia of HHS.
Key Words: Demyelination, Diabetes, Hyperglycaemia, Hyperosmolarity, Pontine myelinolysis
Introduction
Extreme fluctuations in serum sodium concentration and plasma osmolality may have detrimental effects on the central nervous system. Brain oedema could result from the rapid development of hyponatraemia or rapid correction of hypernatraemia [1,2]. On the other hand, the osmotic demyelination syndrome (ODS) mainly develops with the rapid correction of severe hyponatraemia [1]. There is some evidence that hypernatraemia may also lead to the ODS [3,4].
The clinical manifestations of ODS mainly include dysarthria, dysphagia, quadriparesis, behavioural disturbances, lethargy and coma [1]. Seizures are also mentioned with the syndrome, although less frequently [1]. The syndrome entails central pontine myelinolysis (CPM) and extrapontine myelinolysis, either alone or in combination [1]. Diagnosis can be confirmed by finding the demyelinating brain lesions in magnetic resonance images (MRI) or in computed tomography (CT) images, though with much less sensitivity [1]. We present a very rarely reported incident of CPM in association with hyperosmolar hyperglycaemic state (HHS).
Case Report
A 43-year-old woman with poorly controlled type 2 diabetes mellitus wpresented to our hospital in a confused state and fever. Clinically, she had an oral temperature of 39.6°C and was severely dehydrated, with a heart rate of 116 beats/min and a blood pressure of 100/60 mm Hg. On examination, she was lethargic with no focal neurological deficit or neck stiffness. Examination of the heart, chest and abdomen was unremarkable. Blood laboratory investigations showed: sugar 46 mmol/l, sodium 181 mmol/l (135–145 mmol/l), potassium 4.6 mmol/l (3.4–4.9 mmol/l), urea 11 mmol/l (2–6 mmol/l) and creatinine 80 µmol/l (60–80 µmol/l). Arterial blood gas analysis showed: Pao2 8.7 kPa, Paco2 4.2 kPa, pH 7.39, and bicarbonate 20 mmol/l (22–28 mmol/l). Urine showed marked pyuria and negative ketones. Blood and urine cultures were positive for Escherichia coli. Cerebrospinal fluid analysis showed normal biochemistry and cell count with negative microbiological workup. Brain CT was normal. The patient was managed as a case of HHS with Gram-negative sepsis. Immediate treatment with 0.45% saline and intravenous insulin infusions was started together with appropriate intravenous antibiotics. The saline infusion was started initially at 250 ml/h, with modification of rate and concentration so that the fall in serum sodium was not allowed to exceed 0.5 mmol/l per hour (table 1). In the second week of the inpatient course, the patient's lethargy did not improve, in spite of fever subsidence, haemodynamic stabilization and proper correction of the biochemical parameters. She also developed behavioural changes with bouts of agitation and disorientation. Neurological evaluation revealed brisk deep tendon reflexes of the 4 limbs, but plantar responses were normal and there was no weakness. At the end of the second week, a brain MRI was made. It showed a high signal intensity lesion in the posterior central part of the pons in T2-weighted images (fig. 1), consistent with CPM. The patient did not give any history of previous neurological disorder or trauma, and, consequently, she did not have any previous brain CT or MRI studies. A diagnosis of CPM in relation to hypernatraemia of HHS was concluded. No specific treatment was given, and the patient was closely observed. She fully recovered and was discharged in a normal mental and physical state by the end of the fourth week. We could not obtain a repeat brain MRI as the patient was lost to follow-up.
Table 1.
The patient's biochemical parameters through the first week of management with normal values in parentheses
| Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 | |
|---|---|---|---|---|---|---|---|
| Serum sodium (135–145), mmol/l | 181 | 171 | 166 | 168 | 156 | 147 | 145 |
| Serum glucose (4.4–6.1), mmol/l | 46 | 18 | 20 | 17 | 13 | 11 | 11 |
| Serum urea (2–6), mmol/l | 11 | 9 | 8 | 7 | 6 | 6 | 6 |
| Serum potassium (3.4–4.9), mmol/l | 4.6 | 4.2 | 3.9 | 4.1 | 4.8 | 4.2 | 4.0 |
| Room air O2 saturation, % | 96 | 95 | 95 | 95 | 94 | 95 | 96 |
Fig. 1.
