

Summary
The bacteriophage T4 contractile tail (containing a tube and sheath) was the first biological assembly reconstructed in three-dimensions by electron microscopy at a resolution of ~ 35 Å in 1968. A single-particle reconstruction of the T4 baseplate was able to generate a 4.1 Å resolution map for the first two rings of the tube using the overall baseplate for alignment. We have now reconstructed the T4 tail tube at a resolution of 3.4 Å, more than a 1,000-fold increase in information content for the tube from 1968. We have used legacy software (Spider) to show that we can do better than the typical 2/3 Nyquist frequency. A reasonable map can be generated with only 1.5 electrons/Å2 using the higher dose images for alignment, but increasing the dose results in a better map, consistent with other reports that electron dose does not represent the main limitation on resolution in cryo-EM.
eTOC paragraph
Zheng et al. describe a 3.4 Å resolution cryo-electron microscopy reconstruction of the bacteriophage T4 tail tube, used for injecting DNA into bacterial cells. Remarkably, this was accomplished using legacy software. They show that once images are aligned, a reconstruction can be generated with a dose of 1.5 electrons/Å2.
INTRODUCTION
The revolution in cryo-EM (Bai et al., 2015; Egelman, 2016; Kuhlbrandt, 2014; Subramaniam et al., 2016) has meant that near-atomic resolution can now be reached for many more macromolecular complexes with many fewer images much more rapidly. High resolution cryo-EM structures with de novo atomic models are no longer an unusual feat but an almost daily occurrence. While many of these complexes can in principle be crystallized, it has become much easier to solve their structures by cryo-EM, in some cases at higher resolution than what has been achieved by crystallography (Ramakrishnan, 2014). In contrast, if a helical polymer does not have exactly 2, 3, 4 or 6 subunits per turn, it cannot be packed into a crystal so that every subunit is in an identical environment. This is a simple consequence of the finite number of space group symmetries that can exist. Therefore, certain model systems in structural biology, such as tobacco mosaic virus, have only been studied at high resolution using x-ray fiber diffraction (Namba and Stubbs, 1986) or cryo-EM (Ge and Zhou, 2011). Since helical protein and nucleoprotein polymers are ubiquitous in viruses, bacteria, archaea and eukaryotes, the recent advances in cryo-EM means that a huge category of assemblies of great biological interest that were previously viewed as almost intractable structural problems can now be solved at near-atomic resolution. New insights continue to be made into the methods, challenges and limitations in studying such helical assemblies. For example, the intrinisic ambiguities that can exist in determining the correct helical symmetry of a filament (Egelman, 2010) are exacerbated by the possibility that filaments in cryo-EM may be tilted out of the plane perpendicular to the electron beam (Egelman, 2014), in contrast to the assumption made when examining negatively stained samples that they are adsorbed to a substrate that is perpendicular to the beam. While it is possible to solve some helical assemblies at 3.2 Å resolution (Kudryashev et al., 2015), application of similar methods to other assemblies may only yield ~ 6 Å resolution (Kolappan et al., 2016) or worse due to the structural variability within these filaments.
Sequences can diverge to such an extent that two proteins sharing a common fold, and having obvious homology, can have unrecognizable sequence similarity. This has become clear in the case of the contractile tail system of double-stranded DNA (dsDNA) tailed bacteriophage. The tail assembly (containing an inner rigid tube and an outer contractile sheath) of bacteriophage T4 was the first object reconstructed in three-dimensions by electron microscopy (DeRosier and Klug, 1968). The resolution of this reconstruction from negatively stained specimens was ~ 35 Å. It has now become clear from structural studies that this contractile system, used to inject dsDNA into a bacterium, is a homolog of bacterial contractile systems used to transfer toxins, effectors and ions into other cells (Aksyuk et al., 2009; Basler and Mekalanos, 2012; Clemens et al., 2015; Ge et al., 2015; Kudryashev et al., 2015), even though sequence similarities among the components are not readily detected. We return to the T4 tail tube almost 50 years after DeRosier and Klug and show how we can now reach 3.4 Å resolution. Previous cryo-EM studies described this tube as “rather featureless” (Kostyuchenko et al., 2003), which has led to limited resolution in the past using different approaches. However, a large advance was made when a single-particle reconstruction of the T4 baseplate (Taylor et al., 2016) was able to generate a 4.1 Å resolution map for the first two rings of the tube using the overall baseplate for alignment. We now show that high resolution can be achieved for the tube alone with legacy software, and that a reasonable reconstruction can actually be generated using a dose as low as 1.5 electrons/Å2 when the higher dose frames are used for alignment.
