

Abstract
Inflammation mediated by activation of Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling is a major cause of chemotherapy resistance in cancer. We studied the impact of selectively blocking the IL-6 receptor (IL6R) as a strategy to inhibit IL-6-induced STAT activation and to overcome chemoresistance in pancreatic ductal adenocarcinoma (PDAC). To do this, STAT activation was investigated in tumors arising spontaneously in LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1Cre (KPC) mice. Plasma from patients with PDAC was assessed for its ability to activate STAT3/SOCS3 in human monocytes using immunofluorescence microscopy and quantitative gene expression assays. KPC mice and syngeneic mice (wild-type and IL6−/−) implanted with KPC-derived cell lines were treated with an IL6R blocking antibody (anti-IL6R). The impact of treatment on tumor growth in KPC mice and mice with KPC-derived tumor implants was monitored using ultrasonography and calipers, respectively. Tumors were analyzed by immunohistochemistry to detect changes in STAT activation, tumor viability and proliferation. We found that STAT3 was the most activated STAT protein in PDAC tumors from KPC mice. Plasma from patients with advanced PDAC stimulated STAT3/SOCS3 activation in human monocytes. In mice, anti-IL6R antibodies targeted Ly6Chi monocytes, inhibited STAT3 activation in tumor cells and decreased tumor cell proliferation in vivo. IL6R blockade in combination with chemotherapy induced tumor cell apoptosis, tumor regressions and improved overall survival. Overall, we show that IL-6 signaling drives STAT3 activation in tumor cells and mediates chemoresistance in PDAC. Thus, disrupting IL-6 signaling using anti-IL6R antibodies holds promise for improving chemotherapy efficacy in PDAC.
Keywords: IL-6, Stat3, pancreatic cancer, IL-6 receptor, chemoresistance
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is projected to become the second leading cause of cancer-related deaths by 2030 (1). This dismal outlook associated with PDAC is due at least in part to its poor responsiveness to standard cytotoxic therapies, including chemotherapy and radiation. A key determinant of this treatment resistance is the tumor microenvironment, which demonstrates extensive fibrosis and poor vascularity that together result in elevated interstitial fluid pressures capable of impeding drug delivery (2–4). In addition, PDAC recruits a robust inflammatory response, composed of immature myeloid cells, macrophages and granulocytes, which may also limit treatment efficacy (5–7). Inhibiting the recruitment of myeloid cells to tumors using inhibitors of chemokine receptor signaling pathways (e.g. CCL2/CCR2) can enhance the efficacy of chemotherapy and radiotherapy (8–10). Myeloid cell depletion using a colony stimulating factor 1 receptor (CSF1R) inhibitor has also been shown to enhance the efficacy of chemotherapy in PDAC mouse models (11). In addition, strategies designed to deplete elements of the extracellular matrix (e.g. collagen and hyaluronan) have been found to improve drug delivery and the cytotoxic effects of chemotherapy (2, 3). Tumor infiltrating myeloid cells can also be redirected to facilitate depletion of extracellular matrix components and in doing so, enhance the efficacy of chemotherapy (12, 13). Thus, elements of the tumor microenvironment, including fibrosis and inflammation, are key determinants of treatment resistance in PDAC.
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is an important driver of cytokine-mediated cancer inflammation in many human malignancies (14). For PDAC development, activation of the STAT3 pathway in inflammatory cells is necessary to promote pancreatic intraepithelial neoplasia (PanIN) progression (15, 16). Specifically, inhibiting STAT3 activation genetically can block PanIN progression and reduce the development of PDAC in mouse models. However, the development of selective STAT3 inhibitors has been challenging and currently there are no direct STAT3 inhibitors in clinical trials for cancer.
The importance of the JAK/STAT pathway for defining PDAC resistance to chemotherapy has been investigated in mouse models where a non-selective inhibitor of JAK1/2, that blocks signaling via multiple STAT proteins including STAT1, STAT3, STAT5, and STAT6, was shown to improve the activity of chemotherapy (17). Similarly, initial results with the non-selective JAK1/2 inhibitor Ruxolitinib in combination with chemotherapy (versus placebo plus chemotherapy) showed early promising activity in a pre-specified subgroup analysis of PDAC patients with elevated serum C-reactive protein (18). However, two randomized Phase III trials subsequently failed to demonstrate clinical benefit with adding Ruxolitinib to chemotherapy in PDAC patients with metastatic disease selected based on evidence of systemic inflammation (CRP > 10mg/dl) (19). Because non-selective inhibition of the JAK/STAT pathway can produce significant toxicities such as anemia (19, 20) as well as thrombocytopenia, neurotoxicity and increased rates of infection (20), alternative strategies that selectively inhibit JAK1 activity are being investigated for the treatment of PDAC with promising results seen in combination with chemotherapy in a recent early phase clinical study in metastatic PDAC (21). Nonetheless, the role of JAK/STAT signaling in determining chemotherapy efficacy remains ill-defined.
