

Abstract
During development, stress, infection, or normal homeostasis, billions of cells die on a daily basis, and the responsibility of clearing these cellular corpses lies with the phagocytes of innate immune system. This process, termed efferocytosis, is critical for the prevention of inflammation and autoimmunity, as well as modulation of the adaptive immune response. Defective clearance of dead cells is characteristic of many human autoimmune or autoinflammatory disorders, such as systemic lupus erythematosus (SLE), atherosclerosis, and diabetes. The mechanisms that phagocytes employ to sense, engulf, and process dead cells for an appropriate immune response have been an area of great interest. However, insight into novel mechanisms of programmed cell death, such as necroptosis, has shed light on the fact that while the diner (or phagocyte) is important, the meal itself (the type of dead cell) can play a crucial role in shaping the pursuant immune response.
1 Introduction
The phagocytic cells of our innate immune system act as surveyors of the environment, constantly patrolling the body for unwanted, unneeded, and unexpected components and ridding them in a timely and orderly fashion. The ancient, evolutionarily conserved pathway of phagocytosis (“the cellular process of eating”) has been at the vanguard of immunology, developmental biology, and cellular biology since its nineteenth-century discovery (and 1908 Nobel Prize in Physiology and Medicine) by Ilya Metchinkoff and Paul Ehlirch (Krysko and Vandenabeele 2010). While clearance of invading pathogens is indeed a necessary function of phagocytes, the sensing, recognition, and removal of cellular corpses are a critical role that phagocytes play during times of development, cellular homeostasis, and stress (Nagata et al. 2010).
The formation of a “wild-type,” functioning organism is, in actuality, a process wrought with waste. A multitude of extra cells are generated during development, only to unceremoniously undergo programmed cell death (described below) and be cleared by phagocytes (Green 2011). During the development of Caenorhabditis elegans, a total of 1090 cells are generated, and 131 of them are destined for death (Kinchen 2010). Indeed, this cell death is critical for the correct development of the organisms, as animals deficient for a variety of caspases, endoproteases that mediate apoptotic cell death, are grossly malformed and often embryonic lethal (McIlwain et al. 2015). While the generation and subsequent destruction of these cells are necessary for proper development, as well as normal cellular homeostasis, wound healing, and immune responses in the adult organism, the ruin left in its wake would be catastrophic if not for the efficient work of the phagocytic system (Savill et al. 2002; Peter et al. 2010).
Despite the constant turnover of cells through programmed cell death mechanisms (not to mention those induced to die via stress or infection), it is rare to observe apoptotic cells under normal physiological conditions. Considering the average one million adult human cells that undergo apoptosis every second, one must truly appreciate the magnitude of the job facing phagocytes (Ravichandran 2010). Moreover, as a reoccurring and normal event in the life span of an organism, this process of dead cell clearance must occur in a quiescent manner, so as to not inappropriately alert the immune system (Hart et al. 2008).
In this chapter, we will explore efferocytosis not only as a process of cleanup, but also a critical regulator of the immune response. While the manner and efficiency in which dead cell cargo is degraded and processed by the phagocyte are important, the type of dead cell cargo is engulfed can also play a role in how the phagocyte responds.
2 Prepping of the Meal: Types of Cell Death
Death is a part of life. The process of generating, maintaining, and protecting a multicellular organism throughout its lifetime requires the creation and destruction of billions of cells. While damage can certainly cause unwanted cellular death, most cellular death is genetically programmed, and perturbations in these genetic programs can promote cell accumulation, autoimmunity, oncogenesis, attrition, and/or degeneration. Programmed cell death, such as apoptosis, necroptosis, or pyroptosis, is an active mechanism designed to sculpt, control, and aid the body in its development and survival. Like death itself, the innate immune system has tolerant systems in place to manage these morbid, yet necessary events. Death comes in a variety of flavors, some more appetizing than others.
2.1 Apoptosis
The most widely studied form of cell death is apoptosis. Apoptosis (from the Greek meaning “falling off”) is genetically programmed cellular suicide and involves the coordinated dismantling of intracellular components designed to prevent inflammation and limit damage to the surrounding environment (Green 2011). During apoptosis, the plasma membrane forms “blebs,” allowing membraned fragments containing intracellular contents to separate from the larger, dying cells as “apoptotic bodies.” This blebbing is a morphological characteristic of apoptosis and also a key mechanism for confining danger-associated molecular patterns (or DAMPs) and avoid alerting the immune system (Green et al. 2009). Another hallmark of apoptosis is DNA fragmentation and chromatin condensation (Green 2011). These and other characteristics of apoptosis are largely controlled by a family of cysteine proteases with endopeptidase activity called caspases (Taylor et al. 2008).
Caspases exist only in the animal kingdom and are broadly grouped into initiator caspases (caspase-8 and caspase-9), executioner caspases (caspase-3, caspase-6, and caspase-7), and inflammatory caspases (human caspase-1, human caspase-4, and human caspase-5; rodent caspase-1 and rodent caspase-11) (Taylor et al. 2008; Green 2011). The main function of caspases is to cleave proteins to ensure an efficient yet rapid cell death. How then does the cell tolerate such a lethal family of proteases? First of all, caspases only cleave at specific sequence residues that end in aspartate residues (hence the “asp” of caspase) (Kumar 2007). Secondly, caspases exist in inactive forms, requiring dimerization (initiator caspases) or cleavage (executioner caspases) to gain activity (Kumar 2007; Taylor et al. 2008). Thirdly, caspases are not the only proteins involved in regulation of apoptosis (discussed below).
Upstream of apoptotic signaling events, initiator caspases exist as inactive monomers that must be dimerized to become active. This process is known as an “induced proximity model” and results in autocatalytic cleavage and stabilization of the dimer (Muzio et al. 1998; Boatright et al. 2003). Once active, initiator caspases are capable of cleaving numerous targets, most notably the executioner caspases (Green 2011). Executioner caspases exist as inactive dimers and cleavage by proteases, mainly initiator caspases, occurs between the large and small subunits, resulting in a conformational change that brings the two active sites of the executioner caspase dimer together to create a functional mature protease. These active executioner caspases can now cleave and activate other executioner caspases, leading to a rapid feedback loop to facilitate apoptosis (Riedl and Shi 2004).
What triggers this cascade of lethal events? Depending on the adapters and initiator caspases involved, most apoptotic programs fall into either the intrinsic or the extrinsic category. The major mechanism of apoptosis in mammals is the intrinsic or mitochondrial pathway. This pathway is activated by a variety of stress-inducing stimuli, including growth factor deprivation, cytoskeletal disruption, DNA damage, accumulation of unfolded proteins, and hypoxia, as well as developmental signals that instruct cells to die, such as hormones (Brenner and Mak 2009; Green 2011). These signals converge on the mitochondria, where the balancing act between pro-apoptotic and anti-apoptotic members of the BCL2 family orchestrates mitochondrial outer membrane permeabilization (MOMP) to release cytochrome c and other deadly sequestered proteins from within the mitochondria (Tait and Green 2010). The intricacies of BCL2 family interactions are discussed in detail in a number of other sources (Tait and Green 2010; Green 2011; Llambi and Green 2011; Llambi et al. 2011).
The result of MOMP and the release of cytochrome c is the formation of the apoptosome, a multimeric complex comprised of cytochrome c, caspase-9, and the adaptor APAF-1. The formation of the apoptosome activates caspase-9, which leads to the activation of the downstream executioner caspases, such as caspase-3 and caspase-7 (Shiozaki et al. 2002). The importance of the intrinsic apoptotic pathway is highlighted by the developmental phenotypes of gene-targeted mice deficient for components of this pathway. Unsurprisingly, mice deficient for caspase-9, APAF-1, or caspase-3 suffer from large brain outgrowths, characterized by reduced neuronal cell apoptosis, and subsequent perinatal lethality (Colussi and Kumar 1999). Thus, the fate of proper vertebrate development lies in the ability of one organelle, the mitochondria, to know when to maintain its integrity and when to release the mediators of death.
The extrinsic pathway of apoptosis, however, is triggered by signals that engage extracellular death receptors (DR). Signals such as tumor necrosis factor (TNF), CD95-ligand (CD95-L or Fas-L), and TNF-related apoptosis-inducing ligand (TRAIL) bind the DRs TNF receptor-1 (TNFR1), CD95 (or Fas), and TRAIL-R1/2 (DR4/5), respectively (Green 2011). Engagement of a DR ligand with its cognate receptor results in the recruitment of pro-caspase-8 to the death-inducing signaling complex (DISC) formed at the cytoplasmic tail of the engaged DR that also includes the adaptor proteins FADD or TRADD. Recruitment of these caspase-8 monomers results in dimerization and activation, leading to activity of caspase-3 and caspase-7 (Ashkenazi and Dixit 1998; Boatright et al. 2003). Caspase-8 activity can also cleave and activate BCL2 family proteins to trigger the intrinsic apoptotic pathway (discussed above) to induce efficient cell death (Tait and Green 2010; Green 2011).
Perhaps the most important result of apoptosis is the careful organization and packaging of potentially immunogenic cellular component into discreet, tolerogenic pieces. While it is easy to appreciate the appropriate execution of apoptosis in terms of proper development and suicide of infected cells, the real victory is sustained cellular renewal in the absence of immune activation. As described below, not all forms of cell death are tolerated as well.
2.2 Necrosis
While apoptosis is considered a genetically controlled and immunologically silent mechanism of cell death, necrosis has been characterized as a passive type of cell death, resulting in organelle swelling and cellular explosion that uncontrollably releases inflammatory cellular contents (Nikoletopoulou et al. 2013). Mechanistically, classical necrosis is typically not associated with a genetic program and occurs independently of caspase activation (Leist and Jaattela 2001). Unlike apoptosis, which is critical for proper development, necrosis is thought to mediate cellular death in response to catastrophic damage or pathology, including infarction, mechanical trauma, ischemia, frostbite, and animal venom (Raffray and Cohen 1997). Likewise, apoptotic cells that are not efficiently cleared by phagocytes can undergo secondary necrosis, a process that occurs completely independently of any apoptotic machinery.
Various molecules in our cells are pro-inflammatory if they are released from cells. Collectively referred to as damage-associated molecular patterns (DAMPs) or alarmins, they can activate neighboring macrophages and dendritic cells through TLR signaling and other mechanisms (Kono and Rock 2008). Morphologically, necrotic cells are characterized by cellular swelling (oncosis), nuclear distension, and plasma membrane rupture (Green 2011; Nikoletopoulou et al. 2013). This explosion of cellular contents, including proposed DAMPs such as HMGB1, HDGF, nucleotides, metabolites, and uric acid, is typically associated with an inflammatory reaction (Nikoletopoulou et al. 2013). Importantly, DAMP release and recognition might help alert the immune system to a cell-death-inducing pathogen, but if triggered inappropriately, it can also have deleterious effects, including autoimmunity. Importantly, both the extent and type of cell death represent major means of regulating DAMP release. When one considers the trauma that triggers necrotic death, it is unsurprising that inflammation follows, whereas apoptosis, occurring in a structured setting during carefully timed points in development, proceeds without alarm. Our understanding of necrotic cell death is evolving though, as a new programmed form of necrosis, termed necroptosis, has recently been described.
2.3 Necroptosis
Like its name suggests, necroptosis is the marriage between the programmed nature of apoptosis and the morphological features of necrosis. As mentioned previously, mice deficient for caspase-3 or caspase-9 die perinatally and are characterized by an excess of neuronal cells and large brain outgrowths (Colussi and Kumar 1999). Intriguingly, mice deficient for caspase-8, or its anti-apoptotic homologue, FLIP, die at embryonic day 10.5 and have embryonic vascular, cardiac, and hematopoietic defects (Varfolomeev et al. 1998). Additionally, pharmacological inhibition of caspase-8 or knockdown of caspase-8 or FLIP via short interfering RNA (siRNA) sensitizes fibroblasts to TNF-induced necrotic death (Vercammen et al. 1998). This surprising observation, wherein the absence of a pro-apoptotic gene resulted in a deficit of cells, led researchers to examine alternate roles for caspase-8.