RESULTS and DISCUSSION
The images for the tube reconstruction were the same ones previously used for a study of the T4 baseplate (Taylor et al., 2016), and were obtained with a sampling of 1.32 Å/pixel (Figure 1A). A power spectrum (Figure 1B) generated from segments of the tube showed a meridional at ~ 1/(40 Å) indicating an axial rise for the helical unit of ~ 40 Å. All other layer lines appeared to contain Bessel orders that were multiples of 6, consistent with a 6-fold rotational symmetry. The spacing of the “first” layer line (Figure 1B) was ~ 1/(133 Å), showing that the rotation along each helical strand was ~360° *40/(6*133) or ~ 18°. These parameters were us ed in an Iterative Helical Real Space Reconstruction (IHRSR) (Egelman, 2000) and converged to values of 40.2 Å and 18.2°. The most probable out-of-plane tilt angle was ~ 10° (Figure S1A), suggesting that the tubes were likely oriented at this unusual tilt by a preferred orientation of the baseplate in the thin ice. This preferred orientation of the baseplate was confirmed by examining the angular assignments in the Relion (Scheres, 2012) reconstruction of the baseplate (Figure S1B). The large tilt can generate problems in helical reconstructions (Egelman, 2014) if one starts with the initial assumption that all filaments are in a plane perpendicular to the electron beam. One can impose the wrong helical symmetry as a result of such an assumption, leading to an artefactual three-dimensional reconstruction. Since we allowed for out-of-plane tilt of filaments in the processing (see Methods), we avoided this problem.
Figure 1. Cryo-EM analysis of the tail tubes.

(A) An electron micrograph (Taylor et al., 2016) shows the tubes (black arrows) attached to the baseplates (white arrows).
(B) A power spectrum generated from 26,320 overlapping tube segments. The red arrow indicates the first meridional, at a spacing of ~ 1/(40 Å). The yellow arrow indicates the putative “first” layer line, at a spacing of ~ 1/(133 Å), while the blue arrow (at ~ 1/(8.4 Å) indicates the furthest layer line seen. The images have been multiplied by the CTF to boost the SNR, which is why the Thon rings are enhanced.
The initial reconstruction was generated from a motion-corrected stack of 38 out of 40 frames averaged into a single image, where each frame contained a dose of 1.5 electrons/Å2. To minimize both beam-induced image drift and optical instabilities, the first two frames were omitted. While this single image represented a large dose of ~ 60 electrons/Å2, many large sidechains could be seen in the reconstruction from this full dose image (Figure S2), consistent with a previous report that icosahedral viruses could be imaged at fairly high overall resolution using massive electron doses (Grant and Grigorieff, 2015).
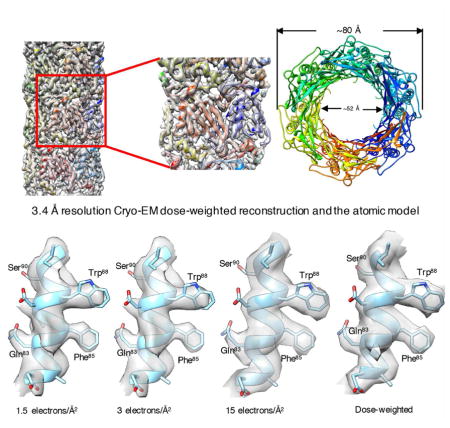
We then decided to explore the dependence of the quality of the reconstruction on the overall dose. First, we produced a reconstruction using drift-corrected frames 3–22 of the stack that corresponded to a dose of 30 electrons/Å2 (the first two frames were again discarded). We then applied the alignment parameters (x, y shifts and Euler angles) of this reconstruction to images that contained progressively fewer frames (sums of the first 10, 5, 3 or 2 frames), as well as to images obtained from only the first frame (frame 3 of the original stack). Somewhat surprisingly, the latter reconstruction that had a dose of only 1.5 electrons/Å2 (following the pre-irradiation of the first two frames with 3 electrons/Å2) looked reasonable (Figure 2A) while the averaged power spectrum of these images showed no visible layer lines due to the extremely poor SNR. The FSC comparison of two independent half-maps generated with this very low dose (Figure S3) gave a resolution of 4.2 Å. It should be noted, however, that no alignment of these images on their own would have been possible, and the alignment parameters were simply those from the 20-frame dataset. The minimum dose needed to properly and iteratively align helical segments, in our experience, varies from specimen to specimen, and is a function of the mass per unit length of filament as well as the presence or absence of internal point group symmetries (such as the C6 symmetry in these tubes). In the absence of such rotational symmetries, and with rather thin filaments, we have seen iterative alignments fail with as much as 10–15 electrons/Å2.
