

Abstract
The recent identification of the somatic GNAQ mutation (c.548G > A) provides insight into the pathogenesis of Sturge–Weber syndrome (SWS). Although the primary SWS brain pathology is the leptomeningeal angiomatosis (LMA), cerebral cortical and white matter abnormalities play a prominent role in the disease manifestations. In some cases, SWS brain involvement is present even without detectable LMA on magnetic resonance imaging (MRI). To expand our understanding of the etiology of SWS brain pathology, surgical SWS brain specimens from nine children (age: 0.8–7.5 years) were carefully separated into LMA and (non-LMA) brain tissue; the latter did not contain any vascular malformation. A custom Competitive Allele-Specific TaqMan PCR (castPCR) assay to detect the mutation in GNAQ was performed in these separated specimens. The mutation was present in all nine LMA and seven of the nine non-LMA brain tissues. LMA tissues were significantly enriched by the mutation, as compared with non-LMA brain (mean: 7.2 ± 2.1% and 1.2 ± 0.4%, respectively; p = 0.008). These results demonstrate that the somatic GNAQ mutation in SWS is not confined to the venous vascular malformation but can directly (although less severely) affect underlying brain parenchyma, not directly affected by LMA, and possibly contribute to SWS brain pathology. Future studies should identify the specific cell type(s) affected by the mutation in the SWS-affected brain parenchyma.
Keywords: Sturge–Weber syndrome, GNAQ mutation, brain tissue, epilepsy, surgery, leptomeningeal angiomatosis
Introduction
Sturge–Weber syndrome (SWS) is a sporadic neurocutaneous disorder characterized by a facial port-wine stain (PWS), leptomeningeal angiomatosis (LMA), and vascular glaucoma.1 Both the PWS and the LMA are vascular developmental malformations, likely caused by a somatic mutation in the GNAQ gene.2 Damage of brain tissue underlying the LMA is thought to be secondary, caused by impaired venous blood flow resulting in chronic ischemia, neuronal loss, atrophy, and calcification, thus leading to cortical and white matter abnormalities on imaging.3–5 In addition to gliotic changes in SWS-affected brain, recent studies also demonstrated the common presence of cortical developmental malformations, hypertrophic pyramidal neurons, and overexpression of angiogenic factors in both neurons and glial cells from SWS surgical tissue.3,5–7 However, it is still not clear whether SWS brain tissue damage is always secondary to LMA-induced hypoxia or could develop or worsen due to primary involvement of non-LMA brain tissue.
Recent studies demonstrated that the GNAQ mutation is enriched in endothelial cell populations in both SWS skin capillary malformations and SWS brain specimens that included LMA.8,9 However, since LMA was not separated from underlying brain tissue, the presence of the mutation in non-LMA brain tissue remained unknown. To address this issue, the goal of this study was to identify the pattern of the somatic GNAQ mutations in surgical SWS brain specimens separated into LMA and non-LMA brain tissue.
Methods
Subjects
Epilepsy surgery brain tissues from nine children with unilateral SWS, who underwent resective surgery for medically refractory epilepsy at the Children’s Hospital of Michigan (Detroit, Michigan, United States) (age at surgery: 0.8–7.5 years), were analyzed in the study (Table 1). Presurgical magnetic resonance imaging (MRI) of all patients showed typical signs of unilateral SWS including leptomeningeal contrast enhancement consistent with LMA (Fig. 1A). The study protocol was approved by the Institutional Review Board at Wayne State University, and informed consent was obtained from the parents.
Table 1.