Here, we examined the effect of targeting the IL-6/IL-6R signaling pathway as a means of selectively inhibiting STAT3 activation in PDAC for enhancing the efficacy of cytotoxic chemotherapy. We found that STAT3 was the dominant STAT protein activated within tumors arising spontaneously in the clinically relevant KrasLSL-G12D/+, Trp53LSL-R172H/+, Pdx1-Cre (KPC) mouse model of PDAC (22). In addition, plasma-derived soluble factors present within the peripheral blood of patients with advanced PDAC were found to activate the STAT3/SOCS3 pathway in monocytes demonstrating systemic activation of this pathway in patients. Anti-IL-6R blocking antibodies were used to disrupt IL-6/STAT3 signaling and were found to rapidly bind to a subset of monocytes that are actively recruited to tumors. Anti-IL-6R treatment also inhibited STAT3 activation, mainly in malignant cells, within the tumor microenvironment – a finding that was reproduced in IL-6 knockout mice demonstrating the importance of host-derived IL-6 in regulating STAT3 signaling in malignant cells. Finally, therapeutic blockade of IL-6R sensitized PDAC tumors to chemotherapy-induced apoptosis leading to improved survival in tumor implantation models and tumor regressions in KPC mice. Together, our data demonstrate a role for IL-6 in regulating chemosensitivity in PDAC. Findings have immediate clinical implications given the availability of a clinical grade FDA approved IL-6R antagonist (i.e. tocilizumab) and warrant clinical evaluation of IL-6R blocking antibodies as a means for enhancing the efficacy of cytotoxic chemotherapy in PDAC.
MATERIALS AND METHODS
Animals
KrasLSL-G12D/+, Trp53LSL-R172H/+, Pdx1-Cre (KPC) mice have been previously described (22). These mice develop pancreatic intraepithelial neoplasia (PanIN), which progresses to invasive PDAC. C57BL/6 (B6) mice (Jackson Laboratories), IL-6 deficient mice (IL-6−/−) on the B6 background (Jackson Laboratories) and normal healthy littermate mice (Trp53LSL-R172H/+, Pdx1-Cre; PC) on the B6 background were used in experiments as controls and for tumor implantation studies. Animal protocols were reviewed and approved by the Institute of Animal Care and Use Committee (IACUC) of the University of Pennsylvania.
Clinical Samples
Plasma was collected by centrifugation of peripheral blood from patients with newly diagnosed chemotherapy-naïve metastatic pancreatic ductal adenocarcinoma. Quantification of IL-6 was performed using Luminex bead array technology (Life Technologies) as previously described (23). Written informed consent was required and the study was conducted in accordance with the Declaration of Helsinki and approved by local institutional review boards.
Cell lines
Previously described murine PDAC cell lines derived from KPC mice backcrossed onto the B6 background were used including 152.PDA (derived August 2012) (10), 7940B.PDA (derived June 2013) (12), and 69.PDA (derived August 2012) (24). Cell lines were authenticated based on histologic analysis of the implanted cell line with comparison to the primary tumor from which the cell line was derived. Cell lines were negative for Mycoplasma contamination (tested on 10/27/2016). Cell lines were grown in DMEM with 10% FBS supplemented with 83 μg/mL gentamicin and L-glutamine.
Animal Experiments
KPC mice were monitored weekly for the presence of spontaneous pancreatic tumor development. Tumors were detected by palpation and confirmed by ultrasonography as previously described (13). Mice with tumors measuring approximately three to five millimeters (mm) in diameter were block randomized and enrolled into studies. For tumor implantation studies, PDAC cell lines were injected subcutaneously or orthotopically into syngeneic B6, B6.PC, or IL-6−/− mice as previously described (24). Tumors were allowed to develop over 14 days to approximately 5 mm in diameter before being enrolled into treatment studies. Tumor volume (mm3) was calculated as V = 1/2 (L × W2) by determining the longest (L) and shortest (W) dimensions using calipers. Mice were treated by intraperitoneal injection of endotoxin-free antibodies purchased from BioXcell including: anti-IL-6Ralpha (clone 15A7) or rat isotype control (clone LTF-2). For survival and tumor growth studies, 0.2 mg of anti-IL-6Ralpha or isotype control antibodies were administered twice weekly. Gemcitabine (Gemzar™, Eli Lilly) pharmaceutical grade powder was purchased through the Hospital of the University of Pennsylvania Pharmacy and resuspended in sterile normal saline at 38 mg/ml 2′-deoxy-2′,2′-difluorocytidine. Gemcitabine (120 mg/kg) was administered by intraperitoneal injection on days 0 and 7 of treatment.