In the absence of caspase-8, ligation of the death receptor pathway of apoptosis, such as the TNF-TNFR pathway, can result in necrotic cell death and requires the kinase activity of receptor-interacting protein kinase-1 (RIPK1) and RIPK3 (Oberst et al. 2011; Weinlich and Green 2014). Strikingly, the embryonic lethality of the caspase-8-deficient mouse is fully rescued by co-ablation of receptor-interacting protein kinase-3 (RIPK3), and caspase-8/RIPK3 double knockout mice are developmentally normal, but develop a severe lymphoaccumulative disorder resembling that of mice or humans lacking Fas or FasL (Bidere et al. 2006; Kaiser et al. 2011; Oberst et al. 2011).
How then does engagement of the extrinsic pathway of apoptosis result in necroptosis? RIPK1 plays a paradoxical role in the survival of the cell. Ligated death receptors, such as TNFR1 (or TLR engagement of TRIF (Feoktistova et al. 2011; Dillon et al. 2014)), promote the recruitment and deubiquitination of RIPK1 to the adaptor TRADD. Together with TRAF2 and the ubiquitin ligases cIAP1, cIAP2, and LUBAC, this complex, termed “Complex I,” can activate the NF-κB signaling pathway, which can induce the expression of FLIP (Zhang et al. 2000; Micheau et al. 2001; Newton 2015). Subsequent formation of a cytoplasmic “Complex II” or “Ripoptosome” containing RIPK1, FADD, and caspase-8 drives apoptotic signaling upon the ligation of death receptors. However, in the absence of extrinsic apoptotic machinery, RIPK1 (or more specifically, the kinase activity of RIPK1) can promote necroptosis via RIPK3 activation. Therefore, while it has been known that FLIP blocks caspase-8-mediated apoptosis, the catalytically active complex of FADD, caspase-8, and FLIP also blocks signaling for necroptosis (Oberst et al. 2011; Newton 2015). Furthermore, inactive RIPK1 can block RIPK3 activity, even when RIPK3 is activated independently of RIPK1 (Oberst et al. 2011).
The activation of RIPK1 and RIPK3 is not the final executioner of the necroptosis pathway, however. Necroptosis also depends on the RIPK3-mediated phosphorylation of the pseudokinase, mixed lineage kinase-like (MLKL) (Kaiser et al. 2013; Rodriguez et al. 2015). Phosphorylated MLKL induces a conformational change that allows for its oligomerization and interaction with the plasma membrane by binding to phosphatidylinositol lipids to directly disrupt membrane integrity (Wang et al. 2014; Rodriguez et al. 2015).
The involvement of RIPK1 adds another layer of complexity. In addition to its role in necroptosis, the kinase activity of RIPK1 is required for a variety of innate immune signaling pathways, such as Toll-like receptors (TLRs), interferons, and the RIG-I-MAVS pathway (Dillon et al. 2014; Weinlich and Green 2014). Whereas ablation of RIPK3 rescued caspase-8-deficient animals, RIPK1/caspase-8 double knockout mice die perinatally at day 1, similar to the RIPK1−/− mice (Dillon et al. 2014). During embryonic development, RIPK1 can trigger TNFR-mediated necroptosis in animals lacking apoptotic machinery, such as FADD, FLIP, or caspase-8, and RIPK1-mediated lethality at later developmental stages that could be mediated by similar signals (termed “signal 1”). Postnatally, RIPK1 can serve two functions: (1) RIPK1 is required to prevent a TNFR1-induced apoptosis, possibly due to its role in NF-κB activation and subsequent FLIP upregulation; and (2) RIPK1 is also required to prevent RIPK3-dependent lethality promoted by other “signal 2,” such as TRIF and IFN (Dillon et al. 2014). The balance and contribution of RIPK1 to cell survival, cell death, and immunity is certainly multifaceted and is discussed in elegant detail in a recent review (Weinlich and Green 2014; Newton 2015).
In addition to the role of RIPK1 in directly regulating inflammatory pathways, necroptosis has been reported to itself be an immunogenic type of cell death, and its inhibition, either genetically or with small-molecule inhibitors, has been demonstrated to lessen disease severity in several mouse models (Weinlich and Green 2014; Newton 2015). Furthermore, necroptosis is very common in vivo, not only in physical traumas, but also mainly in diverse forms of neurodegeneration, and death inflicted by ischemia or infection. Similar to classical necrosis, necroptosis results in the release of intracellular danger signals into the extracellular milieu, and these components can stimulate the immune system. Indeed, animals deficient for the necroptotic pathway are protected from various models of inflammation, such as chemically induced pancreatitis, intestine and skin inflammation, or ischemic reperfusion injury (Linkermann et al. 2013; Weinlich et al. 2013; Wu et al. 2013).
2.4 Pyroptosis
Previously, we discussed the caspases as mediators of apoptosis, but some caspases, such as caspase-1 and caspase-11 (or caspase-5 in humans), have roles in non-apoptotic, biological processes. The best-described function for caspase-1 is its key role in the processing of inactive IL-β and IL-18 into mature inflammatory cytokines. Additionally, excessive caspase-1 activity can cause pyroptosis, a non-apoptotic type of programmed cell death (LaRock and Cookson 2013; Jorgensen and Miao 2015). Execution of pyroptosis differs from apoptosis both at the biochemical and at the morphological level. Although caspase-1 can trigger apoptosis, caspase-1-mediated pyroptosis does not result in the cleavage of substrates of typical caspases. Rather, activated caspase-1 activates caspase-7 (Bergsbaken et al. 2009). Unlike apoptosis, MOMP is typically not associated with pyroptosis. Pyroptosis is characterized by lysis of the plasma membrane and the release of pro-inflammatory intracellular contents. Interestingly, nuclear DNA undergoes extensive fragmentation, similar to that observed in apoptosis, although the mechanism by which this occurs remains unknown (LaRock and Cookson 2013).
Regardless of its pro-inflammatory or pro-pyroptotic outcome, caspase-1 is activated by dimerization at complexes termed inflammasomes that form in the cytosol and detect a diverse repertoire of pathogenic molecules, including bacterial toxins and viral RNA (Henao-Mejia et al. 2012). However, how active caspase-1 kills a cell remains a complete mystery, in the sense that no key substrates have been identified that would account for this rapid deadly event. We can speculate, though, on the evolutionary roots of caspase-1’s dual roles. Bacterial and viral pathogens can subvert caspase-1-mediated IL-1β and IL-18 processing, thus dampening the host inflammatory response and facilitating infection (Bergsbaken et al. 2009; Jorgensen and Miao 2015). Could pyroptosis that represents a strategy by the infected host redirect the activity of caspase-1 toward killing the cell? Alternatively, it is possible that pyroptosis is required for the release of the mature, inflammatory cytokines.
Similar to other types of lytic cell death discussed above, pyroptosis might indeed be pro-inflammatory, as it results in the release of intracellular danger signals. While caspase-1 activation is required for cell death in a variety of experimental settings, including in the immune system, the cardiovascular system, and the central nervous system, mice deficient for caspase-1 develop normally, implying that this protease is redundant in vivo during development (Green 2011; McIlwain et al. 2015). It was recently reported, however, that caspase-1-deficient mice generated from strain 129 embryonic stem cells also harbor a mutation in the caspase-11 locus, and so are in fact caspase-1/caspase-11 double knockout mice (Kayagaki et al. 2011). While it is clear that inflammation mediated by caspase-1 (or caspase-11) is a critical component of pathogen clearance and sepsis, what remains to be elucidated is the role of pyroptosis in vivo inflammatory pathologies, and whether a pyroptotic death is handled or recognized differently by the innate immune system.
3 The Dining Experience: The Mechanisms of Efferocytosis
The meal is prepared, the table is set, but how do the diners know when and where to go? Phagocytes are not typically within close proximity to the dying cells, so they must be recruited and enticed to their cellular meal (Peter et al. 2010). But recruitment is just the beginning. Efferocytosis is a carefully orchestrated process wherein phagocytes, both professional (macrophages and dendritic cells) and non-professional (epithelial cells), migrate toward areas of cell death, recognize and engage cellular corpses via surface molecules specific to dead cells, and internalize, usually in its entirety, a dead cell for degradation and processing (Parnaik et al. 2000; Poon et al. 2014). For apoptotic cells, this is an immunologically tolerable event, as phagocytes recognize cells that have undergone this cell death program as biologically inert and dispose of these corpses before they release their potentially immunogenic intracellular contents, such as DNA (Nagata et al. 2010).
We now recognize that dying cell clearance is an important factor in many different disease models, including SLE, atherosclerosis, and Alzheimer’s disease (Camins et al. 2008; Ravichandran 2010). Thus, there has been a tremendous effort to understand the specific steps by which dying cells are recognized and cleared, as well as how these processes shape the immune response. Efferocytosis can be generally categorized into 4 steps: (1) the recruitment of phagocytes by “find-me” signals; (2) the recognition and engagement of “eat-me” signals; (3) the engulfment of the cellular corpse (Fig. 1); and (4) the processing, degradation, and immune response to the engulfed corpse (Fig. 2).
Fig. 1.
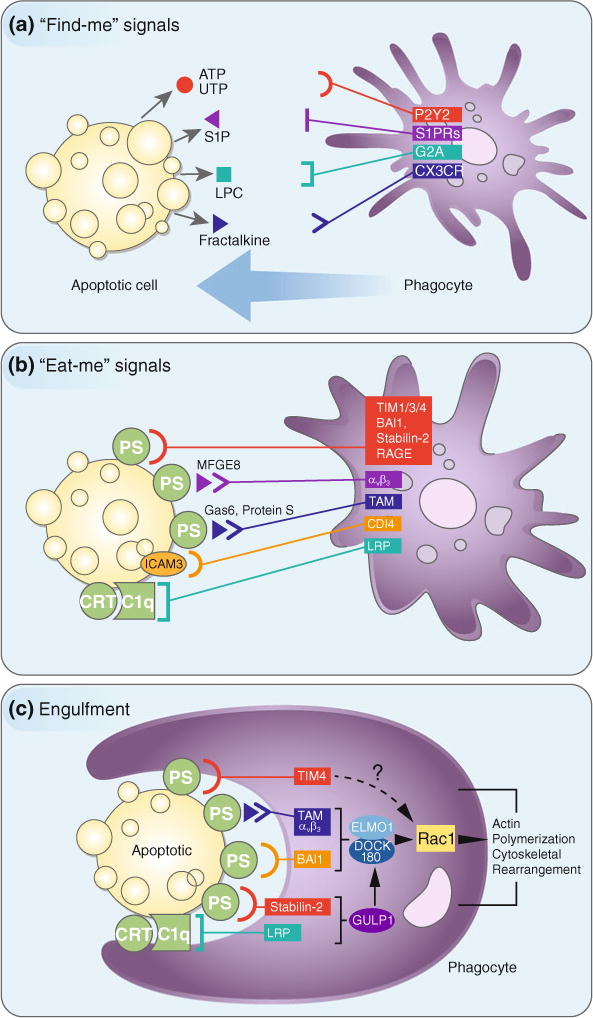
The recruitment of phagocytes to sites of cell death and recognition and engulfment of apoptotic cells by phagocytes. a Apoptotic cells (or other dying cells) release “find-me” signals, such as ATP, UTP, S1P, LPC, or fractalkine, that act to recruit phagocytes to sites of cell death. Phagocytes sense these “find-me” signals via cognate receptors (P2Y2, S1PRs, G2A, and CXCR3, respectively). b Phagocytes employ a system of receptors and bridging molecules to recognize and engage apoptotic cells (or other dying cells) via “eat-me” signals exposed on apoptotic cell surfaces. The most common “eat-me” signal is phosphatidylserine (or PS), which engages the PS-specific receptors, TIM1, TIM3, TIM4, BAI1, stabilin-2, and RAGE, as well as the PS-specific bridging molecules MFG-E8, Gas6, and protein S. These bridging molecules engage other surface engulfment receptors (αvβ3 or TAM) to facilitate uptake. Other “eat-me” signals, such as calreticulin (CRT) and ICAM3, exist and mediate recognition and engulfment via the receptors LRP (via C1q) and CD14, respectively. c Once the engulfment receptors are engaged, actin polymerization and cytoskeletal rearrangement are initiated via activation of the Rac1 pathway. While some engulfment receptors utilize the ELMO1/DOCK180 complex (αvβ3, TAM, stabilin-2, LRP), the mechanism by which TIM4 activates the Rac1 pathway is unknown. Perturbations at any step of this process can result in inflammation and autoimmunity
Fig. 2.
