

SUMMARY
An oncogenic role for CHD4, a NuRD component, is defined for initiating and supporting tumor suppressor gene (TSG) silencing in human colorectal cancer. CHD4 recruits repressive chromatin proteins to sites of DNA damage repair, including DNA methyltransferases where it imposes de novo DNA methylation. At TSGs, CHD4 retention helps maintain DNA hypermethylation-associated transcriptional silencing. CHD4 is recruited by the excision repair protein OGG1 for oxidative damage to interact with the damage-induced base 8-hydroxydeoxyguanosine (8-OHdG), while ZMYND8 recruits it to double-strand breaks. CHD4 knockdown activates silenced TSGs, revealing their role for blunting colorectal cancer cell proliferation, invasion, and metastases. High CHD4 and 8-OHdG levels plus low expression of TSGs strongly correlates with early disease recurrence and decreased overall survival.
In Brief
Xia et al. show that CHD4 is recruited by OGG1 and ZMYND8, respectively, to interact with oxidative DNA damage sites and double-strand breaks. CHD4 recruits repressive chromatin proteins to these sites and helps maintain DNA hypermethylation-associated transcriptional silencing of tumor suppressor genes.
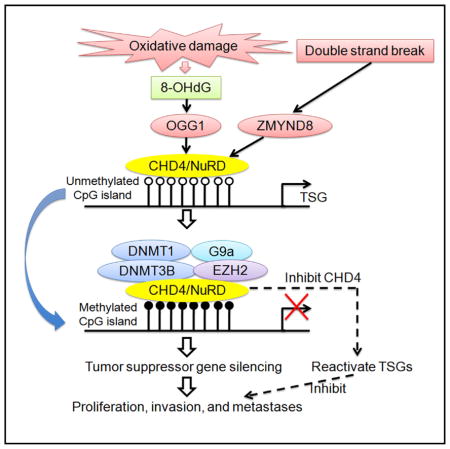
INTRODUCTION
Surviving ongoing DNA damage is a leading risk factor for many human cancers (Grivennikov et al., 2010) and involves need for cells to adapt to DNA damage, such as that induced by increased reactive oxygen species (ROS) (Reuter et al., 2010). These cellular adaptations involve selection of DNA damage repair (DDR) pathways to support the viability of pre-malignant cells and their eventual transformation (Scott et al., 2014).
Epigenetic events play a key role in proper DDR (Sulli et al., 2012). However, in past studies, we have stressed these might also facilitate evolution of cancer epigenetic abnormalities (Ding et al., 2016; O’Hagan et al., 2008; O’Hagan et al., 2011). Within minutes of inducing DNA double-strand breaks (DSBs) or exposing cells to ROS-induced damage, key components of transcriptional repression, including DNA methyltransferases (DNMTs), histone deacetylases (HDACs), and components of the long-term gene silencing polycomb group protein complex (PcG) are recruited to damage sites with consequent silencing of nascent transcription (O’Hagan et al., 2008; O’Hagan et al., 2011). Although normally these are probably transient events to ensure damage repair, gene promoters can retain abnormal DNA methylation and repressive chromatin (O’Hagan et al., 2008). A subset of genes are particularly vulnerable to such retention post oxidative stress (O’Hagan et al., 2011), and these encompass those biased to adopting abnormal, de novo DNA methylation in cancer cells and are developmental genes controlled by PcG in embryonic and adult stem cells (Ohm et al., 2007).
The chromodomain helicase DNA-binding protein 4 (CHD4), a key component of the nucleosome remodeling and histone de-acetylation (NuRD) complex, is essential in DDR and has been linked to oncogenic effects including inducing abnormal stem cell renewal, blunting differentiation, and altering cell-cycle control (Lai and Wade, 2011). Loss of CHD4 function can sensitize tumor cells to oxidative damage and the protein promotes genome stability by helping to regulate p53-dependent cell-cycle checkpoints (Polo et al., 2010). CHD4 can also protect replication forks in a poly(ADP-ribose) polymerase (PARP)-dependent manner, helping BRCA1- and BRCA2-deficient cells survive, perform DDR, and acquire drug resistance (Guillemette et al., 2015; Ray Chaudhuri et al., 2016). One defined role of CHD4 in DSB repair is to recruit repressive chromatin to open chromatin regions in active gene promoters (Chou et al., 2010; Larsen et al., 2010; Polo et al., 2010), serving to protect transcribed regions during repair (Chou et al., 2010; Larsen et al., 2010; Polo et al., 2010). These functions for CHD4 suggest a potential role for this protein to participate in initiating, and subsequently helping, maintain the epigenetic changes previously associated with DDR and onset of abnormal tumor suppressor gene (TSG) silencing in association with abnormal promoter CpG island DNA methylation (O’Hagan et al., 2008; O’Hagan et al., 2011).
We previously linked the known role of CHD4 for recruiting HDAC1 and HDAC2 with maintenance of TSG silencing mediated by abnormal promoter CpG island DNA methylation in cancer cells (Cai et al., 2014). These findings prompted us to query whether there is a link between CHD4 and DDR for recruiting the above-mentioned epigenetic modifier proteins to both single-strand breaks (SSBs), DSBs, and oxidative damage within promoter CpG islands of multiple important TSGs. If so, this recruitment might help mediate the appearance in these genes of repressive histone modifications, DNA methylation, and suppression of nascent gene transcription (O’Hagan et al., 2008; O’Hagan et al., 2011).
RESULTS
CHD4 Induces Recruitment of DNMT1, DNMT3A, and DNMT3B to Chromatin during Oxidative Damage
In reciprocal endogenous co-immunoprecipitations using human CRC and embryonic carcinoma cells, CHD4 interacts with DNMT1, DNMT3A, and DNMT3B with increasing intensity after hydrogen peroxide (H2O2)-induced oxidative damage (Figure S1A). Tightness of binding to chromatin also occurs for CHD4, DNMT1, DNMT3A, and DNMT3B without any change in their total cellular levels (Figures S1B–S1D). Knockdown of CHD4 significantly decreases this tightness of each protein after H2O2 treatment, but this does not occur for knockout of DNMT3A, and DNMT3B, or severe genetic disruption of DNMT1 in CRC cells (DNMT1 hypomorph) (Figures S1B–S1D). Thus, oxidative damage-induced increase in affinity of DNMT1, DNMT3A, and DNMT3B for chromatin is dependent on CHD4.
CHD4 Is Essential for Recruiting DNMT1, DNMT3A, and DNMT3B, with Onset of DNA Methylation, to SSBs and DSBs
In HCT116 cells, by 1 min after laser-induced SSBs and DSBs, endogenous DNMT3A and DNMT3B are detected at the damage sites. These proteins gradually accumulate during the first 5 min, are retained for at least 3 hr, and decrease thereafter (Figure 1A). The endogenous DNMT1 is also recruited with very similar dynamics, but decreases begin after 2 hr with subsequent complete disappearance (Figure 1A). CHD4 knockdown dramatically reduces recruitment of each of the above proteins (Figure 1A), but either knockout of DNMT3A and DNMT3B or severe genetic disruption of DNMT1 (DNMT1 hypomorph) has no effect on CHD4 recruitment (Figure 1B).
Figure 1. The Recruitment of DNMT1, DNMT3A, and DNMT3B to Laser-Induced DNA Damage Sites Is Dependent on CHD4.

(A and B) The indicated cells were micro-irradiated with a 455 nm laser and fixed at the indicated time points. The recruitment of endogenous DNMTs (A) and CHD4 (B) to DNA damage sites and their co-localization with γH2AX was examined by immunofluorescence staining. Representative images at the indicated time points are shown. The graphs below corresponding images represent the percentages of cells with γH2AX micro-irradiation tracks observed that have visible accumulation of DNMTs or CHD4 co-localizing with γH2AX.
(C) The indicated cells were micro-irradiated with a 455 nm laser and fixed at the indicated time points. The accumulation of endogenous 5mc at DNA damage sites and the co-localization with γH2AX was examined by immunofluorescence staining. The graphs represent the percentages of cells with co-localization of γH2AX and accumulation of 5mc at micro-irradiation tracks.
Data are represented as mean ± SEM for triplicate experiments. Scale bars, 10 μm. *p < 0.05. See also Figure S1.
DNMT recruitment can lead to early imposition of DNA methylation at DSBs (O’Hagan et al., 2008), and there is a key role for CHD4 in guiding this process. No 5-methylcytosine (5mc) appears at laser damage sites at 30 min but does so and accumulates over 1 hr to at least 6 hr (Figure 1C). CHD4 knockdown dramatically decreases this accumulation (Figure 1C). CHD4 recruitment of the de novo DNMTs, DNMT3A, and especially DNMT3B, is responsible for this DNA methylation, and DNMT3B knockout dramatically reduces it as severely as CHD4 knockdown (Figure 1C). In contrast, genetic disruption of DNMT1 has no such significant effect (Figure 1C).
CHD4 Tightens EZH2 and G9a to Chromatin and DDR Sites
CHD4 appears essential for recruiting DNMTs and proteins, such as enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), and establishing the repressive chromatin (O’Hagan et al., 2008; O’Hagan et al., 2011). By mass spectrometry, in SW480 cells before and after H2O2 treatment, several NuRD components and known CHD4-interacting proteins including DNMT1, PCNA, HDACs, and PARP1 (Cai et al., 2014; Helbling Chadwick et al., 2009; Pan et al., 2012; Tong et al., 1998) co-immunoprecipitate with CHD4 (Figures S2A–S2C). Also detected are several proteins not previously known to interact with CHD4 but known to interact with DNMTs, including chromatin modifiers (Figure S2D) and DDR proteins (Figure S2E). We further studied EZH2 and euchromatic histone lysine methyl-transferase 2 (EHMT2, also known as G9a) as these catalyze key repressive histone modifications H3K27me3 and H3K9me2, respectively, and both are associated with DNA-methylated gene promoters (Hansen et al., 2008; Shinkai and Tachibana, 2011). Both proteins interact with CHD4 in reciprocal endogenous co-immunoprecipitations (Figure 2A). Oxidative damage dramatically increases this interaction and the tightness of binding of EZH2 and G9a to chromatin (Figures 2A and 2B). CHD4 knockdown, without affecting total protein levels of either protein, dramatically decreases their chromatin tightening, while neither EZH2 nor G9a knockdown affects oxidative damage-induced affinity of CHD4 for chromatin (Figure 2B). Thus, CHD4 is upstream of these EZH2 and G9a interactions.
Figure 2. The Recruitment of EZH2 and G9a to DNA Damage Sites Is Dependent on CHD4.