Histology, immunohistochemistry and immunofluorescence analysis
Immunohistochemistry and immunofluorescence staining were performed on frozen tissue sections. Frozen sections were air dried and fixed with 3% formaldehyde. Primary antibodies against mouse antigens included: rabbit anti-pSTAT1 (Cell Signaling, Beverly, MA), rabbit anti-pSTAT3 (Cell Signaling), rabbit anti-pSTAT5 (Cell Signaling), rabbit anti-pSTAT6 (Abcam), rabbit anti-CC3 (Cell Signaling), and rat anti-Ki67 (Dako).
For immunofluorescence staining, sections were blocked with 10% normal goat serum in PBS + 0.1% Tween-20 (PBST). For detection of intracellular antigens, following fixation with 3% formaldehyde, tissues were incubated with 100% methanol at −20 °C and permeabilized in blocking solution with 0.3% Triton X-100. Tissues were stained with primary antibody in blocking buffer for 60 minutes at room temperature or overnight at 4°C. Sections were washed in PBST and then incubated with Alexa488- or Alexa568-conjugated goat anti-rabbit or goat anti-rat IgG (Life Technologies, Eugene, OR) for 45 minutes at room temperature to visualize the antigen of interest. Nuclei were stained with DAPI.
For immunohistochemistry, endogenous peroxidases were quenched in 0.3% H2O2 in water for 10 minutes and then blocked with 10% normal goat serum in PBST. For detection of intracellular antigens, tissues were incubated with 100% methanol −20 °C and permeabilized in blocking solution with 0.3% Triton X-100. Primary antibody in blocking buffer was applied to tissues for 60 minutes at room temperature or overnight at 4°C. Sections were washed in PBS and then incubated with goat anti-rabbit (Jackson ImmunoResearch, West Grove, PA) or anti-rat biotinylated IgG (BD Biosciences, San Jose, CA). Staining was detected using Vectastain ABC kit (Vector Labs, Burlingame, CA) and sections were counterstained with hematoxylin.
Brightfield images were acquired on a BX43 upright microscope (Olympus) and immunofluorescence images were acquired on an IX83 inverted fluorescence microscope (Olympus). Immunostaining was quantified by analysis of 4–6 high power fields per tissue section. Quantification of phosphorylated STAT proteins in tissues was determined by calculating the number of cells demonstrating pSTAT expression per high power field.
Flow Cytometry
Peripheral blood samples were collected from the tail vein of mice or from human healthy donors. Murine blood samples were centrifuged and red blood cells lysed using ACK Lysis Buffer (Cambrex/BioWhittaker). PDAC tumors were harvested and minced in digestion media containing collagenase (1 mg/mL), dispase (1U/mL) and DNase (150U/mL) and processed as previously described (10). Cell surface molecule analysis of single cell suspensions obtained from peripheral blood or tumor tissue was performed at 4°C for 15 minutes in PBS containing 0.2 mM EDTA with 2% FCS and analyzed on a FACS CANTO (BD Biosciences). Antibodies against mouse antigens were purchased from BD Biosciences unless otherwise specified: CD19 (1D3, APC), CD45 (30-F11, PE-Cy7), F4/80 (eBiosciences, BM8, FITC), Ly6C (AL-21, APC-Cy7), Ly6G (1A8, Percp-Cy5.5), CD3 (BioLegend, 1782, Pacific Blue), IL-6R (BioLegend, D7715A7, PE), IL-6Ralpha (BioXCell, 15A7), biotinylated anti-rat, and streptavidin (PE). Antibodies against human antigens were purchased from BD Biosciences unless otherwise specified: CD56 (NCAM16.2, FITC), CD3 (SK7, PerCP-Cy5.5); CD19 (SJ25C1, PE-Cy7); CD14 (MφP9, APC-Cy7); IL-6Rα (Biolegend, 9C4, PE).
