

Summary
Background
Merkel cell carcinoma is a rare, aggressive skin cancer with poor prognosis in patients with advanced disease. Current standard care uses various cytotoxic chemotherapy regimens, but responses are seldom durable. Tumour oncogenesis is linked to Merkel cell polyomavirus integration and ultraviolet-radiation-induced mutations, providing rationale for treatment with immunotherapy antibodies that target the PD-L1/PD-1 pathway. We assessed treatment with avelumab, an anti-PD-L1 monoclonal antibody, in patients with stage IV Merkel cell carcinoma that had progressed after cytotoxic chemotherapy.
Methods
In this multicentre, international, prospective, single-group, open-label, phase 2 trial, patients with stage IV chemotherapy-refractory, histologically confirmed Merkel cell carcinoma (aged ≥18 years) were enrolled from 35 cancer treatment centres and academic hospitals in North America, Europe, Australia, and Asia. Key eligibility criteria were an ECOG performance status of 0 or 1, measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, adequate haematological, hepatic, and renal function, and immune-competent status (patients with HIV, immunosuppression, haematological malignancies, and previous organ transplantation were excluded). Patient selection was not based on PD-L1 expression or Merkel cell polyomavirus status. Collection of biopsy material or use of archival tissue for these assessments was mandatory. Avelumab was given intravenously at a dose of 10 mg/kg every 2 weeks. The primary endpoint was confirmed objective response (complete response or partial response) assessed according to RECIST version 1.1 by an independent review committee. Safety and clinical activity were assessed in all patients who received at least one dose of study drug (the modified intention-to-treat population). This trial is registered with ClinicalTrials.gov as NCT02155647.
Findings
Between July 25, 2014, and Sept 3, 2015, 88 patients were enrolled and received at least one dose of avelumab. Patients were followed up for a median of 10·4 months (IQR 8·6–13·1). The proportion of patients who achieved an objective response was 28 (31·8% [95·9% CI 21·9–43·1]) of 88 patients, including eight complete responses and 20 partial responses. Responses were ongoing in 23 (82%) of 28 patients at the time of analysis. Five grade 3 treatment-related adverse events occurred in four (5%) patients: lymphopenia in two patients, blood creatine phosphokinase increase in one patient, aminotransferase increase in one patient, and blood cholesterol increase in one patient; there were no treatment-related grade 4 adverse events or treatment-related deaths. Serious treatment-related adverse events were reported in five patients (6%): enterocolitis, infusion-related reaction, aminotransferases increased, chondrocalcinosis, synovitis, and interstitial nephritis (n=1 each).
Interpretation
Avelumab was associated with durable responses, most of which are still ongoing, and was well tolerated; hence, avelumab represents a new therapeutic option for advanced Merkel cell carcinoma.
Funding
Merck KGaA, Darmstadt, Germany.
Introduction
Merkel cell carcinoma is an aggressive skin cancer associated with Merkel cell polyomavirus, exposure to ultraviolet irradiation, immunosuppression, and old age.1,2 Merkel cell carcinoma occurs with an incidence of 0·2–0·4 cases per 100 000 people per year in Europe, 0·79 cases per 100 000 people per year in the USA, and 1·6 cases per 100 000 people per year in Australia.3–5 Global incidence and mortality from Merkel cell carcinoma have risen substantially over the past 30 years.3,4 The median age at diagnosis is approximately 75 years, and 5–12% of the patient population present with metastatic disease.1,4,6,7 The 5-year overall survival rate with metastatic Merkel cell carcinoma ranges from 0–18% based on retrospective analyses.6,8–10 Prospective studies are uncommon in this tumour type, and no approved therapies exist for non-resectable, recurrent, or metastatic Merkel cell carcinoma.
Although Merkel cell carcinoma is a chemosensitive disease, with response rates of 53–61%8,10–13 reported retrospectively for patients with metastatic Merkel cell carcinoma treated in the first-line setting, an overall survival benefit has not been shown.14,15 Responses to chemotherapy are seldom durable.3,14,15 In one report of patients with distant metastatic disease,11 of patients receiving second-line chemotherapy with topotecan (n=7), paclitaxel (n=5), or other regimens (n=18), the objective response was 23% and the median duration of response was 3·3 months.11 In that analysis, median progression-free survival was 2·0 months,11 the progression-free survival rate at 6 months was 13·3% (Nghiem P, unpublished), and the 6-month durable response rate was 6·7% (Nghiem P, unpublished). Chemotherapy is thus considered a treatment option, but not an evidence-based standard of care. Published guidelines recommend enrolment in a clinical trial for patients with metastatic disease.3,14
Research in context.
Evidence before this study
Merkel cell carcinoma is an aggressive skin cancer that is associated with old age, poor prognosis, and lower survival compared with other skin malignancies, including melanoma. No consensus on effective treatment for Merkel cell carcinoma exists. Multiple chemotherapy regimens have been used to treat patients with advanced disease, but responses are short-lived and relapse is common. We searched PubMed on April 7, 2015, and on Jan 27, 2016, for reports published in English since database inception using the search term “Merkel cell carcinoma” combined with “chemotherapy” or the most commonly used chemotherapy drugs. Additionally, we searched congress abstracts published in English from the American Society of Clinical Oncology and the European Society for Medical Oncology from 2010 through 2015 using the term “Merkel cell carcinoma”. Most publications were case reports and retrospective analyses based on institutional or national databases. We identified five cohort studies that assessed patients with distant metastases; of these, only one reported on a confirmed stage IV population. Evidence suggests that Merkel cell carcinoma is a chemosensitive disease but that responses are seldom durable. The reported 5-year overall survival rate is 0–18%. Published guidelines from the National Comprehensive Cancer Network and the European Association of Dermato-Oncology acknowledge the absence of evidence to support chemotherapy as a standard of care and recommend that patients with advanced Merkel cell carcinoma be enrolled in clinical trials of investigative therapies. Additionally, the scientific literature on tumour causes and oncogenesis linked to risk and prognostic factors, including viral infection, ultraviolet-radiation exposure, old age, and immunosuppression, advances the notion that immunotherapy is a promising approach to a crucial unmet medical need.
Added value of this study
This trial investigated avelumab, a fully human IgG1 monoclonal antibody that inhibits PD-L1, in a population of patients with metastatic, chemotherapy-refractory disease. The trial met its primary endpoint, with nearly a third of patients achieving durable objective responses according to Response Evaluation Criteria In Solid Tumors version 1.1 and assessment by an independent review committee. The median duration of response had not been reached after a median follow-up time of 10·4 months. Responses were achieved irrespective of PD-L1 expression or Merkel cell polyomavirus status. Additionally, avelumab was well tolerated, with few grade 3 treatment-related adverse events and no treatment-related grade 4 adverse events or deaths. This is an important advance over chemotherapy, which is associated with a high incidence of toxicity-related morbidity and high disease-related mortality, particularly in patients who are older than 65 years of age with stage IV malignancy. To our knowledge, this is the largest prospective, international, multicentre study of an immune checkpoint inhibitor in Merkel cell carcinoma. On the basis of this study, avelumab received a breakthrough designation, fast-track designation, and orphan drug designation by the US Food and Drug Administration. The European Medicines Agency and the Australian Therapeutic Goods Administration also recognise the orphan drug status of avelumab.