(A) SW480 cells were not treated (Un) or treated with 2 mM H2O2 for 30 min. Immunoprecipitations and immunoblottings were performed using the indicated antibodies.
(B) The indicated cells were not treated (Un) or treated with 2 mM H2O2 for 30 min. Whole-cell extracts and the tight chromatin fractions were analyzed by immunoblotting.
(C–E) The indicated cells were micro-irradiated with a 455 nm laser and fixed at the indicated time points. The recruitment of endogenous EZH2 (C), G9a (D), and CHD4 (E) to DNA damage sites and their co-localization with γH2AX were examined by immunofluorescence staining. The graphs represent the percentages of cells with co-localization of γH2AX with EZH2, G9a, or CHD4 at micro-irradiation tracks. Scale bars, 10 μm.
(F) ChIP analyses of the indicated histone modifications at I-SceI-induced DSBs in SW480 DR-GFP cells infected with the indicated lentivirus. The y axis represents the relative enrichment of the indicated histone modifications compared with the immunoglobulin G (IgG) control.
Data are represented as mean ± SEM for triplicate experiments. *p < 0.05. See also Figure S2.
CHD4 also recruits, starting by 2 min, endogenous EZH2 and G9a to SSB and/or DSB sites, and the proteins continue to accumulate for at least 30 min after micro-irradiation. CHD4 knockdown leads to a dramatic reduction in this recruitment (Figures 2C and 2D). As for the other proteins studied, neither G9a nor EZH2 knockdown affects recruitment of endogenous CHD4 (Figure 2E).
Previously, we found that, for DSBs and ROS-induced damage at gene promoters, active histone modifications are quickly reduced and repressive markers are established (O’Hagan et al., 2008; O’Hagan et al., 2011). Others have also seen silencing of nascent transcription in induced DSBs (Shanbhag et al., 2010). When an exogenous construct is subjected to endonuclease cutting of an engineered I-SceI DNA recognition site (Pierce et al., 1999), CHD4, DNMT1, DNMT3A, DNMT3B, EZH2, and G9a are all enriched with γH2AX near the resultant DSBs (Figures S2F and S2G). Simultaneously, levels of the repressive markers, H3K27me3 and H3K9me2, are significantly increased while active markers, H3K4me3 and H4K16ac, markedly decrease at these sites (Figure 2F). Again, knockdown of CHD4 abolishes these protein-enrichment dynamics (Figure S2G) and preserves enrichment of the active H3K4me3 and H4K16ac markers while not affecting γH2AX enrichment (Figure 2F). Consistent with the earlier laser-induced DNA damage results, knockdown of DNMT1, DNMT3A, DNMT3B, EZH2, or G9a has no significant effect on the enrichment of CHD4 near the DSB sites (Figure S2H). Further, the recruitment of each of these proteins to DSB sites is not dependent on each other (Figure S2H). Similar results were also found for all of the above events in U2OS osteosarcoma cells (Figures S2I–S2K).
The ATPase Activity of CHD4 Is Required for Protein Recruitment to DDR Sites
Importantly, the ATPase activity domain of CHD4 is required for all of the protein-recruitment events described above (Figure 3A). This domain is responsible for the mobility and accessibility of CHD4 to nucleosomes and DNA (Becker and Horz, 2002). When endogenous CHD4 is depleted by Lenti-shCHD4-mediated interference, replacement of Flag-tagged short hairpin RNA (shRNA)-resistant wild-type CHD4, but not ATPase-dead CHD4, rescues the decreased recruitment of DNMTs, EZH2, and G9a to DNA damage sites (Figures 3A and 3B).
Figure 3. The ATPase Activity of CHD4 Is Required for the Recruitment of DNMTs and EZH2 and G9a to DNA Damage Sites.

(A) Cells were infected with Lenti-shCHD4 and lentivirus expressing shRNA-resistant CHD4-WT or ATPase-dead CHD4 (CHD4-DN). After 96 hr, cells were micro-irradiated with a 455 nm laser and fixed at the indicated time points. The recruitment of endogenous proteins to DNA damage sites and their co-localization with γH2AX were examined by immunofluorescence staining. The graphs represent the percentages of cells with co-localization of γH2AX with the indicated proteins at micro-irradiation tracks. Data are represented as mean ± SEM for triplicate experiments. Scale bars, 10 μm. *p < 0.05.
(B) Western blot analyses of the endogenous CHD4 and Flag-tagged CHD4 levels in the indicated cells.
Correlating the Upstream Functions of CHD4 in DDR with Repressive Chromatin, Gene Silencing, and Associated DNA Methylation at TSG Promoters
CHD4-mediated events are key for silencing of important TSGs and to our previous linking of CHD4 to cancer-specific abnormal promoter, CpG island DNA methylation (Cai et al., 2014). In chromatin immunoprecipitation (ChIP) assays, CHD4 is enriched at the CpG island in the promoters of 20 representative genes that are frequently hypermethylated and silenced in human CRC tissues and cell lines (Schuebel et al., 2007; van Engeland et al., 2011) (Figure S3A). In two CRC cell lines, CHD4 knockdown alone reactivates the expression of eight of these genes, E-cadherin (CDH1), WNT inhibitory factor 1 (WIF1), TIMP metallopeptidase inhibitor 2 (TIMP2), TIMP metallopeptidase inhibitor 3 (TIMP3), mutL homolog 1 (MLH1), cyclin-dependent kinase inhibitor 2A (CDKN2A), secreted frizzled related protein 4 (SFRP4), and secreted frizzled related protein 5 (SFRP5) (Figures S3A and S3B). Abnormal silencing of these genes potentially mediates escape from senescence, inhibiting proliferation, anti-WNT activity, and inhibiting invasion and metastases (Baylin and Jones, 2016; van Engeland et al., 2011).
Interactions between CHD4 and all of the proteins under study can be localized to DDR sites in the above genes when pure DSBs are induced in their promoters by a doxycycline-inducible system employing a FokI restriction endonuclease coupled to catalytically dead Cas9 (see Experimental Model and Subject Details) (Figures 4A and S3C). A significant portion of the cells repair the DSB over 24 hr (Figure S3C), during which there is enrichment of γH2AX, CHD4, DNMT1, DNMT3A, DNMT3B, EZH2, and G9a in the vicinity of the induced DSB site (Figures 4B, 4C, and S3D–S3I). In addition, 5mc and repressive markers H3K27me3 and H3K9me2 are increased, whereas active markers H3K4me3 and H4K16ac are decreased near the DSB site at the promoters of all the TSGs (Figures 4B, 4C, and S3D–S3I). CHD4 has an upstream role for these alterations, as its knockdown decreases recruitment of each epigenetic silencing protein and reduces accumulation of 5mc and the repressive markers H3K27me3 and H3K9me2 (Figures 4B–4D and S3D–S3I). This also negates the DSB-induced reduction in RNA transcripts of the eight TSGs (Figure S3J). These data together define a key role for CHD4-mediated recruitment to epigenetic silencing dynamics for DSB damage-induced TSG silencing.
Figure 4. Induction of DSBs at the Promoter CpG Islands of Endogenous TSGs Results in CHD4-Dependent Local Epigenetic Changes and Gene Silencing.

(A) Introduction of the doxycycline-inducible DSB system at the promoter CpG islands of endogenous TSGs. The pCW-dCas9/FokI construct expresses a dCas9/FokI fusion protein containing the FokI restriction endonuclease cleavage domain fused with a catalytically dead Cas9 (top), a working model of the doxycycline-inducible DSB system at the promoter CpG islands of endogenous TSGs (middle), and the time course for doxycycline treatment (bottom).
(B and C) Map of the doxycycline-inducible DSB sites at the endogenous promoter CpG islands of CDH1 (B) and WIF1 (C). Doxycycline-induced DSBs were monitored by a PCR assay with F1, R1 primers spanning the cut site. Four pairs of primers (F2, R2–F5, R5) were used for ChIP assays at the indicated distances from the DSB site (upper panels). ChIP analyses of the indicated protein enrichment, 5mc enrichment and histone modifications near DSB sites at the indicated time points (lower panels). Data are represented as mean ± SEM for triplicate experiments. *p < 0.05.
(D) Summary of the CHD4-mediated recruitment of epigenetic silencing proteins and epigenetic changes in the vicinity of DSB site at the endogenous promoters of eight representative TSGs.
See also Figures S3.
CHD4 binding of nuclear HDAC1 and HDAC2 is tied to its maintaining abnormal silencing of DNA hypermethylated TSGs (Cai et al., 2014). These dynamics are also mediated by CHD4 at damage sites in the above eight TSGs. Both endogenous HDAC1 and HDAC2 are recruited to laser-induced SSB and/or DSBs as early as 1 min, and accumulate for at least 15 min after micro-irradiation. CHD4 knockdown dramatically reduces this HDAC1 and HDAC2 recruitment (Figure S3K). In the doxycycline- inducible DSB system, both HDAC1 and HDAC2 also significantly increase near the DDR sites in TSG promoters, and CHD4 knockdown decreases this recruitment (Figure S3L). Furthermore, double knockdown of HDAC1 and HDAC2 reactivates the expression of these eight TSGs in two CRC cell lines (Figure S3M). In addition, as expected given its maintenance role for DNA methylation, knockdown of DNMT1, but not de novo DNMTs, DNMT3A and DNMT3B or the transcription repressing mediating proteins G9a or EZH2, results in increased expression of DNA hypermethylated genes tested in two different cell lines (Figures S3N and S3O).
CHD4 is involved in the recruitment of epigenetic silencing proteins to promoter CpG islands following oxidative DNA damage. After H2O2 treatment, CHD4, DNMT1, DNMT3A, DNMT3B, EZH2, and G9a are enriched at the promoter CpG islands of the eight TSGs (Figures S3P and S3Q), but not at non-CpG island-containing gene promoters (CXCL8 and NANOG) (Figure S3R). The enrichment of these proteins co-localizes with induced enrichment of the ROS damage product 8-hydroxydeoxyguanosine (8-OHdG). 5mc and the repressive histone modifications H3K27me3 and H3K9me2 are also differentially increased, whereas the active modifications H3K4me3 and H4K16ac are decreased. Again, knockdown of CHD4 reduces the H2O2- induced enrichment of 5mc and repressive histone modifications (Figure S3P) and rescues reductions in RNA transcription (Figure S3S). In addition, H2O2 treatment significantly decreased the transcription of promoter CpG island-containing genes that have high basal expression (MYC, ACTB, RPL13, and RPL10A), which is diminished by knockdown of CHD4, but not for non-CpG island-containing genes (CXCL8, HBD, MYH1, and LAMB4) (Figure S3T).