ELISA to detect rat antibodies
B6 mice received intraperitoneal injections with endotoxin-free antibodies purchased from BioXcell including: anti-IL-6Ralpha (clone 15A7) and rat isotype control (clone LTF-2). Mice were sacrificed at defined time points and peripheral blood was collected via cardiac puncture. Blood was spun at 13,000×g for 15 minutes and serum was collected for analysis. Presence of rat antibodies within the serum was measured using a Rat IgG total ELISA kit (EBioscience, cat no. 88-50490) according to manufacturer recommendations.
RNA Isolation and Quantitative Real-time Polymerase Chain Reaction
Human monocytes from healthy donors were pre-incubated with or without 0.5 mg/mL tocilizumab and then incubated with or without recombinant human IL-6 (0.1ug/ml), normal donor plasma, or patient plasma for an additional 30 minutes in IMDM supplemented with 1.0% human A/B serum. Tocilizumab (Actemra™, Genentech) was purchased through the Hospital of the University of Pennsylvania Pharmacy. Following stimulation, culture medium was removed, cells were lysed directly in TRIzol, and RNA was isolated using a Qiagen RNeasy Kit per manufacturer instructions. cDNA was synthesized from 0.5 – 1.0 μg of RNA per sample (Applied Biosystems, Foster City, CA), and primers for qRT-PCR were designed using the Primer 3 online program (25, 26) and synthesized by Integrated DNA Technologies (SOCS3) or purchased from Applied Biosystems (GAPDH). Relative quantification was measured using either SYBR Green chemistry (Applied Biosystems; SOCS3) or Taqman chemistry (Applied Biosystems; GAPDH). SOCS3 expression was normalized to GAPDH and relative expression was calculated using the ΔCT formula. The fold increase or decrease in expression of treated samples relative to no treatment controls was calculated (ΔΔCT). Primer sequences for human SOCS3 are as follows: (i) Forward: 5′-CAAGGACGGAGACTTCGATT-3′, (ii) Reverse: 5′-AACTTGCTGTGGGTGACCAT-3′.
Statistical Analysis
Statistical analyses were carried out using GraphPad Prism software. Survival comparison testing was performed using the Gehan-Breslow-Wilcoxon test; multiple comparisons testing was performed using one-way ANOVA; all other comparisons were determined by Student’s t-test or Mann-Whitney test.
RESULTS
STAT activation in the tumor microenvironment of invasive PDAC tumors
The JAK/STAT pathway mediates signaling by inflammatory cytokines and is often dysregulated in cancer (14). To investigate JAK/STAT activation in the tumor microenvironment of PDAC, we examined the expression of multiple phosphorylated STAT (pSTAT) proteins in invasive PDAC tumors arising spontaneously in the KrasLSL-G12D/+;Trp53LSL-R172H/+;Pdx1-Cre (KPC) mouse model. With this approach, we detected high expression of pSTAT3Tyr705 and pSTAT5Tyr694 in the tumor microenvironment of late-stage lesions (Figure 1A, B). We also detected pSTAT6Tyr641 at low frequency, whereas pSTAT1Tyr701 expression was negligible (Figure 1A, B). We next evaluated the cell types expressing pSTAT proteins and based on morphology, localized pSTAT6Tyr641 expression primarily to malignant epithelial cells. In contrast, pSTAT5Tyr694 was seen mainly in stromal cells and pSTAT3Tyr705 was found in both malignant epithelial cells and infiltrating stromal cells. We focused our subsequent studies on pSTAT3Tyr705 because it was the most abundantly expressed pSTAT protein detected within PDAC tumors. Using two-color immunofluorescence imaging, we localized pSTAT3Tyr705 expression to multiple cell types within the tumor microenvironment including EpCAM+ malignant epithelial cells, F4/80+ myeloid cells, and αSMA+ fibroblasts (Figure 1C). Thus, the predominant STAT signaling pathways activated within invasive PDAC tissue of KPC mice were STAT3 and STAT5.
Figure 1. IL-6 and STAT activation in spontaneously arising murine PDAC.