Figure 2. Dose limitation in determining the near-atomic resolution structure.

(A) A small section of the map calculated from only the third frame (corresponding to 1.5 electrons/Å2).
(B) The same area in a map created from a weighted-sum of the first 20 frames. The first two frames have been discarded, so the pre-irradiation in (A) is 3 electrons/Å2. A number of bulky sidechains are labelled, showing that they can be positioned quite well in both maps.
A better map (Figure 2B, Figure 3) can be produced using a simple weighted-average of the first 20 frames, similar to the weighting previously described (Grant and Grigorieff, 2015). In this weighting scheme, the first frame receives a weight six times that given to each of the last 10 frames, while the second frame receives a weight of five, etc. While many questions exist about the FSC being the ultimate measure of resolution in cryo-EM (Subramaniam et al., 2016), we used the standard approach and divided the data set into two completely non-overlapping half sets. Each set was used for IHRSR cycles, starting with the full reconstruction that had been filtered to 7 Å resolution. The resulting half-maps yielded a resolution of 3.4 Å using the FSC=0.143 criterion (Figure S3). However, comparison of the two FSC curves (from the 1.5 electrons/Å2 map and from the full dose-weighted map) shows that reducing resolution to a scalar (e.g., 3.4 or 4.2 Å) ignores the fact that there would be considerable differences in the quality of these maps at 6–7 Å resolution.
Figure 3. Overall reconstruction of the T4 tail tube.

(A) An outside view of the T4 tail tube cryo-EM map aligned with aomic model. Three subunits from one ring are shown in yellow, red and magenta. An α-helix (bottom) and a β-sheet (top) from the yellow subunit project out and interface with both the red subunit and subunits in adjacent rings.
(B) A close-up view from the lumen shows how the four β-strands of the red subunit become part of a continuous β-sheet which lines the lumen.
See also Figure S4.
It is clear that despite the two interpolations that have taken place in the image processing (once when e2helixboxer was used to cut filaments from images, and once when the segments are rotated and shifted to be used in the back-projection), and the fact that we have been using legacy software (Spider) written in the 1990s that does not take advantage of more sophisticated approaches, such as Fourier-grid sampling (Yang and Penczek, 2008), we have still been able to do better than 2/3 Nyquist (which would be 4.0 Å for our images). A previous paper (Bammes et al., 2012) showed that it was possible to exceed 2/3 Nyquist in a three-dimensional reconstruction using a direct electron detector, but this was at what would now be considered low resolution.
We then used the 3.4 Å resolution map calculated using the weighing scheme to refine an atomic model of the tube. The starting point was a model for two rings of the tube contained in the baseplate-tube structure (PDB code 5IV5) (Taylor et al., 2016). The model was rebuilt manually with Coot (Emsley et al., 2010), followed by real-space refinement with Phenix (Adams et al., 2010). The refinement statistics are given in Table 1. While the overall rmsd between Cα atoms in the refined model and the starting model was only 0.7 Å, the positions of sidechains are more reliable in the new model. This is apparent in the refinement statistics given in Table S1. While the overall improvement in the model is relatively small, the improvement in the helical map over the single particle map previously used is quite large (Figure S4), showing that with current tools a cryo-EM map of modest quality at 4.1 Å resolution can be used to generate a fairly reliable atomic model.
Table 1.