The Recruitment of CHD4 to Oxidative DNA Damage Sites Depends on 8-Oxoguanine DNA Glycosylase, OGG1
A key protein for repair of oxidative damage is OGG1, the base excision repair (BER) factor that removes the damage product 8-OHdG in GC-rich regions (Radicella et al., 1997). CHD4 forms a complex with OGG1, and H2O2-induced oxidative damage dramatically increases this interaction (Figure 5A). Immunoprecipitation assays using purified OGG1 and CHD4 proteins reveal that OGG1 directly interacts with CHD4 (Figure 5B). Furthermore, oxidative damage-induced increase in affinity of CHD4 for chromatin is dependent onOGG1and the reverse is not true. Knockout of OGG1 significantly decreases the tightness of binding of CHD4 to chromatin after H2O2 treatment, whereas overexpression of OGG1 rescues this decrease (Figure 5C). In contrast, knockdown of CHD4 has no significant effect on the oxidative damage-induced increase in affinity of OGG1 for chromatin (Figure 5D).
Figure 5. The Recruitment of CHD4 to Oxidative DNA Damage Sites Depends on OGG1.

(A) CoIPs of lysates from SW480 cells untreated or treated with 2 mM H2O2 for 30 min were performed with the indicated antibodies.
(B) Purified OGG1 and Flag-CHD4 were incubated with antibodies against Flag or OGG1 in IP buffer. The immunoprecipitated samples were detected by western blot analyses using the antibodies indicated.
(C) After SW480 OGG1 KO cells were transfected with pCMV-Taq or pCMV-OGG1 for 48 hr, the cells were untreated or treated with 2mMH2O2 for 30 min. Whole-cell extracts and the tight chromatin fractions were analyzed by immunoblotting with the indicated antibodies.
(D) Whole-cell extracts and the tight chromatin fractions from SW480 CHD4 KD cells untreated (Un) or treated with 2 mM H2O2 for 30 min were analyzed by immunoblotting as in (C).
(E) Purified OGG1 and Flag-CHD4 were incubated with antibodies against Flag or OGG1 in IP buffer with or without 8-OHdG oligonucleotide. The immuno-precipitated samples were detected by western blot analyses using the antibodies indicated.
(F) Biotin labeled 8-OHdG oligonucleotide incubated with OGG1 and Flag-CHD4 was pulled down using streptavidin beads. Bound proteins were eluted and analyzed by immunoblotting with the indicated antibodies.
(G) SW480 OGG1 KO cells were untreated or treated with 2mMH2O2 for 30 min followed by ChIP for control IgG, 8-OHdG, and CHD4 at the promoter CpG islands of eight representative genes and analyzed by real-time RT-PCR. Data are represented as mean ± SEM for triplicate experiments.
(H) Cells were untreated or treated with 2 mM H2O2 for 30 min. Sequential ChIP analyses were performed to test the co-occupancy of CHD4 and 8-OHdG at the promoter CpG islands of eight TSGs. Data are represented as mean ± SEM for triplicate experiments.
(I) Cells were untreated or treated with 2mMH2O2 for 30 min, and nascent RNA was labeled concurrently. Real-time RT-PCR data are presented as mean ± SEM of the treated over untreated values for triplicate experiments.
(J) Sequential ChIP analyses were performed to test the co-occupancy of CHD4 and 8-OHdG or epigenetic silencing proteins at the promoter CpG islands of eight representative TSGs in fresh frozen human CRC tissues (n = 20) and normal colon epithelial tissues (n = 6). Data are represented as mean ± SEM.
*p < 0.05. See also Figure S4.
8-OHdG added to the immunoprecipitation buffer does not affect the OGG1 and CHD4 interaction, suggesting that this interaction is not DNA dependent (Figure 5E). However, an 8-OHdG oligonucleotide can pull down CHD4 protein in the presence but not the absence of OGG1, suggesting that CHD4 binds this oligonucleotide by interacting with OGG1 (Figure 5F).
Moreover, CRISPR-mediated knockout of OGG1 reduces the H2O2-induced enrichment of CHD4 at the promoter CpG islands of the eight TSGs (Figure 5G). Furthermore, sequential ChIP assays, using antibody against CHD4 in the first immunoprecipitation followed by pulldown with an 8-OHdG antibody, demonstrated co-occupancy of CHD4 and 8-OHdG at TSG promoter CpG islands after H2O2-induced oxidative damage (Figure 5H). Finally, knockout of OGG1 also relieves the H2O2-induced reduction in nascent RNA transcription of the genes (Figure 5I).
Importantly, the CHD4 interactions with OGG1 and with 8-OHdG are recruited by oxidative damage to the eight examined TSGs in primary CRC samples also. In sequential ChIP experiments, when soluble chromatin is first immunoprecipitated with antibody against CHD4 and the immunoprecipitates are subsequently re-immunoprecipitated with antibodies against 8-OHdG or epigenetic silencing proteins, 8-OHdG, DNMT1, DNMT3A, DNMT3B, EZH2, or G9a co-occupy the examined promoter CpG islands with CHD4 (Figure 5J).
As mentioned earlier, the protein zinc-finger MYND-type containing 8 (ZMYND8) has been linked to recruitment of CHD4 to DSBs (Gong et al., 2015). Our ChIP assays show that knockdown of ZMYND8 decreases the enrichment of CHD4 near the DSB sites, whereas knockdown of OGG1 does not (Figures S4A and S4B). In addition, knockdown of ZMYND8 has no effect on the H2O2-induced enrichment of CHD4 at the promoter CpG islands of four examined TSGs (Figure S4C). Our data, therefore, confirm that recruitment of CHD4 depends on different proteins at ROS-induced DNA damage sites and DSBs.
Prognostic Significance of Correlations between 8-OHdG, CHD4 Levels, Promoter DNA Methylation, and Silencing of TSGs in Primary CRC
We then investigated the prognostic importance of the findings described above. ChIP assays using fresh frozen samples for promoter regions of the eight genes examined above reveals enrichment of epigenetic silencing proteins (CHD4, DNMT1, DNMT3A, DNMT3B, EZH2, and G9a) and the oxidative damage marker 8-OHdG in CRC compared with normal colon epithelial tissues (Figure S5A). Furthermore, the enrichment of each of the above proteins in CRC tissues correlates with the enrichment of 8-OHdG in promoter regions of the eight TSGs (Figure S5B). In addition, CHD4 enrichment is positively associated with the enrichment of other epigenetic silencing proteins (DNMT1, DNMT3A, DNMT3B, EZH2, and G9a) at the promoter CpG islands of the eight genes in the above-examined CRC tissues (Figure S5C). These studies then suggest that the correlations we are seeing in cell culture studies between ROS-induced damage and recruitment of repressive chromatin to gene promoter CpG islands also occurs in primary CRC.
The above findings have potentially prognostic importance for patients with CRC. First, the 8-OHdG level, determined by immunohistochemistry analyses, in CRC samples from over 300 patients positively correlate with aggressive tumor behavior (Table S1) and poor prognosis (Figure S5D). Moreover, both negative expression of the above eight genes and the accompanying increased DNA methylation in their promoter regions also associate with these indices of aggressive behavior (Table S2) (Figures 6A–6D and S5E–S5H). Further, co-ordinate high levels of 8-OHdG and low expression and abnormal promoter methylation of the eight TSGs correlate with poorer prognosis (Figures 6A–6D and S5E–S5H), and the presence of promoter methylation in these genes inversely correlates with their expression (Figure S5I). Patients with high levels of 8-OHdG and low expression and abnormal promoter methylation of the eight TSGs in their CRC have the highest CRC recurrence rates and shortest overall survival (Figures 6A–6D and S5E–S5H).
Figure 6. 8-OHdG Is Positively Correlated with the Methylation but Inversely Correlated with the Expression of TSGs in Human CRC Tissues.

(A–D) The top two rows show representative images of immunohistochemistry of 8-OHdG and E-cadherin (A), WIF1 (B), TIMP2 (C), or TIMP3 (D) in human CRC tissues. Scale bars, 200 μm (low magnification) and 50 μm (high magnification). The middle row shows the association between 8-OHdG levels and the methylation or expression of the TSG in human CRC tissues. The lower two rows show Kaplan-Meier analyses of the correlation of the TSG expression, TSG methylation, 8-OHdG/TSG co-expression, or 8-OHdG/TSG methylation status with recurrence and overall survival in patients with CRC.
See also Figure S5, and Tables S1 and S2.
There are additional important correlates. First, in 120 paired samples from the same patients, the mRNA levels of CHD4 are significantly higher in CRC tissues than in the adjacent non-tumor tissues than in colon epithelial tissues from individuals without CRC (Figure 7A). Second, tumors of patients with recurrence of CRC (69 of 120) have higher CHD4 mRNA expression than the levels in tumors from patients without recurrence (51 of 120) (Figure 7A). Third, CHD4 mRNA expression is much higher in primary CRC tissues from patients who developed metastases than in primary CRC tissues from patients who did not (Figure 7A); and, in 20 pairs of specimens, levels are higher in metastatic versus primary lesions (Figure 7A). Supporting these data, in the TCGA database, the mRNA levels of CHD4 are significantly higher in tumors than in non-tumor tissues from patients with several cancer types, including CRC (Figure S6).
Figure 7. Overexpression of CHD4 Is Positively Correlated with the Methylation and Inversely Correlated with Expression of Eight TSGs in Human CRC Tissues.

(A) Real-time RT-PCR analyses of CHD4 mRNA expression in normal colon epithelial tissues (n = 20) and 120 paired, adjacent non-tumor and CRC tissues, in CRC samples from patients with recurrence (n = 69) or without recurrence (n = 51), in CRC samples from patients with metastases (n = 50) or without metastases (n = 50), and in primary colon cancer tissues and paired metastatic colon cancer tissues (n = 20). Data are represented as mean ± SEM. *p < 0.05.
(B) Representative immunohistochemistry staining of CHD4 in adjacent non-tumor tissues and CRC tissues. Scale bars, 200 μm (low magnification) and 50 μm (high magnification).
(C) Kaplan-Meier analyses of the correlation between CHD4 expression and the recurrence or overall survival of patients with CRC.
(D) The association between CHD4 expression and the expression or methylation of TSGs in CRC tissues (upper panels) and Kaplan-Meier analysis of the correlation of CHD4/TSGs co-expression or CHD4/TSGs methylation status with recurrence and overall survival in patients with CRC (lower panels).
See also Figure S6, and Tables S3 and S4.