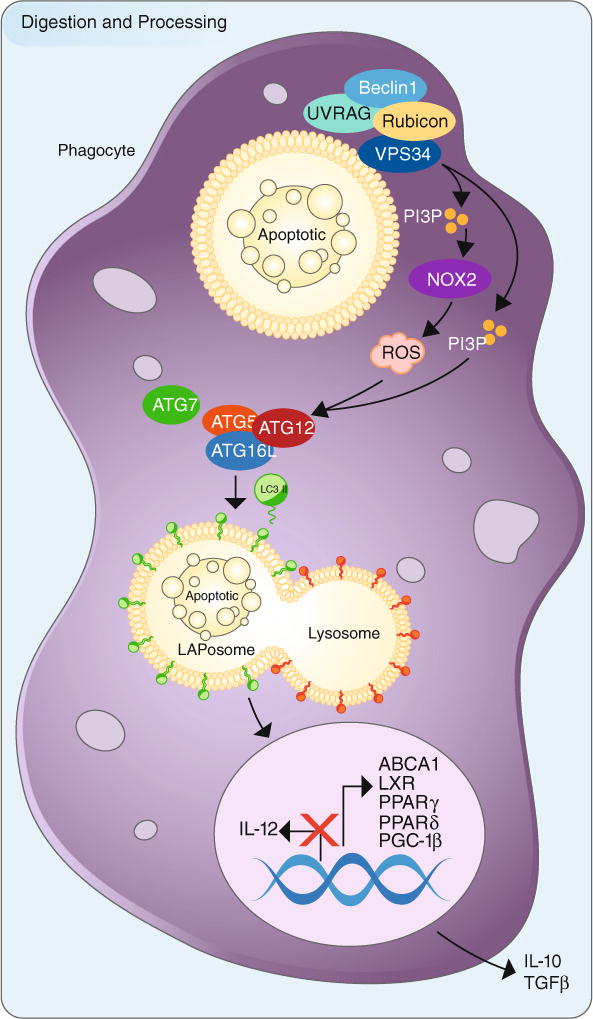
The processing and digestion of engulfed apoptotic cells utilizes LC3-associated phagocytosis and promotes an anti-inflammatory response. Upon engulfment of apoptotic cells (or other dying cells), components of the LC3-associated phagocytosis (LAP) pathway are recruitment to dead cell-containing phagosome (or LAPosome). The Class III PI3 K complex, composed of Beclin-1, VPS34, UVRAG, and Rubicon, is critical to the sustained and localized production of PI(3)P at the LAPosome. PI(3)P serves two roles—the recruitment of the downstream autophagic/LAP machinery (such as ATG5, ATG12, ATG16L, ATG7) and stabilization of the NOX2 complex for the production of ROS. Of note, Rubicon is also required for the stabilization of the NOX2 complex. Both ROS and PI(3)P are required for lipidation and translocation of LC3-II to the single membrane of the LAPosome, and LC3-II is required for fusion to the lysosome and maturation of LAPosome. The anti-inflammatory effects of efferocytosis are mediated by the activity of lipid and cholesterol sensors, such as ABCA1, LXR, PPARγ, PPARδ, and PGC-1β, leading to the production of anti-inflammatory mediators, IL-10 and TGFβ. Pro-inflammatory mediators, such as IL-12, are actively repressed. Perturbations in this process can result in inflammation and autoimmunity
3.1 Perusing the Menu: “Find-Me” Signals
Even in tissues with high rates of cellular turnover, such as the thymus or bone marrow, uncleared apoptotic cells are rarely observed. Our earliest evidence of the phagocytic program of clearing dead cells comes from studies done in C. elegans, wherein phagocytes were recruited to sites of cell death and cleared dying cells before apoptosis (and hence complete death) was even fully executed (Reddien and Horvitz 2000; Hoeppner et al. 2001). Given that phagocytes do not often regularly reside in the tissues they must patrol, apoptotic cells must “advertise” their presence to phagocytes, essentially expediting their own clearance (Elliott et al. 2009).
Apoptotic cells release several distinct molecules, termed “find-me” signals, to attract phagocytic cells via a chemotactic gradient. To date, several potential “find-me” signals have been reported, although their relevance has not always been validated in vivo (Hochreiter-Hufford and Ravichandran 2013). B cells in germinal centers undergo increased rates of apoptosis during affinity maturation, and these apoptotic B cells release the membrane-associated molecule CX3CL1 (or fractalkine) in small vesicles or microparticles. This classical chemokine is sensed by CX3CR1 and mediates the migration of macrophages to the dying cells. However, mice deficient for CX3CR1 do not display a defect in apoptotic cell clearance in their germinal centers, indicating that other factors function to recruit phagocytic cells (Truman et al. 2008).
Lysophosphatidylcholine (LPC) is generated and released by the caspase 3-dependent activation of phospholipase A and was the first “find-me” signal of lipid origin (Lauber et al. 2003). LPS is sensed by the G-protein-coupled receptor G2A, and this interaction can stimulate macrophage recruitment (Peter et al. 2008). Similarly, the lipid sphingosine-1-phosphate (S1P) is secreted by apoptotic cells and sensed by multiple G-protein-coupled receptors S1P-R1-5 to mediate phagocyte chemotaxis (Gude et al. 2008). These lipids, however, are present in the circulation at a concentration higher than that released by apoptotic cells, and their function in vivo has not been assessed. Therefore, their activity is likely to be merely local.
Perhaps the most promising candidates as “find-me” signals are nucleotides. ATP and UTP are released in a caspase-dependent manner via activation of pannexin 1 channels (Chekeni et al. 2010) and are detected by phagocytic cells via purinergic receptors, like P2Y2. Moreover, disruption of the nucleotide/P2Y2 interaction results in an accumulation of apoptotic thymocytes following glucocorticoid treatment in vivo (Elliott et al. 2009). It should be noted that the release of nucleotides from apoptotic cells, while an active process, is significantly smaller (less than 2 % of intracellular ATP levels) than the release that occurs during necrosis (Ravichandran 2010). Furthermore, these released nucleotides are easily degraded by extracellular nucleotidases. Thus, nucleotides most likely act to recruit tissue-resident phagocytes in a short-range capacity.
A number of caveats exist for the theory of “find-me” signals. Many cell types of different origins and function express receptors for these “find-me” signals, yet the vast majority of cells recruited to sites of cell death are macrophages. Is recruitment based on a synergistic effect of these signals or do additional signals exist to regulate migration? Lactoferrin, a glycoprotein released by apoptotic cells, has been shown to act as a “keep-out” signal, excluding neutrophils and eosinophils from sites of cell death (Bournazou et al. 2009, 2010). However, the lactoferrin-deficient animal model has yet to be characterized, so its in vivo role remains unknown. Additionally, the relatively low concentration of these “find-me” signals begs the question—what is the range of chemotactic gradients and how are circulating phagocytes recruited to sites of cell death (Elliott et al. 2009)? While these signals are often caspase-dependent (and hence apoptosis-dependent), these molecules are also released during other forms of cell death, such as necrosis or necroptosis (Iyer et al. 2009), often in greater quantities than in apoptosis. What are the migratory and inflammatory consequences of these disparate levels of potential “find-me” signals? Indeed, some of these “find-me” signals, such as ATP, can also act as a danger-associated molecular pattern (DAMP), thereby alerting the innate immune system. Necrosis, for example, results in the release of uric acid, which acts as both a “find-me” signal and an activating factor of inflammasomes (Kono et al. 2010). Do low levels of “find-me” signals promote macrophage migration, while higher concentrations stimulate a pro-inflammatory response?
Finally, in addition to chemotaxis, what other roles do these “find-me” signals play? There is evidence that sensing of these “find-me” signals, such as ATP or S1P, activates and prime phagocytes, increasing their phagocytic capability (Hanayama et al. 2004). These and other questions remain to be answered as studies unravel the mechanisms behind the active migration of phagocytes to their meal.
3.2 It is All in the Presentation: “Eat-Me” Signals
The ability to distinguish self from nonself is the defining hallmark of our immune system. Likewise, the ability to distinguish living cells from dead cells is critical to development, immunity, and the prevention of unwanted inflammation. How then do phagocytes, actively recruited to sites of cell death by “find-me” signals, target dying cells, while leaving healthy cells unperturbed? It stands to reason that the process of dying transforms healthy cells into targets for engulfment, rendering them distinguishable from living cells.
Lipid bilayers comprise the core structure of the plasma and organelle membranes, and the unique makeup of these different membranes confers distinct protein folding and permeability properties (Leventis and Grinstein 2010). Lipid distribution differs not only among membranes but frequently also between the two leaflets of the bilayer. Notably, the composition of the plasma membrane is an asymmetrical distribution of lipids, wherein the lipid phosphatidylserine (PS) is actively confined to the inner leaflet in viable cells (Balasubramanian and Schroit 2003). However, during apoptosis, PS is rapidly externalized in a caspase-dependent manner. This exposure occurs not only in mammals, but also in C. elegans (Venegas and Zhou 2007) and Drosophila (van den Eijnde et al. 1998). The calcium-mediated cation channel TMEM16F has been shown to mediate lipid scrambling (Suzuki et al. 2010), and recent studies have demonstrated that the scramblase Xkr8 is cleaved by caspase-3 and facilitates PS exposure during apoptosis (Suzuki et al. 2013). The flippase ATP11C normally transports aminophospholipids from the extracellular to the cytoplasmic side. During apoptosis, though, ATP11C is inactivated by caspase-3 cleavage, and PS remains externally exposed (Segawa et al. 2014).
Despite its relatively minor presence in most biological membranes, PS is a lipid of great physiological importance (Leventis and Grinstein 2010). Extracellularly exposed PS is the most well-characterized “eat-me” signal and an essential factor in the recognition and clearance of apoptotic cells (Balasubramanian and Schroit 2003). Phagocytes recognize exposed PS via membrane receptors, such as T cell immunoglobulin mucin receptor 4 (TIM4), brain-specific angiogenesis inhibitor 1 (BAI1), and stabilin-2 (Park et al. 2007, 2008a; Rodriguez-Manzanet et al. 2010). Additionally, there exist bridging molecules, such as milk fat globule-EGF factor 8 (MFG-E8) and Gas6, capable of recognizing PS and being recognized by phagocytic cell surface receptors such as integrin αvβ3, αvβ5, or Tryo3-Axl-Mer (or TAM) receptors (Ishimoto et al. 2000; Hanayama et al. 2002; Zizzo et al. 2012). Engagement of these receptors can result in cytoskeletal rearrangements that facilitate the engulfment of the cellular corpse (discussed below).
While a hallmark of cell death, PS is found extracellularly in low levels on living or activated cells, yet these cells are not engulfed (van den Eijnde et al. 2001). Even forced extracellular levels of PS on viable cells, via constitutively active TMEM16F, do not result in engulfment (Segawa et al. 2011). How then does a phagocyte distinguish a PS-positive dead cell, primed for clearance, from a PS-positive cell that should live to see another day? One answer may lie in the presence of “don’t eat-me” signals, such as CD31, CD47, and CD61. Engagement of these molecules, expressed on viable cells, can negatively regulate phagocytosis, thus signaling to the phagocyte that this cell, while PS-positive, is not intended for clearance (Oldenborg et al. 2000; Elward et al. 2005; Poon et al. 2014).
Further, PS is not the only “eat-me” signal identified. ICAM3, oxidized LDL-like molecules, glycosylated surface proteins, and C1q bound serum proteins have all been described to act as “eat-me” signals (Ravichandran 2010; Poon et al. 2014). The translocation of calreticulin (CRT) from the endoplasmic reticulum to the plasma membrane can also serve as an “eat-me” signal and stimulate engulfment by phagocytes (Gardai et al. 2005). While efferocytosis may be regulated by the balance of “eat-me” and “don’t eat-me” signals or the synergistic effect of multiple “eat-me” signals, it is clear that dead cells actively promote their own clearance to phagocytes that have evolved to recognize and remove such cells from circulation.