Implications of all the available evidence
Our findings and the results from a phase 2 trial of an anti-PD-1 monoclonal antibody in patients with Merkel cell carcinoma who had not received previous systemic therapy provide evidence that these drugs are efficacious and safely administered in both treatment-naive and chemotherapy-refractory settings. These data add substantial support to changing the therapeutic framework for the treatment of advanced Merkel cell carcinoma. Our results showing clinical activity in both virus-related and ultraviolet-radiation-induced tumours provide an impetus for investigating avelumab in other tumour types with similar causes.
Several lines of evidence indicate mechanistic coupling between immunosuppression and Merkel cell carcinoma oncogenesis and thus support immunotherapy as a promising approach. Merkel cell polyomavirus is present in approximately 80% of patients with Merkel cell carcinoma, with an incidence as high as 97% in samples assessed with PCR.2,16,17 The virus integrates into DNA to drive expression of Merkel cell polyomavirus large T antigens, promote tumour proliferation, and disrupt immune responses.2,18 In virus-negative tumours, a mutational burden signature associated with ultraviolet radiation exposure appears to be important for oncogenesis, leading to increased expression of neoantigens, heightened immunogenicity, and probably an increased requirement for immune evasion by the tumour.19–21 Active immunosuppression, occurring in relation to HIV infection, some haematological malignancies, and solid-organ transplantation, is associated with an increased risk of Merkel cell carcinoma; however, patients with Merkel cell carcinoma who are immunosuppressed comprise 8–10% of the total Merkel cell carcinoma population.22,23
PD-L1 is a key therapeutic target in the reactivation of the immune response against multiple cancers.24–26 PD-L1 is often expressed by tumour cells within the tumour microenvironment and binds to the PD-1 receptor on activated T cells, resulting in the inactivation of the T cell. This process appears to be an important mechanism through which tumours inhibit immune responses. A high concentration of tumour-associated PD-L1 might be prognostic of poor outcome, and in some tumour types, a positive predictive marker of therapeutic response to immunotherapy; however, data have shown that PD-L1 overexpression is not a robust biomarker for response, and investigations of its value as a correlative biomarker are ongoing across multiple tumour types.27,28 PD-L1 is expressed by Merkel cell carcinoma cells and by adjacent immune cell infiltrates.29,30 Moreover, tumour-infiltrating CD8-positive and CD4-positive T cells specific to Merkel cell polyomavirus oncoproteins are enriched in some Merkel cell carcinomas in association with enhanced expression of both PD-L1 and the PD-1 receptor.30,31 These patterns of expression of immune-related inhibitory markers provide a rationale for investigating the therapeutic potential of immune checkpoint inhibitors in Merkel cell carcinoma. Anti-tumour activity of pembrolizumab, an antibody that blocks the PD-1/PD-L1 pathway by targeting PD-1, was shown in a phase 2 study32 of patients with stage IIIb and stage IV Merkel cell carcinoma who were treated in a first-line, systemic, chemotherapy-naive setting. These findings in 25 patients support the potential of anti-PD-1/anti-PD-L1 monoclonal antibodies as a therapeutic option for advanced Merkel cell carcinoma.32
Avelumab (proposed non-proprietary name for MSB0010718C) is a fully human anti-PD-L1 IgG1 monoclonal antibody that inhibits PD-L1/PD-1 interactions but leaves intact the PD-L2/PD-1 pathway.33 Antibody-dependent cellular cytotoxicity might contribute to the activity of avelumab, as shown in preclinical models.34 Promising evidence of clinical activity and an acceptable safety profile has been shown in a phase 1 study33,35,36 of avelumab in patients with refractory advanced solid tumours. We aimed to assess the clinical activity and safety of avelumab in patients with metastatic Merkel cell carcinoma progressing after at least one previous line of chemotherapy.
Methods
Study design and participants
In this multicentre, international, prospective, open-label, single-group, phase 2 trial done at 35 cancer treatment centres and academic hospitals in North America, Europe, Australia, and Asia (appendix p 5), we enrolled patients with histologically confirmed stage IV Merkel cell carcinoma refractory to chemotherapy, defined as disease progressed after at least one previous line of chemotherapy for metastatic disease. Eligible patients were adults aged at least 18 years who had an ECOG performance status of 0 or 1, an estimated life expectancy of more than 12 weeks, at least one unidimensional measurable lesion by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1,37 and adequate haem atological function (defined as a white blood cell count of ≥3 × 109 cells per L with an absolute neutrophil count ≥1·5 × 109 cells per L, lymphocyte count ≥0·5 × 109 cells per L, platelet count ≥100 × 109 platelets per L, and haemoglobin ≥9 g/dL), hepatic function (defined as a total bilirubin concentration of ≤1·5 × the upper limit of normal [ULN] range and aspartate aminotransferase and alanine aminotransferase concentrations of ≤2·5 × ULN), and renal function (defined as an estimated creatinine clearance >50 mL/min according to the Cockcroft-Gault formula). Previous therapy with any drug targeting T-cell co-regulatory proteins (ie, immune checkpoint inhibitors) and concurrent anticancer treatment or systemic treatment with corticosteroids or other immunosuppressive drugs were not permitted. Patients who had received any vaccinations for prevention of infectious disease within 4 weeks of trial drug administration were excluded, and vaccination while on trial was also prohibited except for administration of inactivated vaccines (eg, inactivated seasonal influenza vaccine). Patients with HIV, immunosuppression, haematological malignancies, or previous solid-organ transplants were excluded, as were patients with clinically significant comorbidities such as active cardiovascular disease and inflammatory bowel disease. Patient selection was not based on PD-L1 expression or Merkel cell polyomavirus status. Collection of biopsy material or use of archival tissue for these assessments was mandatory. Additional patient eligibility criteria are provided in the appendix (p 6). Patients were enrolled in accordance with an approved protocol, international standards of good clinical practice, institutional review board approval at each site, and institutional safety monitoring, and written informed consent was provided by patients or their legal representatives.
Procedures
Avelumab (EMD Serono, Rockland, MA; Merck KGaA, Darmstadt, Germany) was supplied as a 10 mg/mL solution. Patients received avelumab 10 mg/kg by 1 h intravenous infusion once every 2 weeks until confirmed disease progression, unacceptable toxicity, or occurrence of any other criterion for withdrawal. Radiological tumour assessment by CT or MRI and assessment of photographic scans of skin lesions by the independent review committee to determine response were done every 6 weeks according to RECIST version 1.1. Tumour assessments by CT or MRI and assessment of skin lesions by physical examination were also performed by the investigator every 6 weeks as per RECIST version 1.1. To classify a best overall response as a complete or partial response, confirmation of the response by RECIST version 1.1 was required and preferably was done at regularly scheduled 6-week assessment intervals, but no sooner than 5 weeks after the initial documentation of complete response or partial response. Patients who had a confirmed complete response were treated for a minimum of 6 months and a maximum of 12 months after confirmation, at the discretion of the investigator and in adherence with any criteria for withdrawal. Treatment beyond 12 months in these patients was allowed on the basis of investigator assessment of potential benefit. Confirmation of progressive disease by radiological assessment was required, preferably 6 weeks (but no later) after a diagnosis of progression per RECIST version 1.1. If progression was based on the occurrence of a new lesion in an area not scanned at baseline, a further on-trial scan 6 weeks later was done.