When studied in tissue microarrays for 363 CRC tissues, nuclear levels of CHD4 are higher in tumor versus adjacent non-tumor tissues, and this upregulation correlates with poorer tumor differentiation, higher tumor nodal metastases stage, AJCC stage, shorter overall survival times, and higher recurrence rates (Figures 7B and 7C and Table S3). This CHD4 upregulation, as analyzed in multi-variate analyses is an independent and significant risk factor for recurrence and reduced survival (Table S4).
Taken together, the correlations outlined above for high levels of CHD4, 8-OHdG, reduced expression, and abnormal promoter DNA hypermethylation of eight examined TSGs are associated with increased aggressiveness of CRC (Figure 7D). When CRC tumors are divided into four groups based on these parameters, those with positive CHD4 expression, promoter hypermethylation, and low expression of the genes have the highest recurrence rates and shortest overall survival times (Figure 7D).
CHD4-Mediated Silencing of TSGs Promotes CRC Proliferation, Invasion, and Metastases
It is important to consider whether the silencing of the TSGs studied above may help explain why the relationships between CHD4, ROS damage, and gene silencing have the above prognostic implications. We used SW620 and LoVo cell lines that are known to have high invasion and metastatic capabilities when injected into immune-compromised mice. The possible contribution of all eight TSGs to tumor characteristics were queried in the context of shRNA knockdown of CHD4, which increases the expression of these eight genes in both cell lines (Figures S3A and S3B). Among these, CDH1, WIF1, TIMP2, and TIMP3 are important in invasion and metastases in both SW620 and LoVo cells. Knockdown of CHD4 robustly decreases the migration and invasion of both cell lines (Figures 8A and S7A), which is significantly rescued by preventing the expression of these genes by shRNA (Figure S3B). Decrease of E-cadherin and WIF1 rescue the migration and invasion abilities induced byCHD4 knockdown. Knockdown of TIMP2 and TIMP3 have no significant effect on reduced migration induced by CHD4 knockdown, but rescues decreased invasion induced by CHD4 knockdown (Figures 8A and S7A).
Figure 8. CHD4-mediated Silencing of E-cadherin, WIF1, TIMP2, and TIMP3 Promotes Colon Cancer Metastases.

(A) Transwell assay analyses of the migration and invasion abilities of the indicated CRC cells.
(B–G) SW620 cells infected with the indicated shRNA lentiviruses for the indicated genes were injected into the tail vein of immune-incompetent mice, followed by noninvasive bioluminescence imaging for 9 weeks. Representative bioluminescent imaging (B), bioluminescence signals (C), overall survival (D), incidence of lung colonization (E), the number of lung colonization foci (F), and representative H&E staining of lung tissues (G) from the different groups is shown. Scale bars, 1mm (low magnification) and 100 μm (high magnification).
(H–M) A mouse model of liver metastases was established by intrasplenic injection of the indicated colon cancer cells. Representative bioluminescent imaging 9 weeks after injection (H), bioluminescence signals recorded at the indicated time points (I), overall survival (J), incidence of liver metastases (K), number of tumor foci on the liver surface (L), and representative H&E staining of liver tissues (M) from the different groups is shown. Scale bars, 200 μm.
Data are represented as mean ± SEM. *p < 0.05. See also Figure S7.
Most important, knockdown of CHD4 significantly decreases colonization of the lungs in tail vein injection assays and metastases to the liver in the spleen implantation assay, with corresponding increases in mouse overall survival (Figures 8B–8M and S7B–S7M). Prevention for re-expression of E-cadherin, WIF1, TIMP2, and TIMP3 each individually by shRNA partially rescues these effects induced by CHD4 knockdown (Figures 8B–8M and S7B–S7M).
Next, we investigated the role for MLH1, p16, SFRP4, and SFRP5 in proliferation, viability, ability to grow in soft agar, and tumorigenic potential. Again, shRNA prevention for re-expression of each gene partially rescues the decreases in proliferation and anchorage-independent growth produced by CHD4 knockdown (Figure S7N). Similar findings occur for in vivo tumorigenicity assays, wherein knockdown of CHD4 significantly decreases tumor growth of SW620 and LoVo cells, and simultaneous knockdown of MLH1, p16, SFRP4, or SFRP5 each individually partially rescues this (Figure S7O). Taken together, these studies suggest that CHD4-mediated silencing of these TSGs promotes colon cancer proliferation, invasion and metastases.
DISCUSSION
CHD4, in the context of several types of DNA damage, appears to have a key role for not only initiating but also participating in maintaining epigenetic gene silencing, a hallmark of virtually all human cancers, in association with abnormal promoter CpG island DNA methylation (Shen and Laird, 2013). This role may be integral to some oncogenic properties of CHD4 shown by others (Fu et al., 2011; Nio et al., 2015; Sperlazza et al., 2015). We link CHD4 recruitment to DDR processes induced by laser micro-irradiation to trigger SSBs and DSBs, pure DSBs produced by either site-specific restriction endonuclease cutting or CRISPR-CAS technology, and SSBs in the context of BER repair during oxidative damage. All of these DNA damage responses are important throughout cancer initiation and progression, and our data suggest that each may contribute to the cancer epigenetic abnormalities we have focused upon.
It is important to consider, however, that the dynamics for the initiating and maintenance roles we now attribute to CHD4 for cancer epigenetic abnormalities may differ for the types of DDR events studied. First, in regard to DSBs, studies of others reveal that CHD4 recruitment through ZMYND8 occurs regionally around the damage site, particularly in open chromatin regions for promoters of expressed genes (Gong et al., 2015). We confirm that ZMYND8 is upstream for CHD4 recruitment at DSBs. CHD4 functions in repressing transcription at such DNA damage sites, presumably to protect these regions during the repair process (Chou et al., 2010; Gong et al., 2015). These regional effects would likely affect genes stochastically, and the transcriptional repression and its mediating protein recruitment would be transient and removed following successful repair (Chou et al., 2010; Gong et al., 2015). Retention of the repression, and any imposition of abnormal DNA methylation, could also likely be stochastic and only occasionally produce loss of function for genes important for tumorigenesis. These hypotheses are consistent with findings from our previous studies wherein we induced a DSB in an active, exogenous CpG island promoter, resulting in the disruption of transcription, and the restoration of transcription was dependent on successful repair of the damage (O’Hagan et al., 2008). Although the promoter transcriptional capacity was acutely disrupted by the DSB, and repressive proteins including DNMTs were recruited to the promoter CpG island region, 99% of clones followed over time repaired the DSB and resumed transcriptional capacity with an active chromatin structure. One percent, however, retained all of the repressive characteristics, lacked promoter activity, and evolved promoter CpG island DNA methylation (O’Hagan et al., 2008). However, for endogenous genes, as discussed below for oxidative damage, selection for affected genes could occur and reflect initially low basal transcription level to favor retention of DSB-induced repressive chromatin.
Our previous and present data suggest that the dynamics for effects of oxidative damage during tumor initiation and progression are likely quite different from those for pure DSBs. This may involve much more selection, with respect to tumorigenesis, for loss of function in a large group of important genes. Our previous work (O’Hagan et al., 2011), and now the present work involving CHD4 and SSBs, reveal an acute, genome-wide systemic signaling for DDR after ROS challenge. Thus, there is CHD4-mediated general tightening to chromatin of multiple transcription repressive proteins and increased interaction of CHD4 with BER and targeting to sites of induced 8-OHdG. We previously observed shifting of chromatin-repressive mediating proteins to CpG-rich gene promoters with ROS-induced 8-OHdG (O’Hagan et al., 2011), and we now show the importance of CHD4 in these dynamics. Importantly, there is vulnerability for retention of these transcriptionally repressive events, and evolution of DNA methylation in CpG-rich promoters of genes with low basal expression (O’Hagan et al., 2011) for which we now reveal key roles for CHD4. Our present data for the TSG function of a group genes targeted for this retention in CRC proliferation, invasion, migration, and metastatic properties, attests to this selection process during oxidative damage effects. Certainly, other factors involving proteins in the complex(es) we have studied may play key roles in such a selection. For example we have now observed that induction of a complex between DNMT1 and TET proteins plays a key protective role against abnormal DNA methylation during oxidative-induced DNA damage period, TET proteins actively remove DNA methylation and appear to protect abnormal DNA methylation in normally unmethylated, promoter region CpG islands (Zhang et al., 2017).
Interestingly, for many of the above-discussed genes and promoter CpG islands, a specific role for CHD4 in normally mediating a lowbasal level of expression, but without promoter DNA methylation, has been defined in embryonic stem cells (ESCs) (de Dieuleveult et al., 2016). These promoters are surrounded by “bivalent chromatin modifications” thought to maintain a poised transcription state for developmental genes that must normally retain low expression such that they do not prematurely induce ESCs to commit to lineage determination and differentiation (Bernstein et al., 2006). As we have stressed, these constitute the bulk of genes that are most vulnerable to promoter CpG island DNA hypermethylation in cancer (Easwaran et al., 2012). This chromatin consists of the simultaneous presence of an active histone modification, H3K4me3, at two nucleosomes surrounding transcription start sites (TSS) and a wider zone of the repressive marker H3K27me3 catalyzed by EZH2 surrounding TSS (Bernstein et al., 2006). Others have suggested that the normally low basal expression of these genes with a bivalent chromatin history in ESC and normal adult tissues argues against any importance for the abnormal DNAmethylation that appears in their promoters in cancer cells (Yu et al., 2014). However, our data suggest that it is the DNA methylation block to inducibility of these genes that may be most important. Indeed, signal induction is critical for normal activation of these genes in development and normal cell renewal (Bernstein et al., 2006). Key examples include the fact that failure to induce p16 expression can block onset of senescence and promote retention of cell stemness and proliferation (Baker et al., 2016; Janzen et al., 2006), as can failure to induce expression of genes involved in antagonizing WNT signaling (Baylin and Jones, 2016; Suzuki et al., 2004).