Clinical and GNAQ mutation data of the nine patients
| Patient no. | Gender | Age (y) at surgery | Age (y) at epilepsy onset | Seizure frequency | PWS | MRI (LMA) | Surgery | GNAQ % in LMA | GNAQ % in brain |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 0.8 | 0.2 | 1/wk | L | L FTPO | Hemispherectomy | 0.11 | 0.3 |
| 2 | Female | 1.5 | 0.4 | 5/y | R + L | R POF | Hemispherectomy | 10.3 | 0.5 |
| 3 | Female | 1.5 | 0.4 | 1/mo | R | R FTPO | Hemispherectomy | 14.4 | 2.4 |
| 4 | Male | 2 | 0.8 | 2/wk | L | L TPO | FTPOa resection | 4.8 | 2.9 |
| 5 | Female | 2.5 | 0.3 | 1/mo | L | L PO | TPO resection | 0.5 | 0 |
| 6 | Male | 5 | 3.5 | 2/wk | None | L TPO | TPO resection | 17.9 | 2.1 |
| 7 | Male | 5.3 | 0.2 | 4/d | L | L FTPO | Hemispherectomy | 9.1 | 2.4 |
| 8 | Female | 7.5 | 0.9 | 1/mo | R > L | R PTF | Hemispherectomy | 4.8 | 0 |
| 9 | Male | 9 | 1.8 | 4/y | R | R TPO | TPO resection | 2.6 | 0.16 |
Abbreviations: F, frontal; L, left; LMA, leptomeningeal angiomatosis; MRI, magnetic resonance imaging; O, occipital; P, parietal; PWS, port-wine stain; R, right; T, temporal.
Motor cortex spared.
Fig. 1.
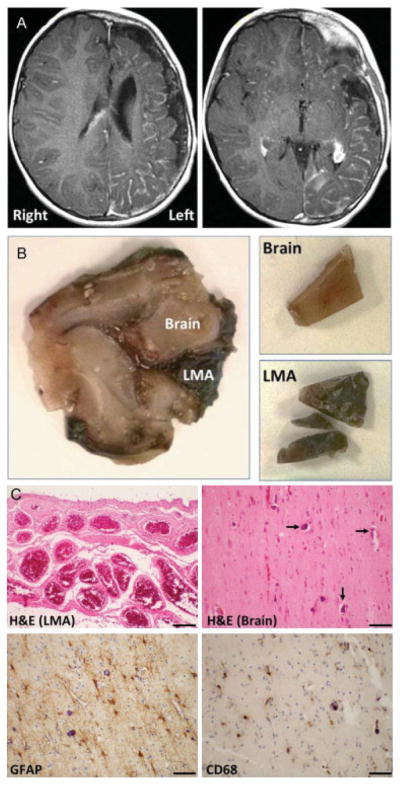
(A) Contrast-enhanced T1-weighted magnetic resonance imaging (MRI) of patient 7 (Table 1) showing extensive, left hemispheric leptomeningeal enhancement and enlarged choroid plexus consistent with severe unilateral Sturge–Weber syndrome. (B) Surgical specimen of the same patient before (left panel) and after separation (right panels) of the leptomeningeal angiomatosis (LMA) and non-LMA brain tissue (brain). Both tissues had detectable GNAQ mutation (9.1% in the LMA and 2.4% in non-LMA brain tissue). (C) Histopathological analysis of LMA and non-LMA brain tissue of the same patient. Hematoxylin and eosin (H&E) staining (top panels) shows both the characteristic LMA (top left) and calcifications (top right, arrows) in non-LMA brain. Denser calcifications were seen in other regions (not shown). Immunostaining for glial fibrillary acidic protein (GFAP, bottom left) demonstrates the reactive gliosis in the brain. Immunostaining for the macrophage/microglia marker CD68 (bottom right) shows scattered macrophages and reactive microglia in a diffuse pattern. Scale bars are 50 μM.
Tissue Processing
Formalin-fixed, paraffin-embedded (FFPE) tissues of surgical specimens underwent routine histopathology examination. Subsequently, tissue blocks containing both brain parenchyma and overlying LMA were selected and warmed to 60°C to liberate the tissue from the paraffin. Stainless-steel Teflon-coated 0.004-inch thick blades (Electron Microscopy Sciences, Hatfield, Pennsylvania, United States) were used to dissect the LMA and adjacent brain (Fig. 1B). Separate blades were used for each tissue block, one blade for the LMA and one for the brain tissue; the latter consisted of mostly white matter, with the tissue edges cut at least 3 mm away from the cortical surface to prevent LMA contamination.