Patients were allowed to stay on treatment beyond observation of progressive disease provided there was no significant clinical deterioration, defined as no new symptoms or worsening of existing symptoms, no change in ECOG performance status to 3 or higher that lasted more than 14 days, and no investigator assessment that a salvage therapy was necessary. Discontinuation from study treatment occurred for any grade 3 or worse adverse event (with the exception of transient [≤6 h] influenza-like symptoms or pyrexia controlled with medical management; fatigue, local infusion-related reaction, headache, nausea, or emesis that resolved to grade ≤1 within 24 h; single laboratory values out of the normal range that were unrelated to study treatment and without clinical correlate [except for elevation in liver enzyme concentrations] that resolved to grade ≤1 within 7 days; and tumour flare, defined as local pain, irritation, or rash localised at sites of known or suspected malignant tissue) or recurring grade 2 treatment-related adverse events. Grade 2 adverse drug reactions were managed by dose modifications (changes in the infusion rate) and dose delays, and those that did not resolve to grade 1 or less by the end of the next cycle led to permanent discontinuation of study treatment. Dose-level reductions were not permitted. However, inter ruptions in delivering the planned dose that resulted in an actual non-zero dose equal to less than 90% of the planned dose were defined as dose reductions within an administration. To mitigate potential infusion-related reactions, all patients were required per protocol to receive premedication with an H1-antihistamine, such as diphenhydramine, and paracetamol 30–60 min before avelumab treatment.
Safety was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE), version 4.0. A customised Medical Dictionary for Regulatory Activities query was used for data retrieval from the clinical database with predefined preferred terms of potential immune-mediated adverse events. Screening procedures included collection of patient demographics and complete medical history, complete physical examination, laboratory assessments, and collection of tumour tissue samples (fresh biopsy or recent biopsy [within 4 weeks] was preferred, but archival material was acceptable). Adverse events, concomitant medications, vital signs, bodyweight, laboratory haematology and haemostaseology, and ECOG performance status were monitored at each visit. Tumour assessments were done every 6 weeks. Blood samples for pharmacokinetics analysis, drug immunogenicity testing, and soluble factor and Merkel cell polyomavirus antibody analyses were collected at specific intervals during trial treatment (appendix p 7). For drug immunogenicity testing, patients who were not positive for the presence of anti-therapeutic antibodies before treatment with avelumab and with at least one positive result by the anti-therapeutic antibody assay were characterised as having a treatment-emergent response and were further assessed for persistence of the antibody response, which was defined as a first and last positive result occurring more than 16 weeks apart or by a positive result at the most recent visit. A positive response to anti-therapeutic antibodies was considered transient if the time between the first and last positive result was less than 16 weeks and further testing showed a negative result at the most recent visit.
Concentrations of PD-L1 protein and Merkel cell polyomavirus large T-antigen expression by tumour cells were measured by immunohistochemical analysis of formalin-fixed, paraffin-embedded blocks or slides of the most recent biopsy or surgical specimen. PD-L1 expression was assessed with a proprietary research-use-only assay (Dako, Carpinteria, CA, USA) based on an anti-PD-L1 rabbit monoclonal antibody clone (clone 73-10; Merck KGaA, Darmstadt, Germany), and PD-L1 positivity was defined in this study as a threshold level of 1% positive tumour cells of any intensity. Merkel cell polyomavirus status on tumour cells by immuno histochemistry was assayed with a monoclonal antibody specific for Merkel cell polyomavirus large T antigen (clone CM2B4; Santa Cruz Biotechnology, Dallas, TX, USA).38
Outcomes
The primary endpoint was confirmed best overall response, defined as complete response, partial response, stable disease, or progressive disease, according to RECIST version 1.1 and assessed by an independent review committee. Secondary endpoints were duration of response (defined as the time from first documented complete or partial response until documented progressive disease or death, whichever occurred first), progression-free survival (defined as time from the first administration of avelumab until documented progressive disease or death, whichever occurred first), overall survival (defined as the time from first administration of avelumab until the date of death), response status by RECIST at 6 and 12 months, safety, population pharmacokinetic profile, and immunogenicity of avelumab. Exploratory endpoints included tumour assessments by investigator using RECIST version 1.1 and modified immune-related response criteria,39 and tumour shrinkage in target lesions from baseline.
Statistical analysis
With a planned sample size of 84 patients and assuming a true objective response (the proportion of patients with a confirmed best overall response of complete or partial response, described in the protocol as objective response rate) of 35%, the study had 87% power to assess clinical activity according to a proportion of patients with an objective response over a threshold of 20% at a one-sided 2·5% significance level. An objective response of 20% at the lower bound was chosen for clinical meaningfulness, given the absence of scientific literature documenting treatment outcomes for second-line patients available at the time of protocol writing. A group sequential testing strategy was used to assess clinical activity—first in an interim analysis 6 months after the start of treatment for the first 56 patients, and then in the primary analysis of the whole study population 6 months after the first treatment of the last patient. The group sequential testing strategy was used to account for repeated significance testing of data collected at predefined time intervals for interim and primary analyses. Safety and clinical activity were analysed in all patients who received at least one dose of avelumab (the modified intention-to-treat [ITT] population). Additionally, a post-hoc sensitivity analysis was done in patients who met key eligibility criteria (ie, distant metastatic, histologically confirmed, chemotherapy-refractory Merkel cell carcinoma and measurable disease at baseline by independent review committee assessment) and had at least one post-baseline assessment. The objective response was reported with corresponding two-sided Clopper-Pearson CIs. A repeated CI for the objective response in the modified ITT analysis set (95·9% CI for the primary analysis) was calculated to account for the group sequential testing approach.40 Time-to-event endpoints—duration of response, progression-free survival, and overall survival—were analysed with Kaplan-Meier methods; median values were calculated with corresponding CI using the Brookmeyer-Crowley method. Post-hoc subgroup analyses of objective response were done on the basis of patient and disease characteristics at baseline: PD-L1 expression of tumour cells and tumour Merkel cell polyomavirus status, the number of previous lines of systemic treatment, disease burden defined by sum of target lesion diameters, and visceral disease status. In this analysis, visceral disease was defined as metastases not isolated to lymph nodes, skin, and soft tissue. The 6-month durable response rate, defined as the proportion of patients with a response of at least 6 months’ duration, was estimated as the product of the objective response and the Kaplan-Meier estimate of 6-months’ durability of response. This way of describing the statistical approach is the most accurate. A 95% CI for the 6-month durable response rate was obtained by applying the standard formula for the variance of a product of independent random variables. Concordance between the independent review committee and the investigator assessment of response was calculated as the proportion of patients classified either as having a response or as not having a response by both methods. Summaries of adverse events were restricted to treatment-emergent adverse events, defined as those with an onset during or after the first dose of trial treatment until 30 days after the last dose of trial treatment but before the start of subsequent anticancer drug therapy. SAS version 9.2 was used for the statistical analysis, and R software package version 2.15.0 was used for the sample size calculations. This trial is registered with ClinicalTrials. gov, number NCT02155647.