Atomic model refinement and validation statistics
| T4 tube | R-type pyocin | |
|---|---|---|
| Cryo-EM data collection | ||
| Microscope | FEI Titan Krios | |
| Voltage (keV) | 300 | |
| Camera | Gatan K2-Summit Gatan Quantum |
|
| Energy Filter | LS (GIF) | |
| Pixel size (Å) | 1.32 | |
| Defocus range (μm) | 0.5–33 | |
| 3D reconstruction | ||
| Number of movies | 333 | |
| Final number of segments | 26,320 | |
| Resolution (Å) | 3.4 | |
| Sharpening B-factor (Å2) | 150 | |
| Coordinate and B-factor refinement | ||
| Refinement high resolution limit (Å) | 3.4 | 3.5 |
| Number of protein atoms (non-H) | 23,472 | 37,890 |
| Mean B-factor protein atoms (Å2) | 126.98 | 46.37 |
| RMSD bonds (Å) | 0.01 | 0.01 |
| RMSD bond angles (°) | 0.78 | 0.95 |
| Map CC (around atoms) | 0.834 | 0.773 |
| Validation | ||
| Ramachandran plot | ||
| Favored (%) | 95.86 | 93.94 |
| Allowed (%) | 4.14 | 6.06 |
| Disallowed (%) | 0.00 | 0.00 |
| Molprobity score | 1.65 | 1.46 |
| All-atom clashscore | 6.56 | 2.66 |
| Rotamer outliers (%) | 0.00 | 0.00 |
The T4 tube is homologous to the tubes of other contractile injection systems that include R-type pyocins (Ge et al., 2015), Clostridium difficile “diffocins” and Photorhabdus virulence cassette (PVC) (Ghequire and De Mot, 2015), Serratia entomophila antifeeding prophage (afp) (Heymann et al., 2013), Pseudoalteromonas luteoviolacea MAC arrays (Shikuma et al., 2014), and the bacterial Type VI Secretion System (T6SS) (Clemens et al., 2015; Kudryashev et al., 2015). These tubes however have evolved to translocate different substrates and the nature of the substrate thus determines the properties of the tube’s channel. Atomic structures of three different tube proteins are now available: T6SS, R-type pyocin and T4. Different T6SS complexes package different effector proteins into their tubes (Silverman et al., 2013), and the T6SS channels are therefore diverse. The main substrates of the T4 and pyocin channels are quite different: negatively charged phage DNA in the case of T4, and an unknown mixture of ions for the pyocin channel. With the structure of T4 tube in hand we decided to compare the surface charge properties of the two structures. However, the deposited atomic model for the pyocin tube (3J9Q) has very poor refinement scores. It is of insufficient quality for electrostatic calculations that are sensitive to proper surface residue placement. We re-refined this model using Rosetta (Wang et al., 2015) and Phenix (Adams et al., 2010) against the corresponding segmented map of EMD-6270 (Ge et al., 2015). As a result, the MolProbity statistics including Ramachandran favored/outliers, favored rotamer, and MolProbity score are improved from 68.5%/7.9%, 88.2%, and 2.87to 93.9%/0%, 100%, and 1.46, respectively (Figure S5; Table S1). The rmsd between the Cα atoms in our refined model and in 3J9Q is only 0.5 Å, showing that while the overall chain trace is the same, secondary structure is now much better defined.
Comparison of the refined pyocin tube subunit with that of the T4 tail tube (Figure 4A) shows that 126 Cα atom pairs of the two proteins are very similar with a rmsd of 2.7 Å, and the Dali server (Hasegawa and Holm, 2009) gives a highly significant Z-score of 8.1 for this pairwise comparison, the two share only about 10% amino acid identity. A striking difference between the two can be seen (Figure 4C and 4E) in a comparison of the electrostatic potentials of the lumen (Baker et al., 2001; Dolinsky et al., 2007). A previous report using the deposited model (3J9Q) found an overall negative charge for the lumen of the tube (Ge et al., 2015). While the pyocin lumen using our refined model (Figure 4E) appears to be almost equally positive, negative and neutral, the T4 tail tube lumen (Figure 4C) is largely electronegative. Such a potential would repel the negatively-charged phosphate groups along the DNA phosphodiester backbone, and serve to “lubricate” the passage of DNA through this lumen as the DNA would be unable to stick to this surface. This would be similar to the reduction in friction provided by magnetic levitation, where a train is repelled from the track along which it travels. A similar electronegative potential has been seen in the lumen of bacterial sex pili (Costa et al., 2016), used for transporting DNA between mating cells, where the electronegative character is provided by the phosphate groups of stoichiometrically bound lipids.