DNA Extraction
Genomic deoxyribonucleic acid (DNA) was extracted from all samples with the ZR Genomic DNA-Tissue MiniPrep (Zymo Research, Irvine, California, United States) according to the manufacturer’s protocol for FFPE tissues. Briefly, tissues were deparaffinized by incubating with xylene at 20°C for 1 hour, twice. Tissue samples were rehydrated by washing with 100, 95, and 75% ethanol followed by one wash with DNAse-free water. Proteinase K digestion was performed at 55°C for 12 hours. DNA was spin-column purified and eluted in DNAse-free water. DNA samples were quantified with a NanoDrop 2000 spectrophotometer (ThermoFisher Scientific, Wilmington, Delaware, United States). The 260/280 ratios for all of the FFPE-derived DNA samples were 1.80 ± 0.07.
GNAQ Mutation Detection
A custom Competitive Allele-Specific TaqMan PCR (castPCR) assay to detect the c.548G > A mutation was obtained (GNAQ_52975_mu and GNAQ_52975_wt; ThermoFisher Scientific, Waltham, MA). The castPCR assay was performed according to the manufacturer’s instructions in a 20 μL reaction volume with 50 ng of genomic DNA and 1× TaqMan Genotyping Master Mix using a StepOnePlus Real-Time PCR System (ThermoFisher). Synthetic GeneArt Strings DNA Fragments (ThermoFisher) corresponding to wild-type and the mutant sequences were used for the assay DNA standards. Data were collected with a manual Cycle Threshold (CT) of 0.25. The raw data were analyzed using the Mutation Detector software (ThermoFisher). A calibration ΔCT value (Eq. 1) was calculated and used to normalize the mutant and reference assays. Normalized sample ΔCT values (Eq. 2) were obtained for each sample, and the % mutation was calculated (Eq. 3).
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
Statistical Analysis
The percentage of DNA containing the mutation in the nine paired LMA and non-LMA brain tissue samples were compared using the Wilcoxon signed-rank test. Based on the sensitivity threshold of 0.1%,10,11 the samples were also classified as positive or negative for the mutation. Percent GNAQ mutation in LMA and non-LMA brain samples were correlated with clinical seizure variables and extent of MRI brain involvement (number of lobes affected) using the Spearman’s rank correlation test. A p-value of less than 0.05 was considered to be significant.
Results
Histopathology in the resected brain tissues showed characteristic LMA (Fig. 1C). Underlying brain parenchyma showed moderate-to-severe gliosis and variable amounts of calcification, involving both cortex and white matter in all specimens (Fig. 1C). CD68 staining showed variably prominent populations of microglia and macrophages, which were extensive in some samples (Fig. 1C). Four patients had moderate-to-severe neuronal loss, three had milder neuronal loss, and two patients had no significant neuronal loss in the specimens. Intraparenchymal capillaries and veins were often prominent, but LMA was confined to the leptomeninges. None of the specimens had evidence of dysplasia or other cortical developmental abnormality.
Using the threshold of 0.1%, all nine LMA and seven out of nine non-LMA brain tissue samples were positive for the mutation (Table 1). The mean percentage mutation in LMA and non-LMA brain tissue was 7.2 ± 2.1 and 1.2 ± 0.4%, respectively, with LMA tissues significantly enriched by the mutation compared with corresponding non-LMA brain tissue (p = 0.008). GNAQ mutation percentage showed no correlation with clinical seizure variables or extent of brain involvement on MRI.