Finally, our basic findings for CHD4 may account for our findings in a large subset of patients with CRC. There is surprisingly strong correlation for levels of CHD4, 8-OHdG, and silencing of target TSGs to the recurrence rate of tumors after initial surgery, for the reduction in overall patient survival, and for the presence of metastases. In turn, these results are important for considering biomarker strategies to monitor the clinical behavior of CRC. It is clear that CHD4 plays a role in ongoing silencing and blocking induction of these genes. Possibly this reflects ongoing damage repair at these loci in cancer cells, but this must be further determined. Our data suggest that developing inhibitors for this function of CHD4 might be an excellent strategy to consider for cancer management. Important for any such drug development is our finding for the key role of the helicase domain of CHD4 for its mediation of abnormal epigenetic events as this presents a potentially druggable target.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-CHD4 | Sigma | Cat#WH0001108M1; RRID: AB_1840675 |
| Rabbit polyclonal anti-CHD4 | Active Motif | Cat#39289; RRID: AB_2614937 |
| Rabbit polyclonal anti-CHD4 | Abcam | Cat#ab72418; RRID: AB_1268107 |
| Rabbit monoclonal anti-DNMT1 | Sigma | Cat#D4692; RRID: AB_262096 |
| Rabbit polyclonal anti-DNMT1 | Santa Cruz | Cat#sc-20701; RRID: AB_2293064 |
| Mouse monoclonal anti-DNMT3A | Novusbio | Cat#NB120-13888; RRID: AB_789607 |
| Rabbit polyclonal anti-DNMT3B | QCB/BioSource International | raised against residues 376–390 (ENKTRRRTADDSATS) |
| Mouse monoclonal anti-EZH2 | Active Motif | Cat#39875; RRID: AB_2561022 |
| Mouse monoclonal anti-EZH2 | Cell Signaling | Cat#3147; RRID: AB_10694383 |
| Mouse monoclonal anti-G9a | Perseus Proteomics Inc | Cat#PP-A8620A-00; RRID: AB_2097650 |
| Mouse monoclonal anti-FLAG | Abcam | Cat#ab49763; RRID: AB_869428 |
| Mouse monoclonal anti-OGG1 | Santa Cruz | Cat#sc-376935 |
| Rabbit polyclonal anti-ZMYND8 | Bethyl Laboratories | Cat#A302-089; RRID: AB_1604282 |
| Rabbit monoclonal anti-IgG | Santa Cruz | Cat#sc-2027; RRID: AB_737197 |
| Mouse monoclonal anti-IgG | Santa Cruz | Cat#sc-2025; RRID: AB_737182 |
| Rabbit monoclonal anti-E-cadherin | Cell Signaling | Cat#3195; RRID: AB_2291471 |
| Rabbit monoclonal anti-WIF1 | Abcam | Cat#ab155101 |
| Rabbit polyclonal anti-WIF1 | Abcam | Cat#ab33281; RRID: AB_2216333 |
| Mouse monoclonal anti-TIMP2 | Abcam | Cat#ab1828; RRID: AB_2256129 |
| Mouse monoclonal anti-TIMP2 | Thermo Fisher | Cat#MA1-774; RRID: AB_889483 |
| Rabbit polyclonal anti-TIMP3 | Abcam | Cat#ab39184; RRID: AB_2204971 |
| Rabbit polyclonal anti-TIMP3 | Thermo Fisher | Cat#PA1-21146; RRID: AB_561715 |
| Rabbit monoclonal anti-MLH1 | Abcam | Cat#ab92312; RRID: AB_2049968 |
| Rabbit monoclonal anti-p16 | Abcam | Cat#ab51243; RRID: AB_2059963 |
| Mouse monoclonal anti-p16 | Thermo Fisher | Cat#MA5-17093; RRID: AB_2538564 |
| Rabbit monoclonal anti-SFRP4 | Abcam | Cat#ab154167 |
| Rabbit polyclonal anti-SFRP5 | Novus Biologicals | Cat#NBP2-20331 |
| Mouse monoclonal anti-γH2AX | Millipore | Cat#05-636; RRID: AB_309864 |
| Mouse monoclonal anti-γH2AX | Santa Cruz | Cat#sc-101696; RRID: AB_2114997 |
| Goat polyclonal anti-LaminB | Santa Cruz | Cat#sc-6216; RRID: AB_648156 |
| Mouse monoclonal anti-GAPDH | Sigma | Cat#WH0002597M1; RRID: AB_1841801 |
| Mouse monoclonal anti-β-actin | Santa Cruz | Cat#sc-47778; RRID: AB_626632 |
| Mouse monoclonal anti-8-OHdG | Millipore | Cat#MAB3560; RRID: AB_94925 |
| Goat polyclonal anti-8-OHdG | Abcam | Cat#ab10802; RRID: AB_297482 |
| Mouse monoclonal anti-5mc | Diagenode | Cat#C15200081; RRID: AB_2572207 |
| Rabbit monoclonal anti-H3 | Millipore | Cat#17-10046; RRID: AB_10618160 |
| Rabbit polyclonal anti-H3K27me3 | Millipore | Cat#17-622; RRID: AB_916347 |
| Mouse monoclonal anti-H3K9me2 | Millipore | Cat#05-1249; RRID: AB_1977256 |
| Rabbit monoclonal anti-H3K4me3 | Millipore | Cat#17-614; RRID: AB_1587135 |
| Rabbit polyclonal anti-H4K16ac | Millipore | Cat#17-10101; RRID: AB_10807418 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| OGG1, human, recombinant | New England Biolabs | Cat#M0241 |
| Lipofectamine® 3000 Transfection Reagent | Thermo Fisher | Cat#L3000015 |
| Complete EDTA free protease inhibitor | Sigma Aldrich | Cat#11873580001 |
| NuPAGE® LDS Sample Buffer (4X) | ThermoFisher | Cat#NP0007 |
| Polybrene | Sigma Aldrich | Cat#H9268 |
| Puromycindihydrochloride | Sigma Aldrich | Cat#P8833 |
| Doxycycline hyclate | Sigma Aldrich | Cat#D9891 |
| Critical Commercial Assays | ||
| QuantiNova SYBR Green PCR Kit | QIAGEN | Cat#208052 |
| QuantiTect Reverse Transcription Kit | QIAGEN | Cat#205311 |
| TRIzol RNA Isolation Reagents | Thermo Fisher | Cat#15596026 |
| Dynabeads® Protein A | ThermoFisher | Cat#10001D |
| Dynabeads® Protein G | ThermoFisher | Cat#10003D |
| EZ DNA Methylation™ Kit | Zymo Research | Cat#D5002 |
| Click-iT® Nascent RNA Capture Kit | Thermo Fisher | Cat#C10365 |
| Wizard® Genomic DNA Purification Kit | Promega | Cat#A1120 |
| MAGnify™ Chromatin Immunoprecipitation System | Thermo Fisher | Cat#492024 |
| Experimental Models: Cell Lines | ||
| SW480 | ATCC | CCL-228 |
| SW620 | ATCC | CCL-227 |
| LoVo | ATCC | CCL-229 |
| HCT116 | ATCC | CCL-247 |
| NCCIT | ATCC | CRL-2073 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 nude | The Jackson Laboratory | Stock No: 000819 |
| Oligonucleotides | ||
| CRISPR/Cas9 gRNA Sequences | This paper | See Table S5 |
| Primers for RT-qPCR | This paper | See Table S6 |
| Primers for Methylation-Specific PCR | This paper | See Table S6 |
| Primers for ChIP-qPCR | This paper | See Table S6 |
| CRISPR guide RNA in the Dox-inducible DSB system | This paper | See Table S7 |
| Recombinant DNA | ||
| lentiCRISPR v2 | Addgene | Cat#52961 |
| pLKO.1 | Addgene | Cat#10878 |
| pMD2.G | Addgene | Cat#12259 |
| psPAX2 | Addgene | Cat#12260 |
| pSQT-dCas9-FokI | Addgene | Cat#53369 |
| pCW-Cas9 | Addgene | Cat#50661 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| sgRNA designer | Broad Institute | http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| ImageJ | Open source | http://rsb.info.nih.gov/ij/index.html |
| SPSS 16.0 | IBM | http://www.ibm.com/analytics/us/en/technology/spss/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Stephen B. Baylin (sbaylin@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
SW480, HCT116, NCCIT, SW620, LoVo cells were obtained from American Tissue Type Culture Collection and maintained at 37 °C and 5% CO2 in McCoy’s5A, McCoy’s5A, RPMI-1640, McCoy’s5A, and DMEM (Dulbecco’s Modified Eagle Medium), respectively. For H2O2 exposure 30% H2O2 (Sigma) was diluted in PBS immediately before adding it to the media and cells were collected 30 min later. DR-GFP U2OS cells with one copy of the DR-GFP gene stably integrated into its genome were gifts from Dr. Maria Jasin (Memorial Sloan Kettering Cancer Center).
Mice
BALB/C nude mice (5 weeks old) were housed under standard conditions and cared for according to the institutional guidelines for animal care. All animal experiments were approved by the Committee on the Use of Live Animals in Teaching and Research (CULATR), Fourth Military Medical University. For the tail vein injection assays, Luciferase tagged SW620 and LoVo (2×106) cells in 200 μl of phosphate-buffered saline (PBS) were injected into the lateral tail vein of nude mice using 25-gauge needles. In weekly intervals, anesthetized mice were injected i.p. with D-luciferin (150 mg/kg) and imaged 10 min after injection using an IVIS 100 Imaging System (Xenogen). The acquisition time was 2 min. The survival of the mice was recorded daily. Nine weeks after tail vein injection, mice were sacrificed and examined for lung metastases using standard histological examination.
A mouse model of liver metastases was established by intrasplenic injection of luciferase tagged SW620 and LoVo cells. Briefly, the spleens were exposed by an incision on the left upper abdomen of mice. For each mouse, 1×106 cells in 100 μl of PBS were injected into the spleen, by which the injected cells were delivered to the liver through the portal circulation. Ten minutes after injection, the spleen was removed and the abdominal cavity was closed. Metastases were monitored by weekly noninvasive IVIS imaging of luciferase activity. The survival of the mice was recorded daily. After 9 weeks post implantation, all mice were sacrificed, and the number of tumor foci on liver surface was calculated. Liver tissues were fixed and stained with H&E to detect the liver metastases.
For the xenograft tumor growth assay, 5×104 cells were injected subcutaneously into the right flank of 5-week-old male BALB/C nude mice. Each group consisted of 10 mice. Tumor formation in nude mice was monitored over a 30-day period, and the tumor volume was measured every 5 days and calculated as: 1/2 (largest diameter)×(smallest diameter)2.
Human Specimens
This study was approved by the ethics committee of the Fourth Military Medical University. All patients provided full consent for the study. Fresh colorectal cancer specimens and matched adjacent tissues were obtained from 363 adult patients who underwent surgery at Xijing Hospital, Fourth Military Medical University (Xi’an, China), between January 2005 and December 2007. None of the patients had received preoperative chemotherapy or radiotherapy. Pathological staging was performed according to the American Joint Committee on Cancer/International Union against Cancer. Patients with stage II, III, and IV disease were treated after surgery with adjuvant chemotherapy, and none of the patients had received postoperative radiotherapy. Histomorphology of all primary tumor specimens and regional lymph nodes was confirmed by hematoxylin–eosin staining by the Department of Pathology, Xijing Hospital. In addition, 20 normal colon epithelial tissues, 120 pairs of frozen fresh colon cancer tissues and peripheral nontumor tissues were collected after surgical resection and stored in liquid nitrogen. These tissue pairs were used to detect the mRNA expression of CHD4. Six normal colon epithelial tissues and 20 fresh frozen colon cancer tissues were collected after surgical resection, and were used to perform ChIP assays.