Role of the funding source
The funder of the study provided the study drug and worked with investigators on the trial design and plan, collection and analyses of data, interpretation of results, and the writing of the manuscript. All authors had access to raw data for review and participated fully in developing, reviewing, and submitting the manuscript for publication. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
As of March 3, 2016, the date of data cutoff for the primary analysis, 125 patients were screened and 88 eligible patients were enrolled and treated with avelumab (figure 1). The date of the first dose in the first patient was July 25, 2014, and the last patient received a first dose on Sept 3, 2015. Baseline characteristics of the patient population are shown in table 1. All patients had distant metastatic disease (M1, defined as metastases beyond regional lymph nodes) at the time of study enrolment, with a median time since diagnosis of metastatic disease of 10·4 months (IQR 6·3–17·2). All patients had at least one previous line of systemic anticancer treatment, including at least one for metastatic disease; 36 (41%) of 88 patients had received two or more previous lines of therapy (appendix p 8). Patients had received a platinum-containing regimen (n=60), an anthracycline-containing regimen (n=6), or another regimen (n=20) in their last previous treatment line (appendix p 9). The primary tumour site was skin in most patients; patients with a non-skin primary site included patients with nodal metastases and unknown primary tumours, at a proportion that is consistent with that reported in the literature (table 1).41 Visceral metastasis was present in 47 (53%) of 88 patients (table 1).
Figure 1.
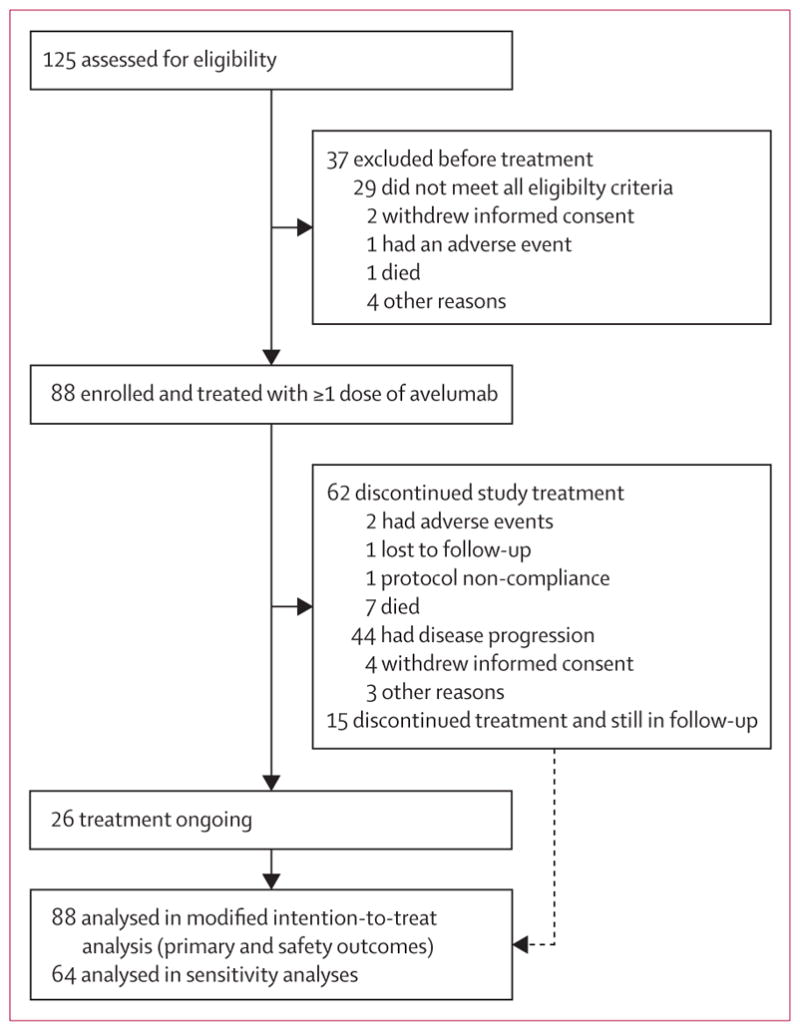
Trial profile
Table 1.
Baseline characteristics
| Patients (n=88) | |
|---|---|
| Age (years) | 72·5 (64·5–77·0) |
| <65 | 22 (25%) |
| ≥65 | 66 (75%) |
|
| |
| Sex | |
| Male | 65 (74%) |
| Female | 23 (26%) |
|
| |
| Pooled region | |
| North America | 51 (58%) |
| Europe | 29 (33%) |
| Rest of world | 8 (9%) |
|
| |
| Site of primary tumour | |
| Skin | 67 (76%) |
| Lymph node | 12 (14%) |
| Other* | 2 (2%) |
| Missing | 7 (8%) |
|
| |
| Metastatic involvement at study entry | 88 (100%) |
|
| |
| Visceral disease at study entry | |
| Present | 47 (53%) |
| Absent | 41 (47%) |
|
| |
| Sum of target lesion diameters (mm) | 79·0 (43·0–138·0) |
|
| |
| ECOG performance status score | |
| 0 | 49 (56%) |
| 1 | 39 (44%) |
|
| |
| Time since first diagnosis (months) | 19·8 (13·7–33·0) |
|
| |
| Time since first diagnosis of metastatic disease (months) | 10·4 (6·3–17·2) |
|
| |
| Number of previous systemic anticancer treatments | |
| 1 | 52 (59%) |
| 2 | 26 (30%) |
| 3 | 7 (8%) |
| ≥4 | 3 (3%) |
|
| |
| Time since last progression of disease (months) | 1·3 (0·8–2·0) |
|
| |
| Lymphocyte count status at study entry | |
| Normal | 35 (40%) |
| Decreased | 53 (60%) |
| Increased | 0 |
|
| |
| Tumour PD-L1 expression† | |
| Positive | 58 (66%) |
| Negative | 16 (18%) |
| Not assessable‡ | 14 (16%) |
|
| |
| Tumour Merkel cell polyomavirus status§ | |
| Positive | 46 (52%) |
| Negative | 31 (35%) |
| Not assessable‡ | 11 (13%) |
|
| |
| Combined PD-L1/Merkel cell polyomavirus status†§ | |
| PD-L1 positive/Merkel cell polyomavirus positive | 36 (41%) |
| PD-L1 positive/Merkel cell polyomavirus negative | 19 (22%) |
| PD-L1 negative/Merkel cell polyomavirus positive | 9 (10%) |
| PD-L1 negative/Merkel cell polyomavirus negative | 7 (8%) |
| Not assessable‡ | 17 (19%) |
Data are median (IQR) or n (%). Percentages that do not sum to 100 are a result of rounding.
Site of primary tumour was cheek mucosa in one patient and rectosigmoid junction in one patient.
PD-L1 positivity was defined as at least 1% of tumour cell membranes with staining of any intensity by immunohistochemistry.
Non-assessable specimens included those that were missing, of poor quality, or otherwise not available to provide results.
Merkel cell polyomavirus status was determined by immunohistochemistry.38
Patients received a median of seven doses (IQR 3–18) of avelumab, and the median duration of treatment was 17 weeks (IQR 7–37). Patients had a median follow-up time of 10·4 months (IQR 8·6–13·1) from first trial treatment to analysis cutoff date. Follow-up was ongoing at the time of this analysis. Samples from 74 patients were assessable for PD-L1 expression and 77 for Merkel cell polyomavirus status by immunohistochemistry; 71 (81%) of 88 patients were assessable for both PD-L1 and Merkel cell polyomavirus status. Of those that were assessable, 58 (79%) were PD-L1 positive (defined as at least 1% positive tumour cells at any staining intensity) and 46 (60%) were Merkel cell polyomavirus positive (table 1).