Figure 4. Comparison between T4 tail tube and pyocin tube.

(A) A subunit from the T4 tail tube (red) is superimposed upon the refined subunit from the R-type pyocin tube (green). A comparison between the molecular surfaces colored according to the electrostatic potential for the T4 tail tube (B,C) and the R-type pyocin tube (D,E). The outer surfaces of the tubes are shown in (B,D) while the surface lining the lumen is shown in (C,E). Negative potential is red, while positive is blue, with neutral white. As expected for a tube transporting DNA, the lumen of the T4 tail tube (C) is quite negative, which would serve to “lubricate” the walls so that DNA would not stick. The red color corresponds to a potential of -10 kT/e whereas the blue color corresponds to a potential of +10 kT/e.
CONCLUDING REMARKS
Here we show that a map carrying high resolution information that is sufficient for building an atomic model can be generated with a dose of only 1.5 electrons/Å2 with the projection alignment information extracted from a higher dose images. Furthermore, this can be accomplished with legacy software. We also show that the lumen of the T4 tube is negatively charged in agreement with its DNA transport function. Finally, we show that the lumen of the re-refined tube of the R-type pyocin, a hypothetical ion channel, does not display an overall preferential charge.
STAR Methods
Image Processing
Images were those described previously for the baseplate (Taylor et al., 2016). The program CTFFIND3 was (Mindell and Grigorieff, 2003) used for determining the defocus and astigmatism. The SPIDER software package (Frank et al., 1996) was used for most other operations. The CTF was corrected by multiplying the images by the theoretical CTF (a Wiener filter in the limit of a very poor SNR), both correcting the phases where needed and improving the SNR. The e2helixboxer routine within EMAN2 (Tang et al., 2007) was used for boxing the tubes from the images.
The filament segments were initially decimated by a factor of 4 from 1.32 Å/px to 5.28 Å/px. They were used in a preliminary IHRSR (Egelman, 2000) reconstruction starting with a solid cylinder as an initial reference, with no out-of-plane tilt considered. To prevent the symmetry parameters (axial rise and rotation per subunit) from diverging from their correct value due to not including the out-of-plane tilt, these parameters were constrained. A search range of 5 pixels in x and y translation for each segment was used. Since the large axial rise (~ 40 Å) was within the search range in y (±26.4 Å), there was no difficulty in bring any segment into alignment with any reference projection. After nine cycles the x- and y-shifts that were found for each segment were converted to nearest integers, and these shifts were then applied to segments that were decimated by a factor of 2 as well as the undecimated segments. Since the shifts were integer, no interpolations were needed and segments were simply shifted by rows or columns. The shifted segments decimated by 2 (to 2.64 Å/px) were then used in IHRSR cycles allowing for out-of-plane tilt, with the constraints relaxed on the helical parameters. Since the segments were now aligned axially, a search range of 3 pixels (±7.92 Å) was adequate even though the large axial rise would otherwise have required a search range of ±20 Å. After 14 cycles, the x- and y-shifts now found for each segment were converted to nearest integers, and these were again applied to the undecimated images. IHRSR cycles were then started with the undecimated images (1.32 Å/px), using a search range of 3 pixels.
Segments were not included in the reconstruction if they met either of two criteria: 1) they were assigned an in-plane rotation angle that was more than 5° from either 0° or 180°; or 2) they were classified as having an out-of-plane tilt of more than 30°. Satisfying either criteria suggested that the particles were misaligned. Every segment was treated completely independently in the IHRSR cycles, as no constraints were imposed upon a segment from the parameters (e.g., in-plane tilt, out-of-plane tilt, azimuthal orientation, polarity) found for surrounding segments. A total of 26,320 overlapping segments were used for the final reconstruction.