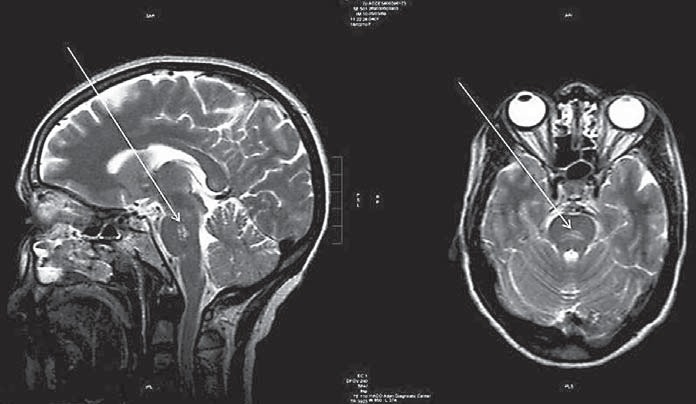
Brain MRI of the patient showing a high-signal intensity lesion (arrows) in the posterior central part of the pons in T2-weighted images.
Discussion
Our patient presented with lethargy that could be attributed to the severe hypernatraemia [2]. Lack of improvement of her condition and the development of an additional neurological deterioration in the form of agitation and behavioural changes led to the suspicion of ODS. Cerebral or subarachnoid haemorrhage due to hypernatraemia and brain oedema due to rapid lowering of serum sodium may also be considered in this setting [2]. In addition to the clinical scenario, imaging with brain CT or MRI could be helpful. However, it should be kept in mind that the radiological findings of the ODS may not become evident within a week or more from the onset of symptoms [1]. Hence, a repeat scan, 10–14 days later, is warranted [1].
There is increasing evidence for the association of ODS with hypernatraemia. Naik and Saroja [4] described extrapontine myelinolysis in 10 cases of post-partum hypernatraemia. ODS has also been linked to hypernatraemia due to hypertonic dehydration [5] and hunger strike [6]. Experimentally, histopathological brain changes compatible with myelinolysis were observed after inducing hypernatraemia in rats by sodium chloride injections [7]. ODS has been only rarely reported in relation to HHS. Mao et al. [3] described a patient with HHS leading to CPM, and considered him as the first documented case. Much earlier, McComb et al. [8] reported lateral pontine myelinolysis and extrapontine myelinolysis in a patient with HHS. Burns et al. [9] reported a case of CPM in a patient with hyperosmolar hyperglycaemia and normal serum sodium. They postulated that CPM is dependent on a hypertonic insult, regardless of sodium abnormality [9]. In a study by McKee et al. [10], CPM was found in autopsies of 7% of burned patients compared to 0.28% of patients of the general autopsy population. Burned patients with CPM had documented episodes of extreme hyperosmolality [10]. The authors suggested that hypernatraemia, hyperglycaemia and azotaemia, alone or combined, accounted for the hyperosmolality in CPM cases [10]. As in our case, most of the published reports showed that ODS occurred in the setting of severe, often prolonged hypernatraemia and/or hyperosmolality [5,7,10].
Pathophysiologically, ODS mainly occurs with the rapid correction of chronic (>48 h duration) hyponatraemia [1]. The brain adapts to hyponatraemia by losing extracellular water into the cerebrospinal fluid and by extruding sodium, potassium and certain organic solutes (osmolytes) out of the brain cells. Both mechanisms result in lowering the brain volume towards normal, thus avoiding brain oedema. In this setting, it is believed that the overly rapid rise in plasma sodium concentration without sufficient time for osmolytes to re-accumulate in brain cells may result in a further fall in brain volume and brain cell injury (osmotic demyelination). The pathophysiology of ODS in the setting of hypernatraemia is not yet known [6]. Myelinolysis seems to be related to the hypertonic challenge imposed by the severe hypernatraemia. The ODS in hyponatraemia correction requires a sequence of events that include forces that swell brain cells (hyponatraemia), then forces that shrink them (process of adaptation), followed by forces that shrink them more (rapidly correcting hyponatraemia without sufficient time for osmolytes to re-accumulate). Consequently, we may theoretically believe that ODS with hypernatraemia perhaps requires fluctuations in osmolality during the course of illness.