Discussion
Even though GNAQ gene mutation is likely the driver mutation in SWS, previous studies did not separate LMA from non-LMA-containing brain tissue to detect the mutation. In this study, we demonstrated that the mutation was present in all LMA samples, and the mutation frequency was higher in the LMA compared with non-LMA brain tissue in all but one SWS surgical sample; however, the majority of the non-LMA brain tissues also had a detectable amount (0.16–2.9%) of the somatic GNAQ mutation. The variability of mutation frequency across tissues could be due to differences in tissue composition, due to variation in tissue sampling, or due to a more fundamental factor such as heterogeneity in mutation origin time.
GNAQ mutation in nonvascular tissue elements has been reported in the facial PWS.12 In that study, 60% of the patients had mutations in the blood vessels only, 20% had mutations in connective tissue without blood vessel involvement, and the remaining 20% had mutations in both blood vessel and connective tissue. In another recent study, observation of GNAQ mutation in white blood cells and buccal swabs in 10% of the samples has been attributed to the common mesodermal origin of blood cells, buccal lymphocytes, and endothelial cells.13 These findings, together with our current data, indicate that while the vascular malformation are enriched in GNAQ mutation, there are also GNAQ mutation-affected cells in nonvascular tissue in a significant proportion of SWS cases. The particular cell type(s) harboring the mutation in the brain remain to be determined, although it has been speculated to involve cells of mesodermal lineage.13 Considering the presence of pyramidal neurons with abnormal morphology and cortical developmental malformations in some SWS patients,5–7 involvement of cells of neural crest origin in a postzygotic somatic mutation should also be explored. Identification of the cell type(s) in non-LMA brain parenchyma harboring the mutation would not only be informative for the timing of the mutation during embryonic development, but mutation in nonvascular cells would also provide a plausible new mechanism of brain damage extending beyond the region of the LMA; this could also explain SWS cases where no evidence of LMA can be detected despite clinical and/or radiological evidence of brain involvement.14
The GNAQ gene mutation is an activating mutation that may lead to increased cell proliferation through increased signaling through the mitogen-activated protein kinase (MAPK) pathway.2 In a previous study, both LMA and, to a lesser degree, cortical blood vessels showed elevated endothelial proliferation, whereas overexpression of vascular endothelial growth factor was found both in blood vessels and in neurons and glia of SWS surgical specimens.3 Whether the ongoing, low-level proliferation in SWS brain is related to the observed GNAQ mutation pattern in non-LMA brain tissue will require further mechanistic studies. Also, the role of mechanistic target of rapamycin (mTOR) pathway activity, hypothesized to play a role in SWS pathology,15 needs to be studied further in both LMA and non-LMA brain tissue.
It should be noted that patients with SWS who undergo epilepsy surgery typically exhibit a more severe form of the disease. Surgical specimens from such patients often contain brain regions with perivascular calcifications and cortical developmental abnormalities along with extensive gliosis,3,5,7,16,17 i.e., histopathology changes that could contribute to both severe epilepsy and neurocognitive deficits. It is possible that patients with a milder clinical phenotype of SWS would show a different pattern of GNAQ mutation in brain and LMA tissue. Nevertheless, the current findings strongly suggest that future studies of cellular origin of the mutation, and associated changes in protein expression and function, should not only focus on vascular elements but should also explore nonvascular cells of the underlying brain tissue to obtain a comprehensive understanding of the mechanisms involved in the pathoembryogenesis of SWS.
Acknowledgments
Funding
The study was partially funded by a grant from the National Institutes of Health (R01 NS041922 to C. J.). The Biobanking and Correlative Sciences Core is supported, in part, by NIH Center grant P30 CA022453 to the Karmanos Cancer Institute at Wayne State University.
Footnotes
Conflict of Interest
The authors report no conflict of interest.