3.3 Savoring the Meal: Phagocytosis of Cellular Corpses
Efferocytosis is an intricately choreographed process requiring action by both the dying cells and the phagocyte. While the dying cell actively recruits phagocytes to sites of cell death via “find-me” signals and advertises its desire to be cleared via “eat-me” signals, the phagocyte facilitates the actual engulfment via engagement of receptors that specifically recognize these signals. As PS is the most characterized “eat-me” signal, PS receptors are the most characterized mechanism for dead cell recognition. While initial thinking hypothesized that a single PS receptor existed to mediate this recognition and phagocytosis, we now know that multiple PS receptors exist (Bratton and Henson 2008; Nagata et al. 2010; Poon et al. 2014). PS can be recognized via bona fide membrane receptors, such as Stablin-2 (Park et al. 2008a), BAI-1 (Park et al. 2007), RAGE (He et al. 2011), and TIM4 (as well as family members TIM1 and TIM3) (Miyanishi et al. 2007; Freeman et al. 2010; Rodriguez-Manzanet et al. 2010). Additionally, bridging molecules, such as MFG-E8 (Hanayama et al. 2004; Hu et al. 2009), protein S, and Gas6 (Rothlin et al. 2007; Lemke and Rothlin 2008), have been demonstrated to simultaneously recognize PS on dead cells and promote engulfment via engagement of their cognate membrane receptors. In order to facilitate phagocytosis, MFG-E8 associates with the integrins αvβ3 or αvβ5, while the Tyro3-Axl-Mer (TAM) family of receptors recognize protein S and Gas6 (Scott et al. 2001; Lemke and Rothlin 2008).
Just as other eat-me signals exist, so do their corresponding recognition receptors. For example, lectins can recognize modified glycoproteins and lipids (Ezekowitz et al. 1990); CD36, with αvβ3, can bind thrombospondin (Fadok et al. 1998b); scavenger receptors like SR-A can bind oxidized LDL-like moieties (Gordon 1999); CD14 can bind ICAM3 (Gregory et al. 1998); and CD91 (or LRP1) binds C1q via calreticulin (Gardai et al. 2005). Here, we will focus on the PS recognition system by phagocytes as a model for dead cell recognition and clearance.
Despite a common goal, PS receptors differ in their expression patterns, mode of PS recognition, and downstream signaling. In addition to professional phagocytes, these receptors are expressed in a variety of tissues, including bone marrow (BAI-1), spleen (BAI-1), brain (BAI-1), lungs (RAGE), kidney cells (TIM-1), and sinusoidal endothelial cells (stabilin-2) (Hochreiter-Hufford and Ravichandran 2013). The tissue specificity of these receptors may help to explain why multiple PS receptors are required for efficient efferocytosis, as different tissues may require specialized PS receptor mechanisms (Camins et al. 2008; Nagata et al. 2010; Poon et al. 2014). For example, defects in BAI-1, highly expressed in glial and neuronal cells, are associated with neurodegenerative disorders (Sokolowski and Mandell 2011), while stabilin-2 expression is highly expressed in endothelial cells within atherosclerotic plaques (Lee et al. 2011).
Likewise, recognition of PS by these molecules occurs via different domains. The TIM family of receptors utilizes an IgV domain (Santiago et al. 2007). BAI-1 binds PS via thrombospondin type 1 repeats (Park et al. 2007), while stabilin-2 contains EGF-like domains that mediate PS recognition (Park et al. 2008a). MFG-E8 binds PS via C1 and C2 domains (Hanayama et al. 2002). Why different receptors or bridging molecules have evolved different PS-binding motifs is unknown and adds an additional layer of complexity to the study of efferocytosis.
Engagement of these PS receptors (or surface receptors engaged by bridging molecules) results in cytoskeletal reorganization to facilitate phagocytosis. Studies to delineate the molecules involved in the engulfment of dead cells in C. elegans and subsequent identification of mammalian homologues have begun to clarify the intracellular signaling events that occur (Reddien and Horvitz 2004). The uptake of dead cells is mediated by the Rho family of small GTPases, including members RhoA, Rac, Rab5, and Cdc42 (Nakaya et al. 2006), which cycle between the resting, inactive GDP-bound state and the active GTP-bound state, mediated by specific guanine nucleotide exchange factors (GEFs).
The physical engulfment of apoptotic cells is morphologically distinct from other types of phagocytosis. Whereas particles engulfed via complement-receptor-mediated phagocytosis seem to be absorbed into the phagocyte, dead cell engulfment involves active membrane ruffling by a process similar to macropinocytosis (Olazabal et al. 2002; Riento and Ridley 2003). Specific Rho family GTPases are conversely activated or inactivated during phagocytosis. Unlike its role in complement-receptor-mediated phagocytosis, it has been demonstrated that RhoA negatively regulates engulfment, as inhibition of this GTPase enhances engulfment. Conversely, overexpression of RhoA inhibits engulfment, and this is dependent on signaling by the RhoA-binding protein, Rho-associated coiled-coil-containing protein kinase (ROCK) (Erwig et al. 2006). An increase in the kinase activity of ROCK affects the status of myosin light chain (MLC) phosphorylation, to promote actomyosin assembly and cell contractility (Riento and Ridley 2003). This decrease in RhoA activation, and in turn the decreased signaling via ROCK, decreases stress fiber formation and probably facilitates cell shape changes during engulfment (Erwig et al. 2006).
Signaling during apoptotic cell engulfment converges on evolutionarily conserved pathway that leads to Rac1 activation, though the molecules used by specific receptors to activate the Rac1 pathway can differ. Unlike RhoA, Rac1 activation translates into membrane ruffles that are necessary to promote phagocytosis (Nakaya et al. 2006). Similarly, CDC42 has been linked to the engulfment of apoptotic cells, although its precise role is unclear (Leverrier et al. 2001)
Engagement of integrins, such as αvβ3 or αvβ5, or Mer by bridging molecules recruits the cytoplasmic protein, CrkII, which then associates with the adaptor proteins ELMO1 and DOCK180 to the phagocytic cup (Albert et al. 2000; Wu et al. 2005). Together, DOCK180 and ELMO1 form a bipartite GEF, which activates Rac1 (Brugnera et al. 2002). BAI1 also requires the activity of the DOCK180/ELMO1 complex for Rac1 activation, but BAI1 is able to recruit and bind ELMO1 independently (Park et al. 2007). The importance of DOCK180 and ELMO1 is highlighted by experiments in C. elegans wherein loss of the ELMO1 homologue (CED-12) results in failure to clear apoptotic cells during development (Wu et al. 2001). Moreover, overexpression of DOCK180 and ELMO1 results in greatly increased Rac1 activity and phagocytic capacity (Brugnera et al. 2002).
Not all “eat-me” signal receptors require the DOCK180/ELMO1 complex for Rac1 activation, though. Stabilin-2 requires the activity of the adaptor protein, GULP, to activate the Rac1 pathway (Park et al. 2008b, 2010b). Likewise, CD91/LRP, which binds calreticulin, interacts with GULP (Su et al. 2002). TIM4, however, contains a very short cytoplasmic region, which is not necessary for Rac1 activation, and currently, the signaling components downstream of TIM4 are unknown (Park et al. 2009).
Regardless of the path, Rac1 activation is a critical point in the engulfment process. GTP-bound Rac acts at sites of apoptotic cell recognition to promote Arp2/3 activation/actin polymerization/cytoskeletal rearrangement via the Scar/WAVE complex (Miki et al. 1998; Castellano et al. 2000). Once engulfment is complete, however, the job of phagocyte is not over. It has merely just begun.
3.4 Aperitifs and Digestifs: Digestion and Immune Response
Once encased inside of the phagocyte, the dead cell is now cargo destined for digestion and degradation. Late-stage RhoA activation is thought to promote apoptotic cell digestion by regulating the acidification of phagosomes (Erwig and Henson 2008). Similarly, GDP-bound small GTPase, Rab5, is recruited to the phagosome and activated by yet-unidentified GEFs. This GTP-bound, active Rab5 promotes activity of the Class III PI3 K, VPS34, which generates PI(3)P on the phagosomal membrane (Kinchen 2010). Recent studies connect Rab5 activation to Rab7 recruitment to the phagosome (Nordmann et al. 2010). Subsequently, the HOPS complex is recruited to the phagosome and activates Rab7, resulting in fusion of the phagosome with the lysosomal network (Kinchen et al. 2008).
Once the phagosome becomes mature via lysosomal fusion, acidic proteases and nucleases get activated and the apoptotic cell targets are degraded into their basic cellular components including fats, sterols, peptides, and nucleotides. DNAse II is a lysosomal enzyme required for the degradation of DNA, and DNAse II deficiency results in an accumulation of undigested DNA fragments within phagocytic cells, as well as polyarthritis and inflammation in joint tissues (Kawane et al. 2001, 2006).
But like any meal, it can have a profound effect on the eater. An area of growing interest is how a phagocyte handles the metabolic stress of ingesting a cellular corpse and essentially doubling its content of cellular components. One such component, cholesterol, can have a profound effect on the phagocyte’s response to engulfed dead cells. In order to maintain their homeostasis in the face of increased cholesterol, phagocytes increase their basal cholesterol efflux activity from the cell (Noelia et al. 2009). Engagement of PS receptors leads to the activation of peroxisome proliferator-activated receptor γ/δ (PPARγ/δ) and liver x receptor (LXR) families, both important regulators of cellular lipid homeostasis and the clearance of apoptotic cells (Mukundan et al. 2009; Roszer et al. 2011). This induction results in the upregulation of phagocytic receptors, such as members of the TAM family, and basal cholesterol efflux machinery, such as 12-transmembrane protein ABCA1 (ATP-binding cassette subfamily A, member 1), to accommodate the increase in cholesterol associated with engulfment (Han and Ravichandran 2011). Moreover, PPARγ−/− and PPARδ−/− macrophages show a defect in apoptotic cell uptake. The dual functions of PPARs and LXRs in both lipid apoptotic cell clearance and lipid homeostasis suggest the interconnectedness between efferocytosis and metabolism.
Apoptotic cell death occurs in healthy organisms as part of normal tissue turnover and thus needs to be immunologically silent with regard to its resolution (Henson and Hume 2006). One of the hallmarks of apoptotic cell clearance is its non-inflammatory and non-immunogenic nature, and cholesterol homeostasis plays a critical role in this tolerant pathway (Hochreiter-Hufford and Ravichandran 2013; Poon et al. 2014). Phagocytes that have engulfed apoptotic cells have been shown to secrete anti-inflammatory cytokines, such as TGFβ and interleukin-10 (IL-10) (Fadok et al. 1998a, 2001). Moreover, the uptake of apoptotic cells actively suppressed pro-inflammatory cytokines, such as tumor necrosis factor (TNF), IL-1 and IL-12 (Kim et al. 2004). Intriguingly, PPARγ and PPARδ are central players in the polarization of M2 macrophages, the phenotype of which is anti-inflammatory. Agonists for both PPARγ and LXR have been shown to inhibit inflammatory responses (Mukundan et al. 2009; Noelia et al. 2009; Poon et al. 2014).
How then do apoptotic cells program their engulfers to tolerate their presence? Engulfment of necrotic cells or opsonization of corpse debris via FcR-mediated phagocytosis does not induce enhanced cholesterol efflux in the phagocytes, despite providing excess cholesterol for the engulfing cells (Kiss et al. 2006). These data suggest it is not the burden of extra cholesterol, but the engagement of ligands on apoptotic cells that induce a “prophylactic” cholesterol efflux from phagocytes. One key ligand seems to be the exposed PS on apoptotic cells. Coculture with mere apoptotic membranes or PS liposomes can induce the cholesterol efflux, anti-inflammatory cytokine production, and suppression of pro-inflammatory genes (Huynh et al. 2002; Kim et al. 2004). Collectively, these data suggest that recognition of apoptotic cells via engagement of “eat-me” signals, specifically PS, contributes to the silent clearance of these cells. Although PS recognition seems to be relevant for such immune tolerance, the roles of other “eat-me” signals and cognate receptor(s), as well as other signaling pathways involved, are unknown.