Confirmed objective responses to avelumab were achieved in 28 (31·8%; 95·9% CI 21·9–43·1) of 88 patients, eight of whom had complete responses and 20 of whom had partial responses according to independent review committee assessment by RECIST version 1.1 (table 2). Responses were noted at the time of the first post-baseline tumour assessment (week 7) in 22 (79%) of 28 patients (figure 2A). Responses were ongoing in 23 (82%) of 28 patients at the time of analysis. By Kaplan-Meier estimates, the proportion of responses with a duration of at least 6 months was 92% (95% CI 70–98). On this basis, the proportion of patients with a durable response, defined in a post-hoc analysis as the proportion of patients with a response lasting at least 6 months, was estimated as 29% (95% CI 20–39). At the time of data cutoff, response duration ranged from at least 2·8 months to at least 17·5 months, and the median duration of response was not reached (95% CI 8·3 months–not estimable). Of all 88 patients in the modified ITT population, 27 (31% [95% CI 21–41]) patients were in response at 6 months after start of treatment. In the 29 of 88 patients with more than 12 months of follow-up, six (21% [95% CI 8–40]) were in response at 12 months after their first dose of avelumab. The exploratory analysis of the percentage change from baseline in target lesions over time and the pattern of responses to avelumab are shown in figure 2B. The change from baseline in the size of target lesions to the smallest post-baseline value is shown in figure 2C. Tumour regression by at least 30% occurred in 29 (33%) of 88 patients, including in one patient for whom early disease progression by RECIST version 1.1 was followed by tumour shrinkage (figure 2C).
Table 2.
Confirmed best overall response
| Confirmed best overall response* (n=88) | |
|---|---|
| Complete response | 8 (9%) |
| Partial response | 20 (23%) |
| Stable disease | 9 (10%) |
| Progressive disease | 32 (36%) |
| Non-complete response/non-progressive disease† | 1 (1%) |
| Non-assessable‡ | 18 (20%) |
| Objective response§ | 31·8%(21·9–43·1) |
Data are n (%) or % (95·9% CI).
Confirmed best overall response was according to independent review committee assessment and Response Evaluation Criteria in Solid Tumors version 1.1.
One patient did not have measurable disease at baseline and thus a best overall response of partial response or stable disease could not be distinguished.
Patients not assessable for a confirmed best overall response had no baseline lesions identified by the independent review committee (n=4), baseline but no post-baseline assessments (n=10; four patients died within 6 weeks after the start of treatment and six additional patients discontinued study treatment in the first 6 weeks), all non-assessable post-baseline assessments (n=2), no post-baseline tumour assessment before the start of new anticancer therapy (n=1), or stable disease of insufficient duration (<6 weeks after start date without further tumour assessment; n=1).
A repeated CI for the objective response in the modified intention-to-treat analysis set (95·9% CI for the primary analysis) was calculated to account for the group sequential testing approach.40
Figure 2. Clinical activity of avelumab.

(A) Time to response, the duration of treatment, and the duration of response to avelumab in 28 patients with a confirmed response. (B) Percentage change in sum of target lesion diameters from baseline over time for all assessable patients (n=65), defined as those patients with baseline tumour assessments and at least one post-baseline assessment. A patient with pseudoprogression is indicated by an asterisk. Upper dotted line represents progression at 20% and lower dotted line represents the RECIST boundary for complete response or partial response at 30%. (C) Plot of tumour regression from baseline as measured by RECIST version 1.1 in all assessable patients (n=65). Upper dotted line represents progression at 20% and lower dotted line represents the RECIST boundary for complete response or partial response at 30%. (D) Kaplan-Meier estimate of progression-free survival in the modified intention-to-treat population (n=88). Vertical lines show censored events. RECIST=Response Evaluation Criteria in Solid Tumors.
Median progression-free survival was 2·7 months (95% CI 1·4–6·9), and the proportion of patients who were progression-free at 6 months was 40% (29–50; figure 2D). Progression-free survival reached a plateau, as shown from the Kaplan-Meier curve, with data still maturing. At the time of analysis, 52 (60%) of 88 patients had a progression-free survival event (progressive disease in 44 [50%] patients and death in eight [9%] patients, including one lost to follow-up). The overall survival rate at 6 months was 69% (95% CI 58–78; appendix p 3), and the median overall survival was 11·3 months (7·5–14·0) based on 43 (49%) of 88 patients with an event.
Post-hoc subgroup analyses are shown in figure 3. Duration of response was generally consistent across subgroups (appendix pp 11–12). An exploratory analysis showed that the proportion of patients who achieved an objective response according to investigator assessment was 31·8% (95% CI 22·3–42·6; eight complete responses and 20 partial responses), in line with the independent review committee assessment. The concordance between independent review and investigator assess ment was 91%. The proportion of patients who achieved an objective response according to the post-hoc sensitivity analysis in 64 patients (defined as those patients who met key eligibility criteria, had measurable disease at baseline by independent review committee assessment, and had at least one post-baseline assessment) was 40·6% (95% CI 28·5–53·6). Exploratory analysis using the modified immune-related response criteria showed that 10 complete responses and 20 partial responses were achieved in the modified ITT population for an objective response of 34·1% (95% CI 24·3–45·0). One patient showed pseudoprogression, indicated in the spider plot (figure 2B). A case of tumour response in a patient treated with avelumab compared with baseline disease before treatment is shown in the appendix (p 4). A partial response by RECIST version 1.1 and a complete response by pathological assessment were reported in this patient; the response in this patient is ongoing, with a duration of 3·9 months at the time of data cutoff and 1·4 months beyond treatment discontinuation.
Figure 3. Objective response by subgroup for select patient characteristics.

Error bars show 95% CI. Disease burden was defined by SLD. SLD=sum of target lesion diameters. ECOG PS=Eastern Cooperative Oncology Group performance status. MCPyV=Merkel cell polyomavirus.
In a post-hoc analysis, among patients whose tumours were assessable for PD-L1 expression, objective responses were achieved in 20 (34·5% [95% CI 22·5–48·1]) of 58 patients who tested positive on the basis of a 1% staining threshold and in three (18·8% [4·0–45·6]) of 16 patients with PD-L1 negative tumours (figure 3). Objective responses also occurred in 12 (26·1% [95% CI 14·3–41·1]) of 46 patients who tested positive for Merkel cell polyomavirus and 11 (35·5% [95% CI 19·2–54·6]) of 31 patients who tested negative for Merkel cell polyomavirus. Key clinical, biomarker, and response characteristics of the patients who achieved a complete response to treatment with avelumab are provided in the appendix (p 10).
Treatment-related adverse events occurred in 62 (70%) of 88 patients; those occurring in more than 10% of patients were fatigue (21 [24%]) and infusion-related reactions (15 [17%]; table 3; appendix pp 18–19). Grade 3 treatment-related adverse events were reported in four (5%) of 88 patients for a total of five events (table 3). This included two patients with lymphopenia and three patients with isolated laboratory abnormalities (elevated blood creatine phosphokinase, blood cholesterol, and hepatic aminotransferase). There were no grade 4 treatment-related adverse events or deaths related to treatment. Adverse events leading to death occurred in eight (9%) of 88 patients: disease progression (n=4), hepatic failure (n=1), ileus (n=1), malignant neoplasm progression (n=1), and pneumonia (n=1). None of these were considered treatment related. Of all 43 deaths, disease progression was reported as the primary reason in 40 cases. Additionally, three deaths occurred for which the primary cause was unknown.
Table 3.