Complete follow-up was available for at least 8 years. During the follow-up period, diagnosis of recurrence and distant metastases was based on imaging methods such as endoscopy, ultrasonography, computed tomography, magnetic resonance imaging, position emission tomography, and, if possible, cytologic analyses and biopsy. Disease-free survival was defined as the time elapsed from surgery to the first occurrence of any of the following events: recurrence of colorectal cancer, colorectal cancer distant metastases, development of second noncolorectal malignancy excluding basal cell carcinoma of the skin and carcinoma-in-situ of the cervix, or death from any cause without documentation of a cancer-related event. Overall survival was defined as the time elapsed from surgery to death of patients with colorectal cancer. Follow-up information of all participants was updated every 3 months by telephone enquiry and questionnaire letters. Deaths of patients were ascertained by reporting from the family and verified by review of public records.
METHOD DETAILS
Lentivirus shRNA Knockdown and CRISPR Knockout
Wild-type and ATP-domain mutant CHD4 were kindly provided by Wenbing Xie (Xie et al., 2012) and subcloned into pLenti-Flag. shRNA-resistant form of CHD4 was generated using Q5® Site-Directed Mutagenesis Kit (NEB) with the oligonucleotides (forward) 5′-AATGGCGTGAATTTAGTACCAATAACCCCTTC-3′ and (reverse) 5′-TTGCACCCAAAACCATCATCATCTTGGAG-3′. The construct was confirmed by DNA sequencing.
All lentiviral shRNA clones were ordered from Sigma. For CRISPR genome-editing, gRNAs were cloned into the lentiCRIPSR v2 vector (Addgene plasmid #52961) through the BsmBI site based on the protocol recommended by Addgene (Cambridge, MA, USA). The complete lists of shRNA and gRNA sequences are shown in Table S5. The production of lentivirus and cell infection were performed according to the pLKO.1 lentiviral vector protocol recommended by Addgene. Briefly, the lentiviral plasmid and packaging plasmids pMD2.G and psPAX2 (Addgene plasmid #12259 and #12260) were transfected into HEK-293T cells with transfection reagent (Lipofectamine®3000, Thermo Fisher Scientific) and OPTI-MEM media (Invitrogen, Waltham, MA, USA). The lentiviruses were harvested twice at day 4 and day 5. Virus were filtered with 0.45 μm filter and stored at −80 °C. The lentiviral infection of target cells was performed in cell culture media with 8 μg/ml polybrene (Sigma H9268). Seventy-two hours after infection, cells were selected for 2 weeks using 2.5 μg/ml puromycin (OriGene). Selected pools of shRNA knockdown cells were used for the following experiments. For CRISPR knockout, subsequently single clones were selected through serial dilution. Clones were verified by western blot and sequencing.
Tight Chromatin and Whole Cell Isolation
Indicated cell pellets were sequentially washed in CEBN buffer [10 mM HEPES pH 7.8, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.2% NP-40, 1× protease inhibitor cocktail (Thermo Scientific), 1× phosphatase Inhibitor cocktail (Sigma), N-ethyl-maleimide (Sigma)], CEB buffer (CEBN buffer without NP-40), soluble nuclear buffer (3 mM EDTA, 0.2 mM EGTA, inhibitors), and 0.45MNaCl buffer (50mMTris pH 8.0, 0.05% NP40, 0.45MNaCl, inhibitors). The remaining pellet was lysed in 4% SDS buffer using a Qiashredder (Qiagen) and referred to as tight chromatin. Band densitometry data for western blots were collected using ImageJ software. Whole cell extracts were prepared from 1/10 of the pellet collected after treatment before beginning the tight chromatin isolation. GAPDH and LaminB immunoblotting serve as cytoplasmic and nuclear controls, respectively, for the designated extractions.
Co-immunoprecipitation
The total nuclear pellet was resuspended in modified RIPA buffer (50 mM Tris PH 7.5, 100 mM NaCl, 3 mM EDTA, 0.5% NP-40, 50 mMNaF), sonicated for three cycles of 30 s using a BioruptorPicosonicator (Diagenode), rotated at 4 °C for 1 hr with 60mM spermine and 20mMspermidine to release chromatin bound proteins, sonicated for two cycles of 30 s, and cleared by high-speed centrifugation. Lysates were rotated with antibody for 4 hr at 4 °C. These antibodies are anti-CHD4 (Sigma, WH0001108M1), anti-DNMT1 (Sigma, D4692), anti-DNMT3A (Novusbio, NB120-13888), anti-DNMT3B (A rabbit polyclonal antibody (QCB/BioSource International) was raised against a fusion protein containing residues 376–390 (ENKTRRRTADDSATS)), anti-EZH2 (Active Motif, 39875), and anti-G9a (Perseus Proteomics Inc, PP-A8620A-00). Protein A/G-magnetic beads (Pierce) were added and the samples were rotated for 3 hr at 4 °C. The beads were washed six times with TNE buffer for 10 min at 4 °C. Complexes were eluted off the beads in loading buffer at 65 °C for 15 min.
Recombinant Flag-tagged human CHD4 (hCHD4) protein was purified from HEK293 cells transfected with (full-length) Flag-hCHD4 plasmid. Purified recombinant hCHD4 and hOGG1 (M0241, NEB) proteins were incubated in IP buffer (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 5 mM MgCl2, 1% NP-40) with or without 8-OHdG oligonucleotides (double strand) at 4 °C for overnight, followed by co-precipitation using anti-flag M2 beads (M8823, Sigma) or protein A/G agarose (sc-2003, Santa Cruz) using antibodies against Flag (ab49763, Abcam) or OGG1 (sc-376935, Santa Cruz). The beads were washed five times using an IP buffer and the immunoprecipitated proteins were analysed by Western blotting. Normal rabbit IgG (sc-2027, Santa Cruz) or normal mouse IgG (sc-2025, Santa Cruz) was used as a negative control. Antibodies were used in the amount of 3 μg per IP. Oligonucleotides for the co-immunoprecipitation are listed in Table S8.
Western Blot Analyses
Proteins from lysed cells were fractionated by SDS-PAGE and transferred to nitrocellulose membranes. Nonspecific binding sites were blocked with 5% milk in TBST (120 mM Tris–HCl (pH 7.4), 150 mM NaCl, and 0.05% Tween 20) for 1 hr at room temperature. Blots were incubated with a specific antibody overnight at 4 °C. Western blotting of β-actin on the same membrane was used as a loading control. The membranes were incubated with primary antibodies anti-CHD4 (Sigma, WH0001108M1), anti-DNMT1 (Sigma, D4692), anti-DNMT3A (Novusbio, NB120-13888), anti-DNMT3B (A rabbit polyclonal antibody (QCB/BioSource International) was raised against a fusion protein containing residues 376–390 (ENKTRRRTADDSATS)), anti-EZH2 (Cell Signaling, #3147), anti-G9a (Perseus Proteomics Inc, PP-A8620A-00), anti-E-cadherin (Cell Signaling, #3195), anti-WIF1 (Abcam, ab155101), anti-TIMP2 (Abcam, ab1828), anti-TIMP3 (Abcam, ab39184), anti-MLH1 (Abcam, ab92312), anti-p16 (Abcam, ab51243), anti-SFRP4 (Abcam, ab154167), anti-SFRP5 (Novus Biologicals, NBP2-20331), anti-γH2AX (Millipore, 05-636), anti-LaminB (Santa Cruz, sc-6216), anti-GAPDH (Sigma, WH0002597M1) and anti-β-actin (Santa Cruz, sc-47778) overnight at 4 °C. The membranes were then washed with PBS 3 times and incubated with an HRP-conjugated secondary antibody. Proteins were visualized using a Dura SuperSignal Substrate (Pierce, USA).
Real-time PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen), and reverse transcription was performed using the Advantage RT-for-PCR Kit (QIAGEN) according to the manufacturer’s instructions. For the real-time PCR analyses, aliquots of double-stranded cDNA were amplified using a SYBR Green PCR Kit (QIAGEN). The cycling parameters were as follows: 95 °C for 15 s, 55–60 °C for 15 s, and 72 °C for 15 s for 45 cycles. A melting curve analyses was then performed. The Ct was measured during the exponential amplification phase, and the amplification plots were analyzed using SDS 1.9.1 software (Applied Biosystems). For the cell lines, the relative expression levels (defined as the fold change) of the target genes were determined by the following equation: 2−ΔΔCt (ΔCt= ΔCttarget −ΔCtGAPDH; ΔΔCt = ΔCtexpressing vector −ΔCtcontrol vector). The expression level was normalized to the fold change that was detected in the corresponding control cells, which was defined as 1.0. For the clinical tissue samples, the fold change of the target gene was determined by the following equation: 2−ΔΔCt (ΔΔCt= ΔCttumor −ΔCtnontumor). This value was normalized to the average fold change in the normal colon epithelial tissues, which was defined as 1.0. All reactions were performed in duplicate. The primer sequences are listed in Table S6.
Nascent RNA Transcription
Nascent transcription assays were performed using the Click-iT Nascent RNA Capture Kit (Thermo Fisher). Cells were labeled with ethynyluridine for 30 min concurrently with the 2 mM H2O2 treatment or Dox treatment if indicated. cDNA was analyzed by qPCR using primers indicated in Table S6.
Chromatin Immunoprecipitation Assay (ChIP)
Cells were cross-linked in 1% formaldehyde at 37 °C for 10 min. After washing with PBS, the cells were resuspended in 300 μl of lysis buffer. The DNA was sheared to small fragments by sonication. Sonicated chromatin was diluted to a final SDS concentration of 0.1% and aliquots were rotated with antibody O/N at 4 °C. The recovered supernatants were incubated with specific antibodies or an isotype control IgG for 2 hr in the presence of herring sperm DNA and Protein A/G Magnetic beads (Thermo Fisher). These antibodies are anti-8OHdG (Millipore, MAB3560), anti-γH2AX (Millipore, 05-636) anti-CHD4 (Sigma, WH0001108M1), anti-DNMT1 (Sigma, D4692), anti-DNMT3A (Novusbio, NB120-13888), anti-DNMT3B (A rabbit polyclonal antibody was raised against a fusion protein containing residues 376–390 (ENKTRRRTADDSATS)), anti-EZH2 (Active Motif, 39875), anti-G9a (Perseus Proteomics Inc, PP-A8620A-00), anti-5mc (Diagenode, C15200081), anti-H3 (Millipore, 17-10046), anti-H3K27me3 (Millipore, 17-622), anti-H3K9me2 (Millipore, 05-1249), anti-H3K4me3 (Millipore, 17-614), and anti-H4K16ac (Millipore, 17-10101). The immunoprecipitated DNA was retrieved from the beads with 1% SDS and a 1.1MNaHCO3 solution at 65 °C for 6 hr. The DNA was then purified using a PCR Purification Kit (QIAGEN, USA). The primers are shown in Table S6.