Despite engaging PS receptors, necrotic, necroptotic, or pyroptotic cells do not elicit an anti-inflammatory response (Martinez et al. 2011, 2013). Moreover, lytic cell death, such as necrosis, necroptosis, and pyroptosis, often results in an increased release of “find-me” signals (Ravichandran 2010). In the absence of efficient efferocytosis, apoptotic cells will undergo secondary necrosis and lyse, causing an inflammatory response (Juncadella et al. 2013). Of note, it is hypothesized that merely caspase activation results in dampening of the immunogenicity of DAMPS, and therefore, apoptotic cells that have transitioned through secondary necrosis are not inflammatory (Kazama et al. 2008; Luthi et al. 2009). However, recent data have shown that apoptotic cells can indeed be inflammatory if they are not phagocytosed in a timely manner (Obeid et al. 2007; Michaud et al. 2011).
How the phagocyte handles the ingested corpse in terms of its processing, degradation, and subsequent influence on the pursuant immune response is an area of growing interest. From the perspective of a single-celled organism, the two ancient systems of phagocytosis and autophagy represent two modes of nutrient acquisition—phagocytosis when extracellular fuel is abundant and autophagy when nutrients are scarce. However, these scenarios become decidedly more complex when one considers the engulfment of pathogens or dead cells (Martinez et al. 2013). The discovery of LC3-associated phagocytosis (LAP) has shed some light on this issue. LAP is a process that marries the evolutionarily conserved pathways of phagocytosis and autophagy into a fundamentally new concept, allowing us to reimagine the impact of the autophagy machinery on innate host defense mechanisms. LAP is triggered wherein an extracellular particle, such as a pathogen, immune complex, or dead cell, is sensed and phagocytosed, and this engulfment recruits some, but not all, members of the autophagy machinery to the cargo-containing, single-membraned vesicle (Sanjuan et al. 2007; Martinez et al. 2011). Engagement of multiple types of receptors, including TLR1/2, TLR2/6, TLR4, FcR, and TIM4 has been shown to induce translocation of autophagy machinery and ultimately LC3 to the cargo-containing phagosome, termed the LAPosome (Florey et al. 2011; Martinez et al. 2011; Henault et al. 2012; Martinez 2015). It is the activity of these autophagic players that facilitates the rapid destruction of the cargo via fusion with the lysosomal pathway. This is not macroautophagy, per se, but a distinct process, and it is triggered upon phagocytosis of particles containing ligands that engage a receptor-mediated signaling pathway.
Molecularly, LAP differs from canonical autophagy in multiple ways. LAP proceeds independently of the preinitiation complex, composed of ULK1/2, FIP200, and ATG13, whereas autophagy requires its activity (Florey et al. 2011; Martinez et al. 2011). Both LAP and autophagy utilize the Class III PI3 K complex and its core components Beclin-1 and VPS34, but LAP exclusively utilizes the UVRAG-containing Class III PI3 K complex (Martinez et al. 2015). In addition to the Class III PI3 K complex, LAP also requires Rubicon (RUN domain protein as Beclin-1 interacting and cysteine-rich containing), which negatively regulates autophagy, via its inhibition of VPS34 (Matsunaga et al. 2009; Zhong et al. 2009) or by blocking GTPase Rab7 activation (Sun et al. 2010). Rubicon associates constitutively with the UVRAG-containing Class III PI3 K complex (Matsunaga et al. 2009), as well as the NOX2 complex, also required for LAP (Martinez et al. 2015). Rubicon is crucial for both the interaction of the Class III PI3 K complex with the LAPosome and subsequent PI(3)P production as well as promoting the production of ROS via the NOX2 complex (Yang et al. 2012; Martinez et al. 2015).
Importantly, LAP can have a profound effect on the immune response to the engulfed material. LAP deficiency results in a failure to efficiently degrade intraphagosomal yeast in vitro (Sanjuan et al. 2007) or clear Aspergillus fumigatus in vitro or in vivo (Martinez et al. 2015). Furthermore, lungs and serum from animals with LAP deficiency display increased levels of pro-inflammatory cytokines when challenged intranasally with Aspergillus fumigatus (Martinez et al. 2015). Additionally, LAP is a critical regulator of the type I interferon response to immune complexes (IC) by plasmacytoid dendritic cells (pDC). Engagement of the FcγR by the IC induces LAP, resulting in LC3 translocation to the IC-containing phagosome in an ATG5- and ATG7-dependent, but ULK1-, FIP200-, and ATG13-independent manner. This failure to translocate LC3 results in a failure to acquire a late-endolysosomal phenotype, and subsequently a failure to form the specialized IRF7-signaling compartment required for TLR9-mediated activation of interferon regulatory factor 7 (IRF7). IFN-α production was completely ablated in ATG7−/−, but not ULK1−/−, pDC in response to DNA-IC, suggesting that LAP could affect the functional immune response elicited by this autoantigen (Henault et al. 2012).
LAP can act as a critical defense mechanism against autoimmune responses. Whereas much has been explored in terms the link between the uptake of dead cells with autoimmunity, how the phagocyte degrades the engulfed corpse is also a critical component to preventing unwanted immune responses. Billions of cells die daily as a result of stress, infection, or normal homeostasis, and it is the responsibility of the phagocytes of the immune system, such as macrophages, to rid the body of these cellular corpses, thus preventing inflammation and autoimmunity (Han and Ravichandran 2011; Martinez et al. 2011). Importantly, LAP has been demonstrated to play a critical role in the efficient clearance of dying cells, as well as promoting the anti-inflammatory response to apoptotic cells. Engagement of the PS receptor, TIM4, results in recruitment of the autophagic machinery to the dead-cell-containing, single-membrane phagosome. Macrophages deficient for ATG7, but not ULK1, fail to recruit LC3 to the phagosome, which results in failures in phagosomal acidification and subsequent corpse degradation. Whereas the phagocytosis of apoptotic cells is generally considered an “immunologically silent” event, ATG7-deficient macrophages produce dramatically increased levels of IL-1β and IL-6 when fed apoptotic cells. Moreover, these ATG7-deficient macrophages produce significantly less anti-inflammatory cytokines, such as IL-10, upon such engulfment (Martinez et al. 2011). How the LAP pathway modulates the immune response to apoptotic cells remains to be elucidated. The process of phagosome maturation and its effect on the immune response are actively being studied, in order to further understand the role of dead cell clearance in the prevention of autoimmunity and other pathologies (discussed below).
4 Food Poisoning: Pathologies Associated with Aberrant Efferocytosis
The lack of detectable apoptotic cells under physiological conditions speaks to the sheer efficiency of efferocytosis. Conversely, but not surprisingly, many different diseases are characterized by the presence of uncleared dead cells or a defect in properly handling engulfed cellular corpses. Furthermore, the unwanted inflammatory response to uncleared dead cells can exacerbate some conditions (Hochreiter-Hufford and Ravichandran 2013; Poon et al. 2014). Non-resolving inflammation contributes to tissue damage and organ dysfunction in a wide array of pathologies. Accumulating evidence indicates that efferocytosis is impaired in many autoimmune and inflammatory disorders.
4.1 Systemic Lupus Erythematosus (SLE)
Perhaps the most common pathological disorder associated with aberrant dead cell clearance is systemic lupus erythematosus (SLE). SLE is a chronic systemic autoimmune disorder affecting the skin, lungs, kidneys, and central nervous system. Patients with SLE display persistence of apoptotic cells within lymph nodes, blood, and skin (Baumann et al. 2002). As discussed earlier, apoptotic cells, while regarded immunologically silent initially, can undergo secondary necrosis if cleared in efficiently. This results in rupture of the protective plasma membrane and the release of intracellular autoantigens, such as DNA, ATP, and HMGB1, normally compartmentalized within an apoptotic cell (Raffray and Cohen 1997; Peter et al. 2010; Nikoletopoulou et al. 2013). SLE patients show a strong correlation between disease progression and the failed clearance of apoptotic cells, as well as increased inflammation (Liu and Davidson 2012). SLE patients also contain elevated levels of DNA or nucleosomes in the circulation, as well as autoantibodies that are specific for nuclear or other “self” components (Rumore and Steinman 1990). These autoantibodies can bind to circulating autoantigens, forming immune complexes that accumulate or deposit in the glomerular and vessel walls of the kidney, causing lupus nephritis (Berden 1997; van Bruggen et al. 1997).
It has been demonstrated that mice with deficiencies for engulfment (and hence clearance) of apoptotic cells, such as MFGE8-, BAI1-, TIM4-, or MerTK-deficient animals, accumulate apoptotic corpses within lymph nodes and develop an SLE-like disease that involves autoantibody formation, splenomegaly, and glomerulonephritis (Cohen et al. 2002; Hanayama et al. 2004; Park et al. 2007; Rodriguez-Manzanet et al. 2010). Furthermore, genetic polymorphisms and aberrant splicing of MFGE8 have been reported in a small subset of patients with SLE, indicating that this pathway of apoptotic cell recognition and clearance could be deregulated in some patients (Hu et al. 2009). Similarly, deficiencies in components of the complement pathway, particularly C1q which plays a key role in apoptotic cell clearance, have been also associated with SLE (Botto et al. 1998).
Not only is phagocytic capacity critical for the prevention of SLE, but the efficient degradation of engulfed dead cells is also an important factor. Mice deficient for DNAse I, critical for the degradation of chromatin, are prone to SLE-like glomerulonephritis (Napirei et al. 2000). Intriguingly, genome-wide association studies have identified polymorphisms in atg5 (Zhou et al. 2011) and possibly atg7 (Clarke et al. 2014), genes involved in both canonical autophagy and LAP (Mizushima 2007; Florey et al. 2011; Martinez et al. 2011, 2015; Henault et al. 2012; Kim et al. 2013), as predisposition markers for SLE.
4.2 Rheumatoid Arthritis
Rheumatoid arthritis is a chronic autoimmune disease associated with progressive joint destruction. It is a systemic inflammatory disorder characterized by increased circulating autoantibodies against citrullinated peptides or the complement protein, C3 (Luban and Li 2010; Kenyon et al. 2011). While there exists little direct evidence that rheumatoid arthritis is caused by defects in efferocytosis, site of inflammation often contain DAMPS, such as HMGB1 or histones H3 and H4, characteristic of uncleared apoptotic cells (Friggeri et al. 2012). These components can bind to phagocytes and inhibit efferocytosis, therefore perpetuating progression to secondary necrosis and the release of immunostimulatory materials (Hurst et al. 1983; Friggeri et al. 2010). Mice deficient for DNAse II, and thus defective for the degradation of engulfed cellular cargo, develop polyarthritis and anemia associated with significant increased inflammatory markers, reminiscent of human rheumatoid arthritis (Kawane et al. 2006) While direct genetic links to efferocytosis have not been described for human rheumatoid arthritis, studies have demonstrated that increasing the levels of bridging molecules for TAM receptor or activating the LXR/PPARγ can have therapeutic benefits in mouse models of inflammatory arthritis (Park et al. 2010a).
4.3 Type 1 Diabetes
Type 1 diabetes (T1D) is a T cell-mediated autoimmune disease that results from destruction of the insulin-producing β-cells in the islets of Langerhans in the pancreas. The clearance of apoptotic cells (a source of self-antigens) by phagocytes, predominantly dendritic cells, induces a tolerogenic response. However, defective clearance of cellular corpses, however, can result in increased rate of β-cells apoptosis, inflammation, and loss of tolerance, as inefficiently cleared apoptotic cells undergo necrosis, releasing danger signals and autoantigens into the extracellular milieu (Vives-Pi et al. 2015).
One of the key factors in T1D is the loss of T cell tolerance to self-antigens. While the mechanisms by which efferocytosis induces selective immunosuppression are not fully understood, recent evidence indicates that impaired clearance of dying cells contributes to immunogenic, not tolerogenic, DC maturation and chronic inflammation. Previous work has demonstrated that macrophages from non-obese diabetic (NOD) mice, which spontaneously develop type 1 diabetes mellitus, have a profound defect in the phagocytosis of apoptotic cells in vitro. NOD mice also display impaired efferocytosis in vivo when challenged with apoptotic stimuli. This defect in apoptotic cell clearance by NOD mice also translated into increased production of antinuclear autoantibodies (ANA) (O’Brien et al. 2006). Another study demonstrated that the tolerogenic behavior of dendritic cells after islet cells efferocytosis is central to silencing autoreactive T cells in type I diabetes (Pujol-Autonell et al. 2013).