Model Refinement
The T4 tube reconstruction map was used for the gp19 tube model refinement. The previous tube model from within the T4-baseplate complex (PDB ID:5IV5) was manually refined in COOT (Emsley et al., 2010) and then real-space refinement was performed in PHENIX (Adams et al., 2010). Rosetta (Wang et al., 2015) was used to refine the deposited pyocin model (PDB ID:3J9Q), followed by real-space refinement in PHENIX. Both models were validated with MolProbity (Chen et al., 2010). The refinement statistics are given in Table 1.
DATA AND SOFTWARE AVAILABILITY
Coordinates of the refined T4 and pyocin tubes have been deposited to the Protein Data Bank under the accession codes 5W5F and 5W5E, respectively. The cryo-EM density map of the T4 tail tube has been deposited to the Eelectron Microscopy Data Bank under the accession code EMD-8767.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Edward Egelman (egelman@virginia.edu).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| T4 tail tube cryo-EM density map | This study | EMD-8767 |
| Refined T4 tail tube atomic model | This study | 5W5F |
| Refined Pyocin tail tube refined model | This study | 5W5E |
| Software and Algorithms | ||
| EMAN2 | Tang et al., 2007 | http://blake.bcm.edu/emanwiki/EMAN2 |
| CTFFIND3 | Mindell and Grigorieff, 2003 | http://grigoriefflab.janelia.org/ctf |
| Spider | Frank et al., 1996 | https://spider.wadsworth.org/spider_doc/spider/docs/spider.html |
| UCSF Chimera | Pettersen et al., 2004 | http://www.cgl.ucsf.edu/chimera |
| Coot | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org |
| Rosetta | Wang et al., 2015 | https://faculty.washington.edu/dimaio/wordpress/software/ |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
Supplementary Material
Highlights.
3.4 Å resolution map for the bacteriophage T4 tail tube.
Can achieve near-atomic resolution reconstruction with a dose of 1.5 electrons/Å2.
Refined T4 tube and Pyocin tube model show improved stereochemistry.
The electronegative lumen of T4 tail tube is used for DNA delivery.
Acknowledgments
The work was supported by NIH GM122510 (to E.H.E.).
Footnotes
AUTHOR CONTRIBUTIONS
E.H.E wrote the manuscript. E.H.E and W.Z. processed the cryo-EM data. W.Z, F.W and N.M.I.T refined the models. R.C.G.-F. and N.M.I.T collected the cryo-EM data. All the authors were involved in revising the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksyuk AA, Leiman PG, Kurochkina LP, Shneider MM, Kostyuchenko VA, Mesyanzhinov VV, Rossmann MG. The tail sheath structure of bacteriophage T4: a molecular machine for infecting bacteria. EMBO J. 2009;28:821–829. doi: 10.1038/emboj.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, McMullan G, Scheres SH. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 2015;40:49–57. doi: 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bammes BE, Rochat RH, Jakana J, Chen DH, Chiu W. Direct electron detection yields cryo-EM reconstructions at resolutions beyond 3/4 Nyquist frequency. Journal of Structural Biology. 2012;177:589–601. doi: 10.1016/j.jsb.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M, Mekalanos JJ. Type 6 secretion dynamics within and between bacterial cells. Science. 2012;337:815. doi: 10.1126/science.1222901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens DL, Ge P, Lee BY, Horwitz MA, Zhou ZH. Atomic Structure of T6SS Reveals Interlaced Array Essential to Function. Cell. 2015;160:940–951. doi: 10.1016/j.cell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa TR, Ilangovan A, Ukleja M, Redzej A, Santini JM, Smith TK, Egelman EH, Waksman G. Structure of the Bacterial Sex F Pilus Reveals an Assembly of a Stoichiometric Protein-Phospholipid Complex. Cell. 2016;166:1436–1444. e1410. doi: 10.1016/j.cell.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosier DJ, Klug A. Reconstruction of three-dimensional structures from electron micrographs. Nature. 1968;217:130–134. doi: 10.1038/217130a0. [DOI] [PubMed] [Google Scholar]
- Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic acids research. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman EH. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- Egelman EH. Reconstruction of helical filaments and tubes. Methods in Enzymology. 2010;482:167–183. doi: 10.1016/S0076-6879(10)82006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman EH. Ambiguities in Helical Reconstruction. eLife. 2014;3:e04969. doi: 10.7554/eLife.04969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman EH. The Current Revolution in Cryo-EM. Biophys J. 2016;110:1008–1012. doi: 10.1016/j.bpj.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. Journal of Structural Biology. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Ge P, Scholl D, Leiman PG, Yu X, Miller JF, Zhou ZH. Atomic structures of a bactericidal contractile nanotube in its pre- and postcontraction states. Nat Struct Mol Biol. 2015;22:377–382. doi: 10.1038/nsmb.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge P, Zhou ZH. Hydrogen-bonding networks and RNA bases revealed by cryo electron microscopy suggest a triggering mechanism for calcium switches. ProcNatlAcadSciUSA. 2011;108:9637–9642. doi: 10.1073/pnas.1018104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghequire MG, De Mot R. The tailocin tale: peeling off phage tails. Trends in microbiology. 2015;23:587–590. doi: 10.1016/j.tim.2015.07.011. [DOI] [PubMed] [Google Scholar]
- Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. eLife. 2015;4:e06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H, Holm L. Advances and pitfalls of protein structural alignment. Curr Opin Struct Biol. 2009;19:341–348. doi: 10.1016/j.sbi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Heymann JB, Bartho JD, Rybakova D, Venugopal HP, Winkler DC, Sen A, Hurst MR, Mitra AK. Three-dimensional structure of the toxin-delivery particle antifeeding prophage of Serratia entomophila. J Biol Chem. 2013;288:25276–25284. doi: 10.1074/jbc.M113.456145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolappan S, Coureuil M, Yu X, Nassif X, Egelman EH, Craig L. Structure of the Neisseria meningitidis Type IV pilus. Nature communications. 2016;7:13015. doi: 10.1038/ncomms13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyuchenko VA, Leiman PG, Chipman PR, Kanamaru S, van Raaij MJ, Arisaka F, Mesyanzhinov VV, Rossmann MG. Three-dimensional structure of bacteriophage T4 baseplate. Nat Struct Biol. 2003;10:688–693. doi: 10.1038/nsb970. [DOI] [PubMed] [Google Scholar]
- Kudryashev M, Wang RY, Brackmann M, Scherer S, Maier T, Baker D, DiMaio F, Stahlberg H, Egelman EH, Basler M. Structure of the Type VI Secretion System Contractile Sheath. Cell. 2015;160:952–962. doi: 10.1016/j.cell.2015.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrandt W. Cryo-EM enters a new era. eLife. 2014;3:e03678. doi: 10.7554/eLife.03678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of Structural Biology. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Namba K, Stubbs G. Structure of tobacco mosaic virus at 3.6 A resolution: implications for assembly. Science. 1986;231:1401–1406. doi: 10.1126/science.3952490. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V. The ribosome emerges from a black box. Cell. 2014;159:979–984. doi: 10.1016/j.cell.2014.10.052. [DOI] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikuma NJ, Pilhofer M, Weiss GL, Hadfield MG, Jensen GJ, Newman DK. Marine tubeworm metamorphosis induced by arrays of bacterial phage tail–like structures. Science. 2014;343:529–533. doi: 10.1126/science.1246794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JM, Agnello DM, Zheng H, Andrews BT, Li M, Catalano CE, Gonen T, Mougous JD. Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Molecular Cell. 2013;51:584–593. doi: 10.1016/j.molcel.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Earl LA, Falconieri V, Milne JL, Egelman EH. Resolution advances in cryo-EM enable application to drug discovery. Curr Opin Struct Biol. 2016;41:194–202. doi: 10.1016/j.sbi.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. Journal of Structural Biology. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Taylor NM, Prokhorov NS, Guerrero-Ferreira RC, Shneider MM, Browning C, Goldie KN, Stahlberg H, Leiman PG. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature. 2016;533:346–352. doi: 10.1038/nature17971. [DOI] [PubMed] [Google Scholar]
- Wang RY, Kudryashev M, Li X, Egelman EH, Basler M, Cheng Y, Baker D, DiMaio F. De novo protein structure determination from near-atomic-resolution cryo-EM maps. Nature methods. 2015;12:335–338. doi: 10.1038/nmeth.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Penczek PA. Cryo-EM image alignment based on nonuniform fast Fourier transform. Ultramicroscopy. 2008;108:959–969. doi: 10.1016/j.ultramic.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.