For sequential ChIP, the first immunoprecipitation was performed as described above except for the elution step, which was performed with SDS lysis buffer (1% SDS, 10 mmol/L EDTA, 50 mmol/L Tris-HCl, pH 8.1, 1×cOmplete Protease Inhibitor Cocktail; Roche Applied Science) for 10 min at 68 °C on a shaker at 1,000 rpm. After removal of beads, the samples were diluted with 1:10 ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mmol/L EDTA, 16.7 mmol/L Tris-HCl, pH 8.1, 167 mmol/L NaCl, 22 μg/mL BSA, 1×cOmplete Protease Inhibitor Cocktail) and incubated with beads, pre-blocked with BSA and herring sperm DNA, for 30 min at 4 °C. After removal of beads, chromatin was incubated with the indicated antibodies overnight at 4 °C followed by further incubation with beads. Beads were washed once with low-salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mmol/L EDTA, 20 mmol/L Tris-HCl, pH 8.1, 150 mmol/L NaCl, 1×cOmplete Protease Inhibitor Cocktail) and twice with high salt immune complex wash buffer (0.1% SDS, 1%Triton X-100, 2 mmol/L EDTA, 20 mmol/L Tris-HCl, pH 8.1, 500 mmol/L NaCl, 1×cOmplete Protease Inhibitor Cocktail). Chromatin was eluted with SDS lysis buffer for 10 min at 68 °C on a shaker at 1,000 rpmand crosslinks were reversed in presence of 300 mmol/L NaCl at 65 °C overnight.
Laser Irradiation and Confocal Microscopy
Briefly, a Nikon Eclipse 2000E spinning-disk confocal microscope with five laser imaging modules and a charge-coupled device (CCD) camera (Hamamatsu) was employed. The setup integrated a Stanford Research Systems (SRS) NL100 nitrogen laser with a Micropoint ablation system (Photonics Instruments). Site-specific DNA damage was induced using the SRS NL100 nitrogen laser adjusted to emit at 455 nm. Positions internal to the nuclei of the indicated cells were targeted using a 60×oil objective lens. Cells were targeted at 5.5% laser intensity to induce DSBs, and images were captured at various time points and analyzed using Volocity, version 5.0, build 6 (Improvision). Experiments were performed using an environmental chamber attached to the microscope to maintain experimental conditions (37 °C, 5% CO2, and 80% humidity). After laser treatment, the cells were incubated at 37 °C for different time points and fixed immediately in freshly prepared 4%formaldehyde (in PBS) for 10 minutes at room temperature. Fixed cells were permeabilized with a PBS solution containing 0.5% Triton X-100 on ice for 10 min. For immunofluorescence staining, cells were incubated at 37 °C for 1 hr with anti-γH2AX (Millipore, 05-636; Santa cruz, sc-101696), anti-DNMT1 (Santa cruz, sc-20701), anti-DNMT3A (Novusbio, NB120-13888), anti-DNMT3B (A rabbit polyclonal antibody (QCB/BioSource International) was raised against a fusion protein containing residues 376–390 (ENKTRRRTADDSATS)), anti-EZH2 (Cell Signaling, #3147), anti-G9a (Perseus Proteomics Inc, PP-A8620A-00), and anti-5mc (Diagenode, C15200081). Cells were incubated with corresponding secondary antibodies (Alexa Fluor goat anti-mouse or Alexa Fluor goat anti-rabbit, Invitrogen). After washing, they were mounted using ProLong Gold antifade reagent with DAPI (Invitrogen). The immunostained cells were visualized and imaged using a Hamamatsu EM-CCD digital camera attached to the Nikon Eclipse TE2000 confocal microscope. Cells (n = 30) were examined for each experimental point. In each experiment, images of cells at various time points were acquired using the same exposure, gain, sensitivity, and contrast settings.
Doxycycline-Inducible DSB System at the Promoter TSGs
Construction of pCW-dCas9-FokI: The pCW-Cas9 (Addgene: #50661) was digested with BamH1, and blunted by DNA polymerase and Klenow fragment. Then, the linearized vector was digested with NheI and the bigger fragment (7.6 kb) was purified. The dCas9-FokI sequence was amplified from the pSQT-dCas9-FokI plasmids (Addgene: #53369). The forward and reverse primers are: 5′-TCTGTCTAGAATGCCTAAGAAGAAGCGGAAG-3′ and 5′-TTGTGGATCCGCTTCACTTGTCATCGTCAT-3′. The PCR products were digested with XbaI and ligated with the 7.6 kb vector fragment. The construct was confirmed by DNA sequencing.
A doxycycline-inducible DSB system at the promoter CpG islands of endogenous TSGs was constructed. First, the FokI restriction endonuclease cleavage domain was fused to a catalytically dead Cas9, which was cloned into a pCW-Cas9 plasmid (Figure 4A). Then SW480 cells were transfected with the plasmid, and a single clone was selected that had a high level of doxycycline-induced dead Cas9/FokI fusion protein expression. Second, these cells were infected with lentivirus expressing gRNAs. After doxycycline induction, a double strand break at the specific site of a gene promoter was generated (Figure 4A). Doxycycline was added to medium for 8 hr followed by washing out the doxycycline and collecting cells at indicated time points (Figure 4A). By western blot analyses, dCas9/FokI fusion protein was induced after 8 hr of doxycycline treatment, and this expression was maintained after an eight hour wash (Figure S3C). By immunofluorescence staining, each cell was shown to express high levels of nuclear flag tagged dCas9/FokI fusion protein at the 8+8 hr time point (Figure S3C). Next, this system was used to produce DSB at the promoter CpG islands of eight representative tumor suppressor genes. Four pairs of primers were designed to detect the local epigenetic changes near the DSB sites, including DSB sites upstream 0.2 kb, 0.4 kb, and 0.6 kb, and DSB sites downstream 0.2 kb (Figures 4B, 4C, and S3D–S3I). To determine the timing of the DSB formation and repair, the dead Cas9/FokI induced cutting was monitored by real-time PCR using a primer pair (F1, R1) flanking the cutting site (Figures 4B, 4C, and S3D–S3I). Using this PCR, only uncut or repaired DNA will result in a PCR product. The PCR product was slightly decreased at the 8 hr time point, followed by a dramatic decrease at the 8+8 hr time point. The PCR product level increased at the 8+24 hr time point suggesting that a significant portion of the cells repair the DSB during this time frame (Figure S3C). All primers are listed in Table S6. Sequence for CRISPR guide RNAs are listed in Table S7.
Construction of Tissue Microarrays and Immunohistochemistry
Colorectal cancer specimens and matched adjacent tissues were used to construct a tissue microarray (Shanghai Biochip Co., Ltd. Shanghai, China). The tissue microarray was stained for 8-OHdG (Abcam, ab10802), CHD4 (Abcam, ab72418), E-cadherin (Cell signaling, #3195), WIF1 (Abcam, ab33281), TIMP2 (Thermo Fisher, MA1-774), TIMP3 (Thermo Fisher, PA1-21146), MLH1 (Abcam, ab92312), p16 (Thermo Fisher, MA5-17093), SFRP4 (Abcam, ab154167), and SFRP5 (Novus Biologicals, NBP2-20331) expression. The array was scored independently by two pathologists for both the staining intensity and the extent of the protein expression across the section.
Immunohistochemistry was performed on 4-μm-thick, routinely processed paraffin-embedded sections. Briefly, after baking on a panel at 60 °C for an hour, the tissue sections were deparaffinized with xylene and rehydrated through gradient ethanol immersion. Endogenous peroxidase activity was quenched by 3% (vol/vol) hydrogen peroxide in methanol for 12 min, followed by three 3-min washes with phosphate-buffered saline (PBS). Then the slides were immersed in 0.01 mol/L citrate buffer solution (pH 6.0) and placed in a microwave oven for 30 min. After washing in PBS (pH 7.4, 0.01 mol/L), the sections were incubated in a moist chamber at 4 °C overnight with the primary antibody diluted in PBS containing 1% (wt/vol) bovine serum albumin. Negative controls were performed by replacing the primary antibody with preimmune mouse serum. After three 5 min washes with PBS, the sections were treated with a peroxidase-conjugated second antibody (Santa Cruz) for 30 min at room temperature, followed by additional three 5 min washes with PBS. Reaction product was visualized with diaminobenzidine for 2 min. Images were obtained under a light microscope (Olympus, Japan) equipped with a DP70 digital camera.
Analyses were performed by two independent observers who were blinded to the clinical outcome. The immunostaining intensity was scored on a scale of 0 to 3: 0 (negative), 1 (weak), 2 (medium) or 3 (strong). The percentage of positive cells was evaluated on a scale of 0 to 4: 0 (negative), 1 (1%–25%), 2 (26%–50%), 3 (51%–75%), or 4 (76%–100%). The final immuno-activity scores were calculated by multiplying the above two scores, resulting an overall scores which range from 0~12. Each case was ultimately considered “negative” if the final score ranges from 0~3, and “positive” if the final score ranges from 4~12.
DNA Methylation Analyses
Genomic DNA was isolated from cells using Genomic DNA Purification kit following the manufacturer’s instructions (Promega). Bisulfite modification of genomic DNA was carried out using the EZDNA methylation Kit (Zymo Research). Briefly, 1μg of genomic DNA was denatured by NaOH (final concentration, 0.2 mol/L) for 10 min at 37 °C. Hydroquinone (10 mmol/L, 30 μl) and 520 μl of 3 mol/L sodium hydroxide (pH 5) were added, and samples were incubated at 50 °C for 16 hr. Modified DNA was purified using Wizard DNA Clean-Up System following the manufacturer’s instructions (Promega) and eluted into 50 μl water. DNA was treated with NaOH (final concentration, 0.3 mol/L) for 5 min at room temperature, ethanol precipitated, and resuspended in 20 μl water. Modified DNA was used immediately or stored at −20 °C. Primer sequences specific to unmethylated and methylated promoter sequences are listed in Table S6. Each methylation-specific PCR reaction incorporated 100 ng of bisulfite-treated DNA as template, 10 pmol/L of each primer, 100 pmol/L deoxynucleoside triphosphate, 10 PCR buffer, and 1 unit of JumpStart Red Taq Polymerase (Sigma-Aldrich, St. Louis, MO) in a final reaction volume of 25 μl. Cycle conditions were as follows: 95 °C 5 min; 35 cycles (95 °C 30 s, 60 °C 30 s, and 72 °C 30 s); and 72 °C 5 min. Methylation-specific PCR products were analyzed with nondenaturing 6% polyacrylamide gel electrophoresis and stained with ethidium bromide.