There is no specific treatment of proven benefit for the ODS [1]. The best supportive care should be given, and there is a possibility of spontaneous recovery. Some small case reports with therapeutic trials of steroids, plasmapheresis and intravenous immunoglobulin, with good outcome, have been published [1]. No randomized studies have been conducted to confirm the efficacy of these treatments [1]. ODS may be prevented by avoiding the rapid correction of chronic hyponatraemia. However, there is no way described so far to prevent the hypernatraemia-related ODS. The prognosis of ODS is generally poor as it may result in permanent neurological sequelae or death [1]. With the rarity of reported cases, there are no available data about the morbidity and mortality of hypernatraemia-related ODS. However, a general look at our patient and other case studies indicates that full recovery may be expected in a considerable proportion of cases [3,4,6].
Conclusion
This case report showed that osmotic demyelination was linked to hypernatraemia and that CPM could result from severe hypernatraemia of HHS. The hypernatraemia-related ODS is mainly reported with severe often prolonged hypernatraemia. Correct diagnosis and good supportive care are necessary for proper management and possible spontaneous recovery.
References
- 1.Martin RJ. Central pontine and extrapontine myelinolysis: the osmotic demyelination syndromes. J Neurol Neurosurg Psychiatry. 2004;75:22–28. doi: 10.1136/jnnp.2004.045906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adrogué HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342:1493–1499. doi: 10.1056/NEJM200005183422006. [DOI] [PubMed] [Google Scholar]
- 3.Mao S, Liu Z, Ding M. Central pontine myelinolysis in a patient with epilepsia partialis continua and hyperglycaemic hyperosmolar state. Ann Clin Biochem. 2011;48:79–82. doi: 10.1258/acb.2010.010152. [DOI] [PubMed] [Google Scholar]
- 4.Naik KR, Saroja AO. Seasonal postpartum hypernatremic encephalopathy with osmotic extrapontine myelinolysis and rhabdomyolysis. J Neurol Sci. 2010;291:5–11. doi: 10.1016/j.jns.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 5.Brown WD, Caruso JM. Extrapontine myelinolysis with involvement of the hippocampus in three children with severe hypernatremia. J Child Neurol. 1999;14:428–433. doi: 10.1177/088307389901400704. [DOI] [PubMed] [Google Scholar]
- 6.Van der Helm-van Mil AH, van Vugt JP, Lammers GJ, Harinck HI. Hypernatremia from a hunger strike as a cause of osmotic myelinolysis. Neurology. 2005;64:574–575. doi: 10.1212/01.WNL.0000150730.80325.11. [DOI] [PubMed] [Google Scholar]
- 7.Soupart A, Penninckx R, Namias B, Stenuit A, Perier O, Decaux G. Brain myelinolysis following hypernatremia in rats. J Neuropathol Exp Neurol. 1996;55:106–113. doi: 10.1097/00005072-199601000-00011. [DOI] [PubMed] [Google Scholar]
- 8.McComb RD, Pfeiffer RF, Casey JH, Wolcott G, Till DJ. Lateral pontine and extrapontine myelinolysis associated with hypernatremia and hyperglycemia. Clin Neuropathol. 1989;8:284–288. [PubMed] [Google Scholar]
- 9.Burns JD, Kosa SC, Wijdicks EF. Central pontine myelinolysis in a patient with hyperosmolar hyperglycemia and consistently normal serum sodium. Neurocrit Care. 2009;11:251–254. doi: 10.1007/s12028-009-9241-9. [DOI] [PubMed] [Google Scholar]
- 10.McKee AC, Winkelman MD, Banker BQ. Central pontine myelinolysis in severely burned patients: relationship to serum hyperosmolality. Neurology. 1988;38:1211–1217. doi: 10.1212/wnl.38.8.1211. [DOI] [PubMed] [Google Scholar]