Treatment-related adverse events in the modified intention-to-treat population
| Grade 1–2 | Grade 3 | |
|---|---|---|
| Fatigue | 21 (24%) | 0 |
|
| ||
| Infusion-related reaction* | 15 (17%) | 0 |
|
| ||
| Diarrhoea | 8 (9%) | 0 |
|
| ||
| Nausea | 8 (9%) | 0 |
|
| ||
| Asthenia | 7 (8%) | 0 |
|
| ||
| Rash | 6 (7%) | 0 |
|
| ||
| Decreased appetite | 5 (6%) | 0 |
|
| ||
| Maculopapular rash | 5 (6%) | 0 |
|
| ||
| Blood creatine phosphokinase increase | 1 (1%) | 1 (1%) |
|
| ||
| Lymphopenia | 0 | 2 (2%) |
|
| ||
| Blood cholesterol increase | 0 | 1 (1%) |
|
| ||
| Aminotransferase increase | 0 | 1 (1%) |
|
| ||
| Potential immune-mediated treatment-related adverse event† | ||
| Hypothyroidism | 3 (3%) | 0 |
| Hyperthyroidism | 2 (2%) | 0 |
| Pneumonitis | 1 (1%) | 0 |
| Type 1 diabetes mellitus | 1 (1%) | 0 |
Any grade in at least 5% of patients or any grade 3 or worse adverse event based on the worst grade per patient; none were grade 4 or 5. The overall summary of safety is shown in the appendix (p 13), and tables listing all treatment-related adverse events occurring in more than one patient and all treatment-emergent adverse events regardless of causality occurring in at least 10% of patients are provided in the appendix (pp 14–19).
An infusion-related reaction in this analysis was based on a composite definition with five different Medical Dictionary for Regulatory Activities terms. Signs and symptoms of a potential infusion-related reaction (eg, fever, chills, or rigors) reported on the day of infusion (but not before dosing) or the following day were queried with investigators to ascertain whether an adverse event of “infusion-related reaction” should be recorded. Of the 15 treatment-related adverse events recorded as an infusion-related reaction, 13 (87%) of 15 resolved on the same day as the infusion. In one patient, resolution of a grade 1 event occurred within 3 days and without the use of concomitant corticosteroid, and in a second patient, a grade 2 event resolved within 6 days, also without the use of corticosteroid. Three patients received corticosteroid treatment for a grade 2 infusion-related reaction that resolved on the same day as the infusion. All other patients were treated with non-steroidal supportive medication.
These events were programmatically derived from a search term list. By manual medical review, potential immune-mediated, treatment-related adverse events were identified in four additional patients: grade 3 increased aminotransferase (n=1); grade 2 diarrhoea (n=1); grade 2 nephritis (n=1); and grade 1 rash (n=1).
With the use of a search-term method, six (7%) of 88 patients had a potential immune-mediated adverse event that was treatment related; all were grade 1 or 2 (table 3). Potential immune-mediated, treatment-related events requiring steroids were identified by medical review in four additional patients; three were grade 1 or 2 and one was grade 3 (table 3). 36 (41%) of 88 patients had a serious adverse event. Those occurring in more than one patient were acute kidney injury (n=4), disease progression (n=4), anaemia (n=3), abdominal pain (n=2), asthenia (n=2), cellulitis (n=2), and general physical health deterioration (n=2). Seven treatment-related serious adverse events occurred in five (6%) of 88 patients: grade 3 aminotransferase elevation (n=1), grade 2 infusion-related reaction (n=1), grade 2 enterocolitis (n=1), grade 2 chondrocalcinosis and grade 2 synovitis (both events occurring in one patient), and two events of interstitial nephritis in one patient (one grade 1 and one grade 2). Two (2%) of 88 patients permanently discontinued treatment because of an adverse event (one patient with grade 2 elevated aminotransferase deemed treatment-related and one patient with grade 3 pericardial effusion deemed not related). One additional patient discontinued study treatment because of a treatment-related grade 1 creatinine elevation, which occurred after the treatment-emergent period and followed an event of grade 2 treatment-related acute interstitial nephritis. Eight (9%) of 88 patients had at least one dose reduction within an administration. At least one dose was delayed in 39 (44%) of 88 patients, with delays of 3 to 6 days in 10 (11%) of 88 patients and 7 or more days in 29 (33%) of 88 patients. Of patients assessable for immunogenicity testing, three (4%) of 79 patients tested positive for treatment-emergent anti-therapeutic antibodies. Two of three immunogenic responses were persistent, and one was transient. Pharmacokinetic data are not yet mature and analyses are ongoing; these data will be reported elsewhere.
Discussion
Avelumab monotherapy in patients previously treated for metastatic disease was well tolerated and achieved rapid and sustained responses in 28 (32%) of 88 patients; another nine patients (10%) achieved stable disease. These data provide evidence of therapeutic activity in patients with metastatic, chemotherapy-refractory Merkel cell carcinoma. Merkel cell carcinoma is an aggressive cutaneous malignancy associated with poor survival outcomes in patients with metastatic disease. Although Merkel cell carcinoma is a rare cancer, the proportion of patients with recurrent disease exceeds 40%,6 and the incidence of disease-associated mortality is approximately three times that of melanoma.7,18 Chemotherapy has been shown to produce responses in this population, but they are seldom durable.3,14,15 In a recent observational study, the proportion of patients with chemotherapy-refractory metastatic disease who responded to chemotherapy in the second-line setting was 23%, with a 6-month durable response rate of 6·7%.11
Patients in this study all had distant metastases that had been treated with at least one previous line of therapy for metastatic disease, indicating the highest unmet medical need and population with the poorest prognosis among patients with Merkel cell carcinoma. The median duration of response was not reached in this study, and most patients (23 [82%] of 28) had ongoing responses at a median follow-up of 10·4 months (IQR 8·6–13·1). By contrast, responses to chemotherapy reported in the scientific literature are typically short-lived. The response duration and durable response rate reported in this study already exceed what can be achieved with chemotherapy.11 On the basis of Kaplan-Meier analysis, the 6-month estimate of durability was 92%, and the observed plateau of the Kaplan-Meier curve for progression-free survival was driven by durable responses. The 6-month durable response rate was 29%, and the 6-month progression-free survival rate was 40%. Complete responses occurred in patients who had visceral disease, had a high disease burden based on size of target lesions, and were heavily pretreated. Additionally, a complete response was ongoing in one patient for 9·5 months after this patient had ended treatment with avelumab. These data suggest that meaningful clinical benefit was achieved with avelumab treatment with a median time to response of 6 weeks.
Responses to avelumab were observed irrespective of PD-L1 expression or Merkel cell polyomavirus status, suggesting that avelumab might achieve a therapeutic benefit in patients whose disease response and cause are driven by different underlying mechanisms. Specific mechanisms related to the interplay of viral antigen, ultraviolet-based mutagenesis, and PD-L1 expression in the tumour microenvironment are not well understood. However, mutational landscape analyses suggests that Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative signatures might represent viral-dependent and ultraviolet-induced subtypes, respectively.19–21 Furthermore, the patterns of responsiveness to anti-PD-L1 treatment with respect to viral status suggest that PD-L1 expression might be driven by mechanisms of immune evasion and by increased mutagenesis and neoantigen expression in patients with viral-negative tumours. Our results showing clinical activity in both virus-related and ultraviolet-radiation-induced tumours provide an impetus for investigating avelumab in other tumour types with similar causes.