Impaired wound healing is a common complication encountered by patients with both type 1 and type 2 diabetes mellitus. A large number of neutrophils are recruited to the wound site but must be cleared adequately by macrophages to initiate the next stage of wound healing. Macrophages isolated from diabetic wounds display dysfunctional efferocytosis, resulting in increased dead cell burden at the wound site, increased inflammation, and delayed wound healing (Maruyama et al. 2007; Khanna et al. 2010). While it has been demonstrated that diabetes is associated with compromised efferocytosis and high levels of pro-inflammatory cytokines, the mechanisms underlying these defects have not been characterized.
4.4 Atherosclerosis
Atherosclerosis is associated with chronic inflammation of the vascular wall, predominantly caused by the recruitment and accumulation of monocytes, macrophages, and dendritic cells (DCs). These phagocytic cells engulf oxidized lipids, causing the lipid-laden cells to undergo apoptosis, wherein they themselves become engulfed by neighboring phagocytes (Tabas 2005; Ley et al. 2011). While efficient efferocytosis can compensate during the initial onset of atherosclerosis, mature atherosclerotic lesions (known as plaques) are characterized by the presence of foam cells, macrophages that have taken up necrotic cells at the core of plaques and failed to stimulate cholesterol efflux (Vucic et al. 2012; Poon et al. 2014). Pathologically, this results in further reduced clearance of apoptotic cells, secondary necrosis, expansion of plaque lesions, and eventual plaque rupture, leading directly to acute coronary syndromes and stroke in humans (Schrijvers et al. 2005; Tabas 2005).
Mouse models with defects in the efferocytosis machinery, such as MerTK-, MFG-E8-, or C1q-deficient models, display an accumulation of apoptotic cellular debris within plaques and exacerbated atherosclerosis (Tabas 2005; Bhatia et al. 2007; Thorp et al. 2008). Apoptotic cell uptake via interaction with ICAM3 can be bound and inhibited by oxidized lipids present in plaques (Miller et al. 2003).
While the correlation between decreased efferocytosis and atherosclerotic lesions is not incompletely understood, several clinical observations linked to possible mechanisms have been described. HMG-CoA reductase inhibitors (also known as statins) are commonly used pharmacological agents for the treatment of atherosclerosis and vascular disease. In addition to lowering cholesterol and inflammation, an additional mechanism of action of statins is the inhibition of RhoA, a negative regulator of engulfment that is highly expressed in atherosclerotic lesions (Loirand et al. 2006).
Downstream of engulfment, activation of the nuclear receptors liver X receptors (LXRs) and peroxisome proliferator-activated receptors (PPARs) are critical to promoting tolerance, as well as by upregulating MerTK expression. Moreover, synthetic agonists to LXR or PPARs have been demonstrated to be beneficial to the treatment of atherosclerosis (Noelia et al. 2009). Intricately linked to the pathological progression of atherosclerosis is cholesterol efflux. Uptake of apoptotic cells by phagocytes stimulates cholesterol efflux, primarily though upregulation of ABCA1, a critical molecule that transports free cholesterol from within the cells to lipid-poor apoA1 that is then modified in the plasma for transport to the liver and excretion (Oram and Heinecke 2005; Cuchel and Rader 2006). Indeed, decreased ABCA1 activity in mouse models promotes inflammation and atherosclerotic lesions. Conversely, overexpression of ABCA1 can dampen the inflammatory response and reverses the disease (Tang et al. 2009; Zhu et al. 2010). Finally, mice with deficiencies in the autophagic machinery, shared by the LAP pathway, such as LysM-Cre Atg5flox/flox mice, display more atherosclerotic lesions, suggesting the possibility that LAP-mediated processing is required for the prevention of inflammation (Liao et al. 2012; Razani et al. 2012).
4.5 Lung Inflammation
Upon the induction of lung inflammation, neutrophils, the most abundant cell type involved in the innate immune response, are quickly recruited airway to airway. Neutrophils, however, as a short-lived population, undergo apoptosis in order to prevent the release of histotoxic contents and subsequent damage to surrounding tissues. Clearance of these apoptotic neutrophils by phagocytes is central to the successful resolution of the inflammatory response (Fox et al. 2010). Both mouse and human models of chronic obstructive pulmonary disease and cystic fibrosis have demonstrated that impaired efferocytosis of dying neutrophils during inflammation can lead to a prolonged inflammatory response. Chronic obstructive pulmonary disease (COPD) is a common, yet complex disease, highly associated with cigarette smoke. Chronic inflammation, extracellular matrix destruction, and increased airway epithelial cell and neutrophil apoptosis are reported in COPD models, and alveolar macrophages from COPD patients demonstrate a decreased phagocytic capacity for apoptotic cells (Hodge et al. 2003). Cystic fibrosis (CF) lung disease is characterized by early, protracted inflammation associated with a massive influx of neutrophils and other inflammatory cells, and the efficient clearance of these inflammatory cells (now apoptotic) by phagocytes is critical to the resolution of inflammation. Similar to COPD patients, alveolar macrophages from CF patients display defective efferocytosis, possibly attributable to necrotic neutrophil-derived proteases capable of cleaving PS receptors on the surface of phagocytes (Vandivier et al. 2002a, b).
Human allergic asthma is a chronic inflammatory disorder of the airways and is characterized by airway inflammation, persistent airway hyperresponsiveness (AHR), and intermittent, reversible airway obstruction. “Airway remodeling,” including airway fibrosis, goblet cell hyperplasia, and other structural changes, is thought to be the result of chronic inflammation and serves to exacerbate the asthma symptoms (Nials and Uddin 2008). Alveolar macrophages from patients with sever asthma or poorly controlled asthma are defective in clearing apoptotic cells (Huynh et al. 2005; Fitzpatrick et al. 2008). Similarly, patients with non-eosinophilic asthma have increased numbers of neutrophils in the airways, and alveolar macrophages from these patients show an impaired ability to phagocytose apoptotic cells (Simpson et al. 2013).
While the exact mechanisms underlying defective phagocytosis in patients with severe asthma are not yet understood, the use of corticosteroids, the most common treatment in asthma, induces eosinophil apoptosis as well as eosinophil engulfment by macrophages in vitro, via the binding of protein S to apoptotic eosinophils and the upregulation of MerTK on the surface of macrophages (Liu et al. 1999; McColl et al. 2009). Unsurprisingly, this treatment is significantly less effective in non-eosinophilic asthma and other neutrophil-dominated lung inflammatory disorders (Vago et al. 2012; Poon et al. 2014).
4.6 Neurodegenerative Disorders
In the peripheral immune system, phagocytes tasked with efferocytosis are mainly comprised of macrophages, monocytes, and dendritic cells. In the brain, however, microglia act as resident macrophages to accomplish this function. Microglia have phenotypical similarities to peripheral macrophages, in that they express and utilize PS receptors for the recognition and uptake of dead cells (Witting et al. 2000). Multiple neurodegenerative disorders have been associated with defective efferocytosis, such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease (Mattson 2000). The most direct link of efferocytosis to neurodegenerative diseases is MFG-E8. Microglia express MFG-E8, and treatment of microglia cultures with fractalkine (CX3CL1) increases MFG-E8 mRNA levels (Leonardi-Essmann et al. 2005; Fuller and Van Eldik 2008). Inhibition of MFG-E8 in microglia cells resulted in decreased engulfment of apoptotic neurons (Fuller and Van Eldik 2008). Of note, it has also been reported that MFG-E8 also mediates the phagocytosis of viable, LPS-activated neurons, resulting in death of the engulfed neuron (Fuller and Van Eldik 2008; Fricker et al. 2012). Finally, MFG-E8 levels in the brains of the Tg2576 mouse model of Alzheimer’s disease were severely decreased with age compared to wild-type controls (Fuller and Van Eldik 2008). Taken together, MFG-E8 seems to play a central role in the clearance of dead cells in the brain, and deficiencies in MFG-E8 can contribute to the onset and severity of Alzheimer’s disease.
Other proteins that have tangential roles in efferocytosis have been implicated in neurodegenerative disorders. Apolipoprotein E (apoE) is a cholesterol transport protein expressed in liver, central nervous system, vascular smooth muscle cells, adrenals, macrophages, and adipocytes. The E4 isoform of apoE (apoE4) is a major genetic risk factor for multiple inflammatory metabolic diseases, including atherosclerosis and Alzheimer’s disease. Peritoneal macrophages isolated from APOE4 mice demonstrate defects in efferocytosis and increased inflammatory response, presumably through ER stress (Cash et al. 2012). How the cells of the brain mediate the clearance of potentially damaging dead cells, as well as other debris, is currently an area of great interest.
5 Conclusions
The controlled cell death program of apoptosis is an integral part of maintaining development and cell turnover, yet like a large meal, too much of a good thing can be detrimental. The sheer magnitude of the task undertaken by professional phagocytes to keep an organism free of potentially dangerous dead cells, and thus unwanted autoimmunity and inflammation, is a daunting one, but one performed with exquisite accuracy under normal conditions. Indeed, many autoimmune conditions have been clearly linked to defects in efferocytosis, in terms of recognition, engulfment, and digestion of its cellular corpse cargo. While the field of efferocytosis is relatively young, these new insights into the underlying mechanisms of dead cell clearance will provide invaluable opportunities to attack autoimmune and autoinflammatory diseases at its core. Bon appetit!