In Vitro Migration and Invasion Assays
A 24-well transwell plate (8-μmpore size, Corning, USA) was used to measure the migratory and invasion capacity of each tested cell line. For transwell migration assays, 5×104 cells were plated in the top chamber lined with a non-coated membrane. For invasion assays, chamber inserts were coated with 200 mg/ml of Matrigel and dried overnight under sterile conditions. Then, 1×105 cells were plated in the top chamber. The mean of triplicate assays for each experimental condition was used. The average number of cells in five fields per membrane was counted in triplicate inserts. The relative invasion/migration was expressed as the number of treated cells to control cells.
MTT, Colony Formation Assay, and Soft Agar Colony Formation Assay
The proliferation of colon cancer cells in vitro was measured using the MTT assay. 5000 stably infected cells were seeded into each well of 96 well plates. Six wells of each group were detected every day. 100 μl fresh medium containing MTT 0.5 mg/ml was put into each cell and incubated at 37 °C for 4 hr, then the medium was replaced by 100 μl of DMSO and shaken at room temperature for 10 min. The absorbance was measured at 490 nm.
For colony formation assays, 500 cells were seeded into 35 mm dishes and shaken. Then the cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2 in air for 2 weeks. Subsequently, the medium was removed and the cells stained with crystal violet, and the dishes were imaged with a light microscope (Olympus, Japan) equipped with a DP70 digital camera. Only positive colonies (diameter > 40 um) in the dishes were counted and compared.
For soft agar colony formation assay, 1×104 cells were suspended in 1 ml of soft agar mixture (2×cell culture medium, 20% FBS and 0.7% agarose) and were subsequently overlaid on the agar base. After 2–3 weeks, colonies (≥ 10 cells) were counted under the microscope in 10 fields per well. Three independent experiments were performed in duplicate.
QUANTIFICATION AND STATISTICAL ANALYSIS
The quantitative data were compared between groups using the Student’s t-test. Categorical data were analyzed using the Fisher’s exact test. The cumulative recurrence and survival rates were determined using the Kaplan-Meier method and log-rank test. The Cox proportional hazards model was used to determine the independent factors that influence survival and recurrence based on the variables that had been selected from the univariate analyses. A value of p < 0.05 was considered to be significant. All the analyses were performed using the SPSS software (version 16.0).
Supplementary Material
Significance.
CHD4, a key component of the NuRD complex, has been tied to mediating transcriptional repression during double-strand break DNA damage repair. We now show CHD4 recruitment to single-strand break repair associated with oxidative damage. At these sites, CHD4 plays an upstream role for recruitment of DNA methyltransferases, key transcriptional repressor proteins, and the initiation of abnormal de novo DNA methylation. CHD4 retention at the above damage sites in abnormally DNA hypermethylated CpG island promoter regions can maintain TSGs silencing in human colorectal cancer (CRC) cells. These activities define an oncogenic role for CHD4, which we show has important biologic and translational connotations for CRC cells and for prognostic implications in patients with this cancer.
Highlights.
CHD4 facilitates DNA hypermethylation
CHD4 knockdown reactivates hypermethylated genes
Inhibiting CHD4 blunts tumorigenesis
High level of CHD4 in tumor indicates poor prognosis
Acknowledgments
Research was supported by grants from The National Institutes of Environmental Health Sciences (NIEHS) grant RO1 ES011858 (S.B.B.), the Hodson Trust (S.B.B.), and The National Natural Science Foundation of China No. 81522031 (L.X.) and No. 81272652 (L.X.). The study was also supported in part by internal funds from the National Center for Toxicological Research, U.S. Food and Drug Administration (L.Y.). The information in this article is not a formal dissemination of information by the FDA and does not represent agency position or policy. We thank Kathy Bender for help with manuscript preparation. S.B.B. is a consultant for MDxHealth. M.S.P. is licensed to MDxHealth in agreement with Johns Hopkins University (JHU). S.B.B. and J.H.U. are entitled to royalty shares received from sales.
Footnotes
Supplemental Information includes seven figures and eight tables and can be found with this article online at http://dx.doi.org/10.1016/j.ccell.2017.04.005.
AUTHOR CONTRIBUTIONS
L.X. led design and participated in many of the experiments and W.H. performed animal experiments; L.X., M.M.S., and M.B. performed the laser micro-irradiation experiments; K.W., D.F., and Y.N. performed immunohistochemistry and ChIP assays; Y.C. and L.Y. performed mass spectrometry analyses; Y.W.Z., H.L., C.A.Z., W.X., and R.C.Y. performed TCGA data analyses and cell culture. L.X., F.V.R., and S.B.B. predominantly wrote and edited the manuscript.
References
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016;8 doi: 10.1101/cshperspect.a019505. http://dx.doi.org/10.1101/cshperspect.a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker PB, Horz W. ATP-dependent nucleosome remodeling. Annu Rev Biochem. 2002;71:247–273. doi: 10.1146/annurev.biochem.71.110601.135400. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Cai Y, Geutjes EJ, de Lint K, Roepman P, Bruurs L, Yu LR, Wang W, van Blijswijk J, Mohammad H, de Rink I, et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene. 2014;33:2157–2168. doi: 10.1038/onc.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci USA. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Dieuleveult M, Yen K, Hmitou I, Depaux A, Boussouar F, Bou Dargham D, Jounier S, Humbertclaude H, Ribierre F, Baulard C, et al. Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells. Nature. 2016;530:113–116. doi: 10.1038/nature16505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding N, Bonham EM, Hannon BE, Amick TR, Baylin SB, O’Hagan HM. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J Mol Cell Biol. 2016;8:244–254. doi: 10.1093/jmcb/mjv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran H, Johnstone SE, Van Neste L, Ohm J, Mosbruger T, Wang Q, Aryee MJ, Joyce P, Ahuja N, Weisenberger D, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012;22:837–849. doi: 10.1101/gr.131169.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Qin L, He T, Qin J, Hong J, Wong J, Liao L, Xu J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011;21:275–289. doi: 10.1038/cr.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong F, Chiu LY, Cox B, Aymard F, Clouaire T, Leung JW, Cammarata M, Perez M, Agarwal P, Brodbelt JS, et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription- associated DNA damage that promotes homologous recombination. Genes Dev. 2015;29:197–211. doi: 10.1101/gad.252189.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette S, Serra RW, Peng M, Hayes JA, Konstantinopoulos PA, Green MR, Cantor SB. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015;29:489–494. doi: 10.1101/gad.256214.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10:1291–1300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- Helbling Chadwick L, Chadwick BP, Jaye DL, Wade PA. The Mi-2/NuRD complex associates with pericentromeric heterochromatin during S phase in rapidly proliferating lymphoid cells. Chromosoma. 2009;118:445–457. doi: 10.1007/s00412-009-0207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer. 2011;11:588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ, Rendtlew Danielsen JM, Menard P, Sand JC, Stucki M, et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol. 2010;190:731–740. doi: 10.1083/jcb.200912135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nio K, Yamashita T, Okada H, Kondo M, Hayashi T, Hara Y, Nomura Y, Zeng SS, Yoshida M, Sunagozaka H, et al. Defeating EpCAM(+) liver cancer stem cells by targeting chromatin remodeling enzyme CHD4 in human hepatocellular carcinoma. J Hepatol. 2015;63:1164–1172. doi: 10.1016/j.jhep.2015.06.009. [DOI] [PubMed] [Google Scholar]
- O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer cell. 2011;20:606–619. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan MR, Hsieh HJ, Dai H, Hung WC, Li K, Peng G, Lin SY. Chromodomain helicase DNA-binding protein 4 (CHD4) regulates homologous recombination DNA repair, and its deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitor treatment. J Biol Chem. 2012;287:6764–6772. doi: 10.1074/jbc.M111.287037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29:3130–3139. doi: 10.1038/emboj.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1997;94:8010–8015. doi: 10.1073/pnas.94.15.8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuebel KE, Chen W, Cope L, Glockner SC, Suzuki H, Yi JM, Chan TA, Van Neste L, Van Criekinge W, van den Bosch S, et al. Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet. 2007;3:1709–1723. doi: 10.1371/journal.pgen.0030157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott TL, Rangaswamy S, Wicker CA, Izumi T. Repair of oxidative DNA damage and cancer: recent progress in DNA base excision repair. Antioxid Redox Signal. 2014;20:708–726. doi: 10.1089/ars.2013.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperlazza J, Rahmani M, Beckta J, Aust M, Hawkins E, Wang SZ, Zu Zhu S, Podder S, Dumur C, Archer K, et al. Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to genotoxic agents and reduces tumor formation. Blood. 2015;126:1462–1472. doi: 10.1182/blood-2015-03-631606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulli G, Di Micco R, d’Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012;12:709–720. doi: 10.1038/nrc3344. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature. 1998;395:917–921. doi: 10.1038/27699. [DOI] [PubMed] [Google Scholar]
- van Engeland M, Derks S, Smits KM, Meijer GA, Herman JG. Colorectal cancer epigenetics: complex simplicity. J Clin Oncol. 2011;29:1382–1391. doi: 10.1200/JCO.2010.28.2319. [DOI] [PubMed] [Google Scholar]
- Xie W, Ling T, Zhou Y, Feng W, Zhu Q, Stunnenberg HG, Grummt I, Tao W. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc Natl Acad Sci USA. 2012;109:8161–8166. doi: 10.1073/pnas.1201262109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DH, Waterland RA, Zhang P, Schady D, Chen MH, Guan Y, Gadkari M, Shen L. Targeted p16(Ink4a) epimutation causes tumorigenesis and reduces survival in mice. J Clin Invest. 2014;124:3708–3712. doi: 10.1172/JCI76507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Wang Z, Xie W, Cai Y, Xia L, Easwaran H, Luo J, Yen RC, Li Y, Baylin SB. Acetylation enhances TET2 function in protecting against abnormal DNA methylation during oxidative stress. Mol Cell. 2017;65:323–335. doi: 10.1016/j.molcel.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.