A rationale for immune checkpoint inhibition as a promising approach is further supported by evidence of anti-tumour activity with the anti-PD-1 antibody pembrolizumab in first-line metastatic Merkel cell carcinoma.32 This phase 2 study of 25 assessable patients with Merkel cell carcinoma receiving a first-line systemic therapy for unresectable or metastatic disease and with at least one tumour assessment during treatment reported an objective response of 56% (95% CI 35–76) by RECIST version 1.1. The patients in the first-line pembrolizumab study had stage IIIb and stage IV disease, whereas the patients in this study had only stage IV disease and had been treated in the second-line setting for chemotherapy-refractory disease. Median follow-up time in the first-line study was 7·6 months, compared with 10·4 months in the avelumab second-line study. Because durability of response is an indicator of clinical activity, the longer follow-up time shows a robust signal of benefit with avelumab. Differences observed in response based on PD-L1 expression or Merkel cell polyomavirus status in the two studies might relate to the different patient populations or the number of patients included in the trials; however, both studies reported responses in patients regardless of PD-L1 and Merkel cell polyomavirus status. Although the population in the pembrolizumab study32 differed from that in the current study of avelumab across several dimensions—less advanced disease and a more heterogeneous patient population, less heavily pretreated in a first-line setting, fewer patients, and shorter follow-up time—the two studies, taken together, reinforce the notion that targeting the PD-L1/PD-1 axis is an effective therapeutic strategy.
The safety profile of avelumab was manageable and consistent with anti-PD-L1/PD-1 antibodies in other tumour types.42 Most adverse events related to avelumab were low grade, consisting mostly of fatigue and infusion-related reactions. In particular, all infusion-related reactions were low grade (CTCAE grade 1 or 2) and, in most cases, resolved on the same day with only supportive medications; none resulted in discontinuation of study treatment. Four patients had grade 3 treatmentrelated adverse events, one of which led to permanent discontinuation (elevated aminotransferases). The grade 3 treatment-related adverse events were all related to laboratory abnormalities, which is consistent with safety data reported for pembrolizumab in first-line Merkel cell carcinoma.32 No grade 4 treatment-related events or deaths related to study treatment occurred. The number of potential immune-mediated adverse events related to avelumab was low, and the events were manageable.
The patients in this study had completed previous chemotherapy and represent a particularly challenging population. Previous reports in patients with metastatic Merkel cell carcinoma treated with chemotherapy have shown high rates of serious dose-limiting toxicities— including sepsis, neutropenia, and renal toxicity—and treatment-related death, especially in elderly patients.10,11 The median age of our population was 72·5 years, and no treatment-related deaths were noted. Thus, avelumab was safe in patients with previously treated, metastatic Merkel cell carcinoma.
This study does have limitations, which include the non-randomised study design and the small sample size. Merkel cell carcinoma is a rare disease with a rapid natural history in a population with often substantial comorbidities, which makes large randomised clinical trials difficult. To our knowledge, this study is the largest trial of metastatic Merkel cell carcinoma ever reported. Although this trial was not randomised to directly assess anti-PD-L1 therapy compared with chemotherapy in the second-line setting, our findings suggest that treatment with anti-PD-L1 achieves durable disease remission or stabilisation and is well tolerated. Additionally, this trial was designed a priori to assess the clinical activity of avelumab in patients with metastatic Merkel cell carcinoma after progressing on first-line chemotherapy. This study shows a confirmed objective response per RECIST version 1.1 criteria of 31·8% with a 95·9% CI excluding 20%. As a result, the study met its predefined primary objective of clinical activity.
We noted objective responses to avelumab in all subgroups analysed. We noted a higher proportion of patients with a response in subgroups who had received fewer lines of previous therapy compared with those who had received more lines of previous therapy. One possible explanation for this observation is that patients who received fewer lines of cytotoxic therapy might be more likely to have fully functioning immune systems than those who had received more lines of therapy, and thus might respond in a more robust way to immunotherapy with a checkpoint inhibitor. Our findings in the second-line and later-line setting, together with the results for pembrolizumab in first-line patients, indicate that anti-PD-L1/PD-1 therapies could become the standard of care in treatment-naive and advanced Merkel cell carcinoma. Additionally, these studies support the clinical activity and safety of anti-PD-L1/PD-1 monotherapy in the treatment framework. Combination approaches with anti-PD-L1/PD-1 antibodies and other immunotherapies have been initiated. Our findings show that avelumab represents a new therapeutic option for advanced Merkel cell carcinoma.
Supplementary Material
Acknowledgments
We thank the patients and their families, the investigators, co-investigators, and the study teams at each of the participating centres and at Merck KGaA, Darmstadt, Germany; EMD Serono, Billerica, MA, USA; and Quintiles, Durham, NC, USA. This trial was sponsored by Merck KGaA, Darmstadt, Germany, and is part of an alliance between Merck KGaA and Pfizer. Medical writing support was provided by ClinicalThinking, Hamilton, NJ, and funded by Merck KGaA, Darmstadt, Germany and Pfizer.
Footnotes
See Online for appendix
Contributors
HLK, JHL, KC, AvH, J-MC, and PN conceived and designed the study. HLK, OH, SB, JHL, KC, LM, AvH, J-MC, and PN collected and assembled the data. All authors analysed and interpreted the data, wrote the manuscript, and approved the final version of the manuscript.
Declaration of interests
HLK reports personal fees from Alkermes, Amgen, EMD Serono, Prometheus, and Sanofi; non-financial support from Merck; grants from Bristol-Myers Squibb, outside the submitted work. OH reports personal fees from Merck, outside the submitted work. PT reports personal fees from Merck Sharp & Dohme, Bristol-Myers Squibb, Roche, Novartis, and GlaxoSmithKline, outside the submitted work. SPD’A reports personal fees from EMD Serono and Amgen, outside the submitted work. CL reports personal fees from Novartis, Bristol-Myers Squibb, Roche Glycart, Amgen, and Merck Sharp & Dohme, outside the submitted work. MM reports personal fees from AstraZeneca, Novartis, Pfizer, Celgene, and NeoPharm, outside the submitted work. KDL reports institutional research funding from EMD Serono, outside the submitted work. KC is an employee of EMD Serono and holds stock in Bristol-Myers Squibb. LM is an employee of EMD Serono. AvH is an employee and stockholder at Merck KGaA. J-MC is an employee of EMD Serono. PN has been reimbursed for travel, accommodation, or expenses from EMD Serono, outside the submitted work. All other authors declare no competing interests.