References
- 1.Bodensteiner JB, Roach ES. Overview of Sturge-Weber syndrome. In: Bodensteiner B, Roach ES, editors. Sturge-Weber Syndrome. Mt Freedom, NJ: The Sturge-Weber Foundation; 2010. pp. 19–32. [Google Scholar]
- 2.Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comati A, Beck H, Halliday W, Snipes GJ, Plate KH, Acker T. Upregulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha in leptomeningeal vascular malformations of Sturge-Weber syndrome. J Neuropathol Exp Neurol. 2007;66(01):6–97. doi: 10.1097/nen.0b013e31802d9011. [DOI] [PubMed] [Google Scholar]
- 4.Juhász C, Haacke EM, Hu J, et al. Multimodality imaging of cortical and white matter abnormalities in Sturge-Weber syndrome. AJNR Am J Neuroradiol. 2007;28(05):900–906. [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto AL, Chen L, Friedman R, et al. Sturge-Weber syndrome: brain magnetic resonance imaging and neuropathology findings. Pediatr Neurol. 2016;58:25–30. doi: 10.1016/j.pediatrneurol.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Maton B, Krsek P, Jayakar P, et al. Medically intractable epilepsy in Sturge-Weber syndrome is associated with cortical malformation: implications for surgical therapy. Epilepsia. 2010;51(02):257–267. doi: 10.1111/j.1528-1167.2009.02304.x. [DOI] [PubMed] [Google Scholar]
- 7.Wang DD, Blümcke I, Coras R, et al. Sturge-Weber syndrome is associated with cortical dysplasia ILAE Type IIIc and excessive hypertrophic pyramidal neurons in brain resections for intractable epilepsy. Brain Pathol. 2015;25(03):248–255. doi: 10.1111/bpa.12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Couto JA, Huang L, Vivero MP, et al. Endothelial cells from capillary malformations are enriched for somatic GNAQ mutations. Plast Reconstr Surg. 2016;137(01):77e–82e. doi: 10.1097/PRS.0000000000001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang L, Couto JA, Pinto A, et al. Somatic GNAQ mutation is enriched in brain endothelial cells in Sturge-Weber syndrome. Pediatr Neurol. 2017;67:59–63. doi: 10.1016/j.pediatrneurol.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Didelot A, Le Corre D, Luscan A, et al. Competitive allele specific TaqMan PCR for KRAS, BRAF and EGFR mutation detection in clinical formalin fixed paraffin embedded samples. Exp Mol Pathol. 2012;92(03):275–280. doi: 10.1016/j.yexmp.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Applied Biosystems Product Bulletin. Accurate and sensitive somatic mutation detection powered by castPCR™ technology. Available at https://tools.thermofisher.com/content/sfs/brochures/cms_095916.pdf.
- 12.Tan W, Nadora DM, Gao L, Wang G, Mihm MC, Jr, Nelson JS. The somatic GNAQ mutation (R183Q) is primarily located within the blood vessels of port wine stains. J Am Acad Dermatol. 2016;74(02):380–383. doi: 10.1016/j.jaad.2015.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchiyama Y, Nakashima M, Watanabe S, et al. Ultra-sensitive droplet digital PCR for detecting a low-prevalence somatic GNAQ mutation in Sturge-Weber syndrome. Sci Rep. 2016;6:22985. doi: 10.1038/srep22985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39(04):591–620. doi: 10.1016/s0031-3955(16)38367-5. [DOI] [PubMed] [Google Scholar]
- 15.Shirazi F, Cohen C, Fried L, Arbiser JL. Mammalian target of rapamycin (mTOR) is activated in cutaneous vascular malformations in vivo. Lymphat Res Biol. 2007;5(04):233–236. doi: 10.1089/lrb.2007.1012. [DOI] [PubMed] [Google Scholar]
- 16.Di Trapani G, Di Rocco C, Abbamondi AL, Caldarelli M, Pocchiari M. Light microscopy and ultrastructural studies of Sturge-Weber disease. Childs Brain. 1982;9(01):23–36. doi: 10.1159/000120032. [DOI] [PubMed] [Google Scholar]
- 17.McCartney E, Squier W. Patterns and pathways ofcalcification in the developing brain. Dev Med Child Neurol. 2014;56(10):1009–1015. doi: 10.1111/dmcn.12493. [DOI] [PubMed] [Google Scholar]