References
- Albert ML, Kim JI, Birge RB. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol. 2000;2(12):899–905. doi: 10.1038/35046549. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Balasubramanian K, Schroit AJ. Aminophospholipid asymmetry: a matter of life and death. Annu Rev Physiol. 2003;65:701–734. doi: 10.1146/annurev.physiol.65.092101.142459. [DOI] [PubMed] [Google Scholar]
- Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Berden JH. Lupus nephritis. Kidney Int. 1997;52(2):538–558. doi: 10.1038/ki.1997.365. [DOI] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, Boyle JJ, Haskard DO. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol. 2007;170(1):416–426. doi: 10.2353/ajpath.2007.060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidere N, Su HC, Lenardo MJ. Genetic disorders of programmed cell death in the immune system. Annu Rev Immunol. 2006;24:321–352. doi: 10.1146/annurev.immunol.24.021605.090513. [DOI] [PubMed] [Google Scholar]
- Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11(2):529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19(1):56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Bournazou I, Pound JD, Duffin R, Bournazos S, Melville LA, Brown SB, Rossi AG, Gregory CD. Apoptotic human cells inhibit migration of granulocytes via release of lactoferrin. J Clin Invest. 2009;119(1):20–32. doi: 10.1172/JCI36226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazou I, Mackenzie KJ, Duffin R, Rossi AG, Gregory CD. Inhibition of eosinophil migration by lactoferrin. Immunol Cell Biol. 2010;88(2):220–223. doi: 10.1038/icb.2009.86. [DOI] [PubMed] [Google Scholar]
- Bratton DL, Henson PM. Apoptotic cell recognition: will the real phosphatidylserine receptor(s) please stand up? Curr Biol. 2008;18(2):R76–R79. doi: 10.1016/j.cub.2007.11.024. [DOI] [PubMed] [Google Scholar]
- Brenner D, Mak TW. Mitochondrial cell death effectors. Curr Opin Cell Biol. 2009;21(6):871–877. doi: 10.1016/j.ceb.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, Macara IG, Madhani H, Fink GR, Ravichandran KS. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4(8):574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- Camins A, Pallas M, Silvestre JS. Apoptotic mechanisms involved in neurodegenerative diseases: experimental and therapeutic approaches. Methods Find Exp Clin Pharmacol. 2008;30(1):43–65. doi: 10.1358/mf.2008.30.1.1090962. [DOI] [PubMed] [Google Scholar]
- Cash JG, Kuhel DG, Basford JE, Jaeschke A, Chatterjee TK, Weintraub NL, Hui DY. Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J Biol Chem. 2012;287(33):27876–27884. doi: 10.1074/jbc.M112.377549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano F, Montcourrier P, Chavrier P. Membrane recruitment of Rac1 triggers phagocytosis. J Cell Sci. 2000;113(Pt 17):2955–2961. doi: 10.1242/jcs.113.17.2955. [DOI] [PubMed] [Google Scholar]
- Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467(7317):863–867. doi: 10.1038/nature09413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, Vyse TJ. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196(1):135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi PA, Kumar S. Targeted disruption of caspase genes in mice: what they tell us about the functions of individual caspases in apoptosis. Immunol Cell Biol. 1999;77(1):58–63. doi: 10.1046/j.1440-1711.1999.00788.x. [DOI] [PubMed] [Google Scholar]
- Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006;113(21):2548–2555. doi: 10.1161/CIRCULATIONAHA.104.475715. [DOI] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189–1202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP, Gasque P. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J Biol Chem. 2005;280(43):36342–36354. doi: 10.1074/jbc.M506579200. [DOI] [PubMed] [Google Scholar]
- Erwig LP, Henson PM. Clearance of apoptotic cells by phagocytes. Cell Death Differ. 2008;15(2):243–250. doi: 10.1038/sj.cdd.4402184. [DOI] [PubMed] [Google Scholar]
- Erwig LP, McPhilips KA, Wynes MW, Ivetic A, Ridley AJ, Henson PM. Differential regulation of phagosome maturation in macrophages and dendritic cells mediated by Rho GTPases and ezrin-radixin-moesin (ERM) proteins. Proc Natl Acad Sci U S A. 2006;103(34):12825–12830. doi: 10.1073/pnas.0605331103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezekowitz RA, Sastry K, Bailly P, Warner A. Molecular characterization of the human macrophage mannose receptor: demonstration of multiple carbohydrate recognition-like domains and phagocytosis of yeasts in Cos-1 cells. J Exp Med. 1990;172(6):1785–1794. doi: 10.1084/jem.172.6.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998a;101(4):890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Warner ML, Bratton DL, Henson PM. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3) J Immunol. 1998b;161(11):6250–6257. [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Guthrie L, Henson PM. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. J Immunol. 2001;166(11):6847–6854. doi: 10.4049/jimmunol.166.11.6847. [DOI] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AM, Holguin F, Teague WG, Brown LA. Alveolar macrophage phagocytosis is impaired in children with poorly controlled asthma. J Allergy Clin Immunol. 2008;121(6):1371–1378. doi: 10.1016/j.jaci.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011;13(11):1335–1343. doi: 10.1038/ncb2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun. 2010;2(3):216–227. doi: 10.1159/000284367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–189. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M, Neher JJ, Zhao JW, Thery C, Tolkovsky AM, Brown GC. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci. 2012;32(8):2657–2666. doi: 10.1523/JNEUROSCI.4837-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friggeri A, Yang Y, Banerjee S, Park YJ, Liu G, Abraham E. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol. 2010;299(6):C1267–C1276. doi: 10.1152/ajpcell.00152.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friggeri A, Banerjee S, Xie N, Cui H, De Freitas A, Zerfaoui M, Dupont H, Abraham E, Liu G. Extracellular histones inhibit efferocytosis. Mol Med. 2012;18:825–833. doi: 10.2119/molmed.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller AD, Van Eldik LJ. MFG-E8 regulates microglial phagocytosis of apoptotic neurons. J Neuroimmune Pharmacol. 2008;3(4):246–256. doi: 10.1007/s11481-008-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Gordon S. Macrophage-restricted molecules: role in differentiation and activation. Immunol Lett. 1999;65(1–2):5–8. doi: 10.1016/s0165-2478(98)00116-3. [DOI] [PubMed] [Google Scholar]
- Green DR. Means to an end: apoptosis and other cell death mechanisms. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2011. [Google Scholar]
- Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9(5):353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory CD, Devitt A, Moffatt O. Roles of ICAM-3 and CD14 in the recognition and phagocytosis of apoptotic cells by macrophages. Biochem Soc Trans. 1998;26(4):644–649. doi: 10.1042/bst0260644. [DOI] [PubMed] [Google Scholar]
- Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, Barbour SE, Milstien S, Spiegel S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008;22(8):2629–2638. doi: 10.1096/fj.08-107169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han CZ, Ravichandran KS. Metabolic connections during apoptotic cell engulfment. Cell. 2011;147(7):1442–1445. doi: 10.1016/j.cell.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417(6885):182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304(5674):1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- Hart SP, Dransfield I, Rossi AG. Phagocytosis of apoptotic cells. Methods. 2008;44(3):280–285. doi: 10.1016/j.ymeth.2007.11.009. [DOI] [PubMed] [Google Scholar]
- He M, Kubo H, Morimoto K, Fujino N, Suzuki T, Takahasi T, Yamada M, Yamaya M, Maekawa T, Yamamoto Y, Yamamoto H. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011;12(4):358–364. doi: 10.1038/embor.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Strowig T, Flavell RA. Inflammasomes: far beyond inflammation. Nat Immunol. 2012;13(4):321–324. doi: 10.1038/ni.2257. [DOI] [PubMed] [Google Scholar]
- Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, Coyle AJ, Kolbeck R, Green DR, Sanjuan MA. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–997. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 2006;27(5):244–250. doi: 10.1016/j.it.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5(1):a008748. doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81(4):289–296. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x. [DOI] [PubMed] [Google Scholar]
- Hoeppner DJ, Hengartner MO, Schnabel R. Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature. 2001;412(6843):202–206. doi: 10.1038/35084103. [DOI] [PubMed] [Google Scholar]
- Hu CY, Wu CS, Tsai HF, Chang SK, Tsai WI, Hsu PN. Genetic polymorphism in milk fat globule-EGF factor 8 (MFG-E8) is associated with systemic lupus erythematosus in human. Lupus. 2009;18(8):676–681. doi: 10.1177/0961203309103027. [DOI] [PubMed] [Google Scholar]
- Hurst NP, Nuki G, Wallington T. Functional defects of monocyte C3b receptor-mediated phagocytosis in rheumatoid arthritis (RA): evidence for an association with the appearance of a circulating population of non-specific esterase-negative mononuclear phagocytes. Ann Rheum Dis. 1983;42(5):487–493. doi: 10.1136/ard.42.5.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109(1):41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh ML, Malcolm KC, Kotaru C, Tilstra JA, Westcott JY, Fadok VA, Wenzel SE. Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am J Respir Crit Care Med. 2005;172(8):972–979. doi: 10.1164/rccm.200501-035OC. [DOI] [PubMed] [Google Scholar]
- Ishimoto Y, Ohashi K, Mizuno K, Nakano T. Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem. 2000;127(3):411–417. doi: 10.1093/oxfordjournals.jbchem.a022622. [DOI] [PubMed] [Google Scholar]
- Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106(48):20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. 2015;265(1):130–142. doi: 10.1111/imr.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, Ravichandran KS. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493(7433):547–551. doi: 10.1038/nature11714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471(7338):368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawane K, Fukuyama H, Kondoh G, Takeda J, Ohsawa Y, Uchiyama Y, Nagata S. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science. 2001;292(5521):1546–1549. doi: 10.1126/science.292.5521.1546. [DOI] [PubMed] [Google Scholar]
- Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443(7114):998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29(1):21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon KD, Cole C, Crawford F, Kappler JW, Thurman JM, Bratton DL, Boackle SA, Henson PM. IgG autoantibodies against deposited C3 inhibit macrophage-mediated apoptotic cell engulfment in systemic autoimmunity. J Immunol. 2011;187(5):2101–2111. doi: 10.4049/jimmunol.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna S, Biswas S, Shang Y, Collard E, Azad A, Kauh C, Bhasker V, Gordillo GM, Sen CK, Roy S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE. 2010;5(3):e9539. doi: 10.1371/journal.pone.0009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21(5):643–653. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, Ferguson TA. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154(2):365–376. doi: 10.1016/j.cell.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen JM. A model to die for: signaling to apoptotic cell removal in worm, fly and mouse. Apoptosis. 2010;15(9):998–1006. doi: 10.1007/s10495-010-0509-5. [DOI] [PubMed] [Google Scholar]
- Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, Hengartner MO, Ravichandran KS. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol. 2008;10(5):556–566. doi: 10.1038/ncb1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss RS, Elliott MR, Ma Z, Marcel YL, Ravichandran KS. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol. 2006;16(22):2252–2258. doi: 10.1016/j.cub.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8(4):279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest. 2010;120(6):1939–1949. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Vandenabeele P. Clearance of dead cells: mechanisms, immune responses and implication in the development of diseases. Apoptosis. 2010;15(9):995–997. doi: 10.1007/s10495-010-0524-6. [DOI] [PubMed] [Google Scholar]
- Kumar S. Caspase function in programmed cell death. Cell Death Differ. 2007;14(1):32–43. doi: 10.1038/sj.cdd.4402060. [DOI] [PubMed] [Google Scholar]
- LaRock CN, Cookson BT. Burning down the house: cellular actions during pyroptosis. PLoS Pathog. 2013;9(12):e1003793. doi: 10.1371/journal.ppat.1003793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113(6):717–730. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- Lee GY, Kim JH, Oh GT, Lee BH, Kwon IC, Kim IS. Molecular targeting of atherosclerotic plaques by a stabilin-2-specific peptide ligand. J Control Release. 2011;155(2):211–217. doi: 10.1016/j.jconrel.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2(8):589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8(5):327–336. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi-Essmann F, Emig M, Kitamura Y, Spanagel R, Gebicke-Haerter PJ. Fractalkine-upregulated milk-fat globule EGF factor-8 protein in cultured rat microglia. J Neuroimmunol. 2005;160(1–2):92–101. doi: 10.1016/j.jneuroim.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- Leverrier Y, Lorenzi R, Blundell MP, Brickell P, Kinnon C, Ridley AJ, Thrasher AJ. Cutting edge: the Wiskott-Aldrich syndrome protein is required for efficient phagocytosis of apoptotic cells. J Immunol. 2001;166(8):4831–4834. doi: 10.4049/jimmunol.166.8.4831. [DOI] [PubMed] [Google Scholar]
- Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31(7):1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15(4):545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Hackl MJ, Kunzendorf U, Walczak H, Krautwald S, Jevnikar AM. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant. 2013;13(11):2797–2804. doi: 10.1111/ajt.12448. [DOI] [PubMed] [Google Scholar]
- Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. 2012;18(6):871–882. doi: 10.1038/nm.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C, Dransfield I, Savill J, Rossi AG. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol. 1999;162(6):3639–3646. [PubMed] [Google Scholar]
- Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21(1):12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44(4):517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98(3):322–334. doi: 10.1161/01.RES.0000201960.04223.3c. [DOI] [PubMed] [Google Scholar]