References
- 1.Agelli M, Clegg LX. Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol. 2003;49:832–41. doi: 10.1016/s0190-9622(03)02108-x. [DOI] [PubMed] [Google Scholar]
- 2.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lebbé C, Becker JC, Grob JJ, et al. Diagnosis and treatment of Merkel cell carcinoma. European consensus-based interdisciplinary guideline. Eur J Cancer. 2015;51:2396–403. doi: 10.1016/j.ejca.2015.06.131. [DOI] [PubMed] [Google Scholar]
- 4.Fitzgerald TL, Dennis S, Kachare SD, Vohra NA, Wong JH, Zervos EE. Dramatic increase in the incidence and mortality from Merkel cell carcinoma in the United States. Am Surg. 2015;81:802–06. doi: 10.1177/000313481508100819. [DOI] [PubMed] [Google Scholar]
- 5.Youlden DR, Soyer HP, Youl PH, Fritschi L, Baade PD. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993–2010. JAMA Dermatol. 2014;150:864–72. doi: 10.1001/jamadermatol.2014.124. [DOI] [PubMed] [Google Scholar]
- 6.Allen PJ, Bowne WB, Jaques DP, Brennan MF, Busam K, Coit DG. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol. 2005;23:2300–09. doi: 10.1200/JCO.2005.02.329. [DOI] [PubMed] [Google Scholar]
- 7.Lemos BD, Storer BE, Iyer JG, et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: analysis of 5823 cases as the basis of the first consensus staging system. J Am Acad Dermatol. 2010;63:751–761. doi: 10.1016/j.jaad.2010.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tai PT, Yu E, Winquist E, et al. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: case series and review of 204 cases. J Clin Oncol. 2000;18:2493–99. doi: 10.1200/JCO.2000.18.12.2493. [DOI] [PubMed] [Google Scholar]
- 9.Santamaria-Barria JA, Boland GM, Yeap BY, Nardi V, Dias-Santagata D, Cusack JC., Jr Merkel cell carcinoma: 30-year experience from a single institution. Ann Surg Oncol. 2013;20:1365–73. doi: 10.1245/s10434-012-2779-3. [DOI] [PubMed] [Google Scholar]
- 10.Voog E, Biron P, Martin JP, Blay JY. Chemotherapy for patients with locally advanced or metastatic Merkel cell carcinoma. Cancer. 1999;85:2589–95. doi: 10.1002/(sici)1097-0142(19990615)85:12<2589::aid-cncr15>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 11.Iyer JG, Blom A, Doumani R, et al. Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. 2016 doi: 10.1002/cam4.815. published online July 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma D, Flora G, Grunberg SM. Chemotherapy of metastatic Merkel cell carcinoma: case report and review of the literature. Am J Clin Oncol. 1991;14:166–69. doi: 10.1097/00000421-199104000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Satpute SR, Ammakkanavar NR, Einhorn LH. Role of platinum-based chemotherapy for Merkel cell tumor in adjuvant and metastatic settings. Proc Am Soc Clin. 2014;32(suppl 5S) abstr 9049. [Google Scholar]
- 14.National Comprehensive Cancer Network. [accessed Aug 17, 2016];NCCN clinical practice guidelines in oncology: Merkel cell carcinoma. 2016 https://www.nccn.org/professionals/physician_gls/pdf/mcc.pdf.
- 15.Desch L, Kunstfeld R. Merkel cell carcinoma: chemotherapy and emerging new therapeutic options. J Skin Cancer. 2013;2013:327150. doi: 10.1155/2013/327150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos-Juanes J, Fernandez-Vega I, Fuentes N, et al. Merkel cell carcinoma and Merkel cell polyomavirus: a systematic review and meta-analysis. Br J Dermatol. 2015;173:42–49. doi: 10.1111/bjd.13870. [DOI] [PubMed] [Google Scholar]
- 17.Rodig SJ, Cheng J, Wardzala J, et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J Clin Invest. 2012;122:4645–53. doi: 10.1172/JCI64116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488–97. doi: 10.1007/s11912-011-0197-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong SQ, Waldeck K, Vergara IA, et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. 2015;75:5228–34. doi: 10.1158/0008-5472.CAN-15-1877. [DOI] [PubMed] [Google Scholar]
- 20.Goh G, Walradt T, Markarov V, et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget. 2016;7:3403–15. doi: 10.18632/oncotarget.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harms PW, Vats P, Verhaegen ME, et al. The distinctive mutational spectra of polyomavirus-negative Merkel cell carcinoma. Cancer Res. 2015;75:3720–27. doi: 10.1158/0008-5472.CAN-15-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375–81. doi: 10.1016/j.jaad.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulson KG, Iyer JG, Blom A, et al. Systemic immune suppression predicts diminished Merkel cell carcinoma-specific survival independent of stage. J Invest Dermatol. 2013;133:642–46. doi: 10.1038/jid.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 25.Hirano F, Kaneko K, Tamura H, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–96. [PubMed] [Google Scholar]
- 26.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–97. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–56. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 28.Aguiar PN, Jr, Santoro IL, Tadokoro H, et al. The role of PD-L1 expression as a predictive biomarker in advanced non-small-cell lung cancer: a network meta-analysis. Immunotherapy. 2016;8:479–88. doi: 10.2217/imt-2015-0002. [DOI] [PubMed] [Google Scholar]
- 29.Lipson EJ, Vincent JG, Loyo M, et al. PD-L1 expression in the Merkel cell carcinoma microenvironment: association with inflammation, Merkel cell polyomavirus and overall survival. Cancer Immunol Res. 2013;1:54–63. doi: 10.1158/2326-6066.CIR-13-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Afanasiev OK, Yelistratova L, Miller N, et al. Merkel polyomavirus-specific T cells fluctuate with Merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin Cancer Res. 2013;19:5351–60. doi: 10.1158/1078-0432.CCR-13-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iyer JG, Afanasiev OK, McClurkan C, et al. Merkel cell polyomavirus-specific CD8(+) and CD4(+) T-cell responses identified in Merkel cell carcinomas and blood. Clin Cancer Res. 2011;17:6671–80. doi: 10.1158/1078-0432.CCR-11-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nghiem PT, Bhatia S, Lipson EJ, et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med. 2016;374:2542–52. doi: 10.1056/NEJMoa1603702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heery CR, O’Sullivan Coyne G, Madan RA, et al. Phase I open-label, multiple ascending dose trial of MSB0010718C, an anti-PD-L1 monoclonal antibody, in advanced solid malignancies. Proc Am Soc Clin Oncol. 2014;32(suppl 5S) abstr 3064. [Google Scholar]
- 34.Boyerinas B, Jochems C, Fantini M, et al. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res. 2015;3:1148–57. doi: 10.1158/2326-6066.CIR-15-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heery CR, O’Sullivan Coyne G, Marte JL, et al. Pharmacokinetic profile and receptor occupancy of avelumab (MSB00110718C), an anti-PD-L1 monoclonal antibody, in a phase I, open-label, dose escalation trial in patients with advanced solid tumors. Proc Am Soc Clin Oncol. 2015;33(suppl) abstr 3055. [Google Scholar]
- 36.Kelly K, Patel MR, Infante JR, et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with metastatic or locally advanced solid tumors: assessment of safety and tolerability in a phase I, open-label expansion study. Proc Am Soc Clin Oncol. 2015;33(suppl) abstr 3044. [Google Scholar]
- 37.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1. 1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 38.Shuda M, Arora R, Kwun HJ, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer. 2009;125:1243–49. doi: 10.1002/ijc.24510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 40.Jennison C, Turnbull BW. Group sequential methods with applications to clinical trials. Boca Raton, FL: Chapman & Hall/CRC; 2000. [Google Scholar]
- 41.Veness MJ, Perera L, McCourt J, et al. Merkel cell carcinoma: improved outcome with adjuvant radiotherapy. ANZ J Surg. 2005;75:275–81. doi: 10.1111/j.1445-2197.2005.03353.x. [DOI] [PubMed] [Google Scholar]
- 42.Weber JS, Yang JC, Atkins MB, Disis ML. Toxicities of immunotherapy for the practitioner. J Clin Oncol. 2015;33:2092–99. doi: 10.1200/JCO.2014.60.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.