- Luban S, Li ZG. Citrullinated peptide and its relevance to rheumatoid arthritis: an update. Int J Rheum Dis. 2010;13(4):284–287. doi: 10.1111/j.1756-185X.2010.01553.x. [DOI] [PubMed] [Google Scholar]
- Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, Taylor RC, Kersse K, Vandenabeele P, Lavelle EC, Martin SJ. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31(1):84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci USA. 2011;108(42):17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martinez J, Verbist K, Wang R, Green DR. The relationship between metabolism and the autophagy machinery during the innate immune response. Cell Metab. 2013;17(6):895–900. doi: 10.1016/j.cmet.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Mallireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, Kanneganti TD, Virgin HW, Green DR. Molecular characterization of LC3-associated phagocytosis (LAP) reveals distinct roles for Rubicon, NOX2, and autophagy proteins. Nature Cell Biol. 2015 doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Maruyama K, Asai J, Ii M, Thorne T, Losordo DW, D’Amore PA. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am J Pathol. 2007;170(4):1178–1191. doi: 10.2353/ajpath.2007.060018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1(2):120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- McColl A, Bournazos S, Franz S, Perretti M, Morgan BP, Haslett C, Dransfield I. Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. J Immunol. 2009;183(3):2167–2175. doi: 10.4049/jimmunol.0803503. [DOI] [PubMed] [Google Scholar]
- McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2015;7(4) doi: 10.1101/cshperspect.a026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, di Virgilio F, Zitvogel L, Kroemer G. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334(6062):1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21(16):5299–5305. doi: 10.1128/MCB.21.16.5299-5305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17(23):6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem. 2003;278(3):1561–1568. doi: 10.1074/jbc.M209634200. [DOI] [PubMed] [Google Scholar]
- Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, Goh YP, Eagle AR, Dunn SE, Awakuni JU, Nguyen KD, Steinman L, Michie SA, Chawla A. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15(11):1266–1272. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem. 1998;273(5):2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140(5):619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S. Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J Biol Chem. 2006;281(13):8836–8842. doi: 10.1074/jbc.M510972200. [DOI] [PubMed] [Google Scholar]
- Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25(2):177–181. doi: 10.1038/76032. [DOI] [PubMed] [Google Scholar]
- Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25(6):347–353. doi: 10.1016/j.tcb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Nials AT, Uddin S. Mouse models of allergic asthma: acute and chronic allergen challenge. Dis Model Mech. 2008;1(4–5):213–220. doi: 10.1242/dmm.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833(12):3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Noelia A-G, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31(2):245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordmann M, Cabrera M, Perz A, Brocker C, Ostrowicz C, Engelbrecht-Vandre S, Ungermann C. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr Biol. 2010;20(18):1654–1659. doi: 10.1016/j.cub.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien BA, Geng X, Orteu CH, Huang Y, Ghoreishi M, Zhang Y, Bush JA, Li G, Finegood DT, Dutz JP. A deficiency in the in vivo clearance of apoptotic cells is a feature of the NOD mouse. J Autoimmun. 2006;26(2):104–115. doi: 10.1016/j.jaut.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Olazabal IM, Caron E, May RC, Schilling K, Knecht DA, Machesky LM. Rho-kinase and myosin-II control phagocytic cup formation during CR, but not FcgammaR, phagocytosis. Curr Biol. 2002;12(16):1413–1418. doi: 10.1016/s0960-9822(02)01069-2. [DOI] [PubMed] [Google Scholar]
- Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288(5473):2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85(4):1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450(7168):430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008a;15(1):192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- Park SY, Kang KB, Thapa N, Kim SY, Lee SJ, Kim IS. Requirement of adaptor protein GULP during stabilin-2-mediated cell corpse engulfment. J Biol Chem. 2008b;283(16):10593–10600. doi: 10.1074/jbc.M709105200. [DOI] [PubMed] [Google Scholar]
- Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19(4):346–351. doi: 10.1016/j.cub.2009.01.042. [DOI] [PubMed] [Google Scholar]
- Park MC, Kwon YJ, Chung SJ, Park YB, Lee SK. Liver X receptor agonist prevents the evolution of collagen-induced arthritis in mice. Rheumatology (Oxford) 2010a;49(5):882–890. doi: 10.1093/rheumatology/keq007. [DOI] [PubMed] [Google Scholar]
- Park SY, Kim SY, Kang KB, Kim IS. Adaptor protein GULP is involved in stabilin-1-mediated phagocytosis. Biochem Biophys Res Commun. 2010b;398(3):467–472. doi: 10.1016/j.bbrc.2010.06.101. [DOI] [PubMed] [Google Scholar]
- Parnaik R, Raff MC, Scholes J. Differences between the clearance of apoptotic cells by professional and non-professional phagocytes. Curr Biol. 2000;10(14):857–860. doi: 10.1016/s0960-9822(00)00598-4. [DOI] [PubMed] [Google Scholar]
- Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, Wesselborg S, Lauber K. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem. 2008;283(9):5296–5305. doi: 10.1074/jbc.M706586200. [DOI] [PubMed] [Google Scholar]
- Peter C, Wesselborg S, Herrmann M, Lauber K. Dangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis. 2010;15(9):1007–1028. doi: 10.1007/s10495-010-0472-1. [DOI] [PubMed] [Google Scholar]
- Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14(3):166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol-Autonell I, Ampudia RM, Planas R, Marin-Gallen S, Carrascal J, Sanchez A, Marin A, Puig-Domingo M, Pujol-Borrell R, Verdaguer J, Vives-Pi M. Efferocytosis promotes suppressive effects on dendritic cells through prostaglandin E2 production in the context of autoimmunity. PLoS ONE. 2013;8(5):e63296. doi: 10.1371/journal.pone.0063296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffray M, Cohen GM. Apoptosis and necrosis in toxicology: a continuum or distinct modes of cell death? Pharmacol Ther. 1997;75(3):153–177. doi: 10.1016/s0163-7258(97)00037-5. [DOI] [PubMed] [Google Scholar]
- Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207(9):1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15(4):534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Horvitz HR. CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol. 2000;2(3):131–136. doi: 10.1038/35004000. [DOI] [PubMed] [Google Scholar]
- Reddien PW, Horvitz HR. The engulfment process of programmed cell death in caenorhabditis elegans. Annu Rev Cell Dev Biol. 2004;20:193–221. doi: 10.1146/annurev.cellbio.20.022003.114619. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5(11):897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4(6):446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2015 doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Manzanet R, Sanjuan MA, Wu HY, Quintana FJ, Xiao S, Anderson AC, Weiner HL, Green DR, Kuchroo VK. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci U S A. 2010;107(19):8706–8711. doi: 10.1073/pnas.0910359107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszer T, Menendez-Gutierrez MP, Lefterova MI, Alameda D, Nunez V, Lazar MA, Fischer T, Ricote M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011;186(1):621–631. doi: 10.4049/jimmunol.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Rumore PM, Steinman CR. Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J Clin Invest. 1990;86(1):69–74. doi: 10.1172/JCI114716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- Santiago C, Ballesteros A, Martinez-Munoz L, Mellado M, Kaplan GG, Freeman GJ, Casasnovas JM. Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity. 2007;27(6):941–951. doi: 10.1016/j.immuni.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2(12):965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25(6):1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411(6834):207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci U S A. 2011;108(48):19246–19251. doi: 10.1073/pnas.1114799108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344(6188):1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- Shiozaki EN, Chai J, Shi Y. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc Natl Acad Sci U S A. 2002;99(7):4197–4202. doi: 10.1073/pnas.072544399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JL, Gibson PG, Yang IA, Upham J, James A, Reynolds PN, Hodge SA, SR Group Impaired macrophage phagocytosis in non-eosinophilic asthma. Clin Exp Allergy. 2013;43(1):29–35. doi: 10.1111/j.1365-2222.2012.04075.x. [DOI] [PubMed] [Google Scholar]
- Sokolowski JD, Mandell JW. Phagocytic clearance in neurodegeneration. Am J Pathol. 2011;178(4):1416–1428. doi: 10.1016/j.ajpath.2010.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su HP, Nakada-Tsukui K, Tosello-Trampont AC, Li Y, Bu G, Henson PM, Ravichandran KS. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP) J Biol Chem. 2002;277(14):11772–11779. doi: 10.1074/jbc.M109336200. [DOI] [PubMed] [Google Scholar]
- Sun Q, Westphal W, Wong KN, Tan I, Zhong Q. Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A. 2010;107(45):19338–19343. doi: 10.1073/pnas.1010554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468(7325):834–838. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341(6144):403–406. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25(11):2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284(47):32336–32343. doi: 10.1074/jbc.M109.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/−mice. Arterioscler Thromb Vasc Biol. 2008;28(8):1421–1428. doi: 10.1161/ATVBAHA.108.167197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112(13):5026–5036. doi: 10.1182/blood-2008-06-162404. [DOI] [PubMed] [Google Scholar]
- Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC, Pinho V, Teixeira MM, Sousa LP. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J Leukoc Biol. 2012;92(2):249–258. doi: 10.1189/jlb.0112008. [DOI] [PubMed] [Google Scholar]
- van Bruggen MC, Kramers C, Walgreen B, Elema JD, Kallenberg CG, van den Born J, Smeenk RJ, Assmann KJ, Muller S, Monestier M, Berden JH. Nucleosomes and histones are present in glomerular deposits in human lupus nephritis. Nephrol Dial Transplant. 1997;12(1):57–66. doi: 10.1093/ndt/12.1.57. [DOI] [PubMed] [Google Scholar]
- van den Eijnde SM, Boshart L, Baehrecke EH, De Zeeuw CI, Reutelingsperger CP, Vermeij-Keers C. Cell surface exposure of phosphatidylserine during apoptosis is phylogenetically conserved. Apoptosis. 1998;3(1):9–16. doi: 10.1023/a:1009650917818. [DOI] [PubMed] [Google Scholar]
- van den Eijnde SM, van den Hoff MJ, Reutelingsperger CP, van Heerde WL, Henfling ME, Vermeij-Keers C, Schutte B, Borgers M, Ramaekers FC. Transient expression of phosphatidylserine at cell-cell contact areas is required for myotube formation. J Cell Sci. 2001;114(Pt 20):3631–3642. doi: 10.1242/jcs.114.20.3631. [DOI] [PubMed] [Google Scholar]
- Vandivier RW, Fadok VA, Hoffmann PR, Bratton DL, Penvari C, Brown KK, Brain JD, Accurso FJ, Henson PM. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest. 2002a;109(5):661–670. doi: 10.1172/JCI13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandivier RW, Fadok VA, Ogden CA, Hoffmann PR, Brain JD, Accurso FJ, Fisher JH, Greene KE, Henson PM. Impaired clearance of apoptotic cells from cystic fibrosis airways. Chest. 2002b;121(3 Suppl):89S. doi: 10.1378/chest.121.3_suppl.89s. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9(2):267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- Venegas V, Zhou Z. Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol Biol Cell. 2007;18(8):3180–3192. doi: 10.1091/mbc.E07-02-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187(9):1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Pi M, Rodriguez-Fernandez S, Pujol-Autonell I. How apoptotic beta-cells direct immune response to tolerance or to autoimmune diabetes: a review. Apoptosis. 2015;20(3):263–272. doi: 10.1007/s10495-015-1090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic E, Calcagno C, Dickson SD, Rudd JH, Hayashi K, Bucerius J, Moshier E, Mounessa JS, Roytman M, Moon MJ, Lin J, Ramachandran S, Tanimoto T, Brown K, Kotsuma M, Tsimikas S, Fisher EA, Nicolay K, Fuster V, Fayad ZA. Regression of inflammation in atherosclerosis by the LXR agonist R211945: a noninvasive assessment and comparison with atorvastatin. JACC Cardiovasc Imaging. 2012;5(8):819–828. doi: 10.1016/j.jcmg.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. 2014;56(4):469–480. doi: 10.1016/j.molcel.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich R, Oberst A, Dillon CP, Janke LJ, Milasta S, Lukens JR, Rodriguez DA, Gurung P, Savage C, Kanneganti TD, Green DR. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 2013;5(2):340–348. doi: 10.1016/j.celrep.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witting A, Muller P, Herrmann A, Kettenmann H, Nolte C. Phagocytic clearance of apoptotic neurons by microglia/brain macrophages in vitro: involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J Neurochem. 2000;75(3):1060–1070. doi: 10.1046/j.1471-4159.2000.0751060.x. [DOI] [PubMed] [Google Scholar]
- Wu YC, Tsai MC, Cheng LC, Chou CJ, Weng NY. C. elegans CED-12 acts in the conserved crkII/DOCK180/Rac pathway to control cell migration and cell corpse engulfment. Dev Cell. 2001;1(4):491–502. doi: 10.1016/s1534-5807(01)00056-9. [DOI] [PubMed] [Google Scholar]
- Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118(Pt 3):539–553. doi: 10.1242/jcs.01632. [DOI] [PubMed] [Google Scholar]
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, Yang Z, Wu SQ, Chen L, Han J. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23(8):994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS, Lee JS, Rodgers M, Min CK, Lee JY, Kim HJ, Lee KH, Kim CJ, Oh B, Zandi E, Yue Z, Kramnik I, Liang C, Jung JU. Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe. 2012;11(3):264–276. doi: 10.1016/j.chom.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12(3):301–311. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, Su Y, Li ZG, Zhang H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70(7):1330–1337. doi: 10.1136/ard.2010.140111. [DOI] [PubMed] [Google Scholar]
- Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ, Hiltbold EM, Fessler MB, Parks JS. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51(11):3196–3206. doi: 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. 2012;189(7):3508–3520. doi: 10.4049/jimmunol.1200662. [DOI] [PMC free article] [PubMed] [Google Scholar]