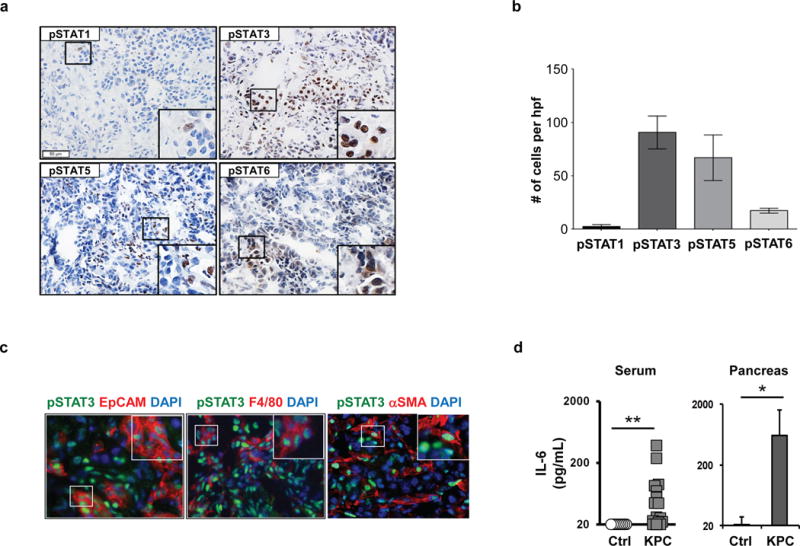
(A) Shown are representative images of immunohistochemical staining to detect expression of pSTAT1, pSTAT3, pSTAT5, and pSTAT6 in primary pancreatic tumors arising spontaneously in KPC mice. (B) Quantification of pSTAT expressing cells detected by immunohistochemistry. (C) Representative images of 3-color immunofluorescence microscopy to detect pSTAT3 protein expression (green) in EpCAM+ malignant cells, F4/80+ myeloid cells, and αSMA+ myofibroblasts (red). (D) IL-6 protein levels (pg/ml) detected in serum and pancreatic tissue from tumor-bearing KPC and control littermate mice. *, p<0.05; **, p<0.01; Mann-Whitney test.
Serum-derived soluble factors from PDAC patients activate STAT3 signaling in peripheral blood monocytes
Previous studies in the KC model of pancreatic intraepithelial neoplasia (PanIN) have implicated IL-6 as a key mediator of STAT3 activation during PanIN progression and PDAC development (15). However, the role of IL-6/STAT3 in regulating the biology of invasive PDAC remains poorly understood, despite its well-recognized association with cachexia, advanced tumor stage, and poor survival (27–29). In our studies using KPC mice bearing late-staged tumors with comparison to age-matched control littermate mice, we detected increased levels of IL-6 protein in both sera and tumor tissue (Figure 1D).
Elevated IL-6 levels in the peripheral blood have been previously reported to be a potential prognostic factor for chemotherapy-naïve patients with PDAC (30). However, increased IL-6 levels are not malignancy-specific and can also be seen in benign inflammatory conditions of the pancreas (e.g. pancreatitis) (31, 32). Consistent with these prior reports, we detected high levels of IL-6 protein in the plasma of a subset of patients with newly diagnosed metastatic PDAC (Figure 2A). IL-6 induces STAT3 phosphorylation by binding to IL-6Ralpha and glycoprotein 130 (gp130) (33). Among human peripheral blood mononuclear cells, we found IL-6Ralpha expression on CD14+ monocytes as well as CD3+ T cells, but not CD19+ B cells or CD56+ NK cells (Supplemental Figure 1 and 2). Because monocytes are actively recruited to PDAC tumors and their infiltration into tumor tissue is associated with treatment resistance, we hypothesized that plasma-derived soluble factors may alter monocyte phenotype and in doing so, regulate treatment resistance in PDAC. Consistent with this hypothesis, we detected increased expression of pSTAT3 in monocytes exposed to plasma collected from patients compared to healthy volunteers (Figure 2B). We next examined the impact of patient versus healthy volunteer plasma on monocyte expression of SOCS3, a downstream target of STAT3 signaling (33). Here, we also found that patient (versus healthy volunteer) plasma induced a significant increase in SOCS3 expression that was greatest in patients with high plasma levels of IL-6 (>40 pg/mL) (Figure 2B). However, tocilizumab, a human anti-IL6R blocking antibody, was unable to inhibit increases in SOCS3 expression detected in monocytes treated with patient plasma (Figure 2D). Moreover, we found that IL-6, at concentrations detected in the peripheral blood of patients, was unable to significantly induce STAT3 phosphorylation and SOCS3 expression in peripheral blood monocytes (Supplemental Figure 3). Together, these findings indicate that plasma-derived soluble factors, present in patients with advanced stage PDAC, can stimulate STAT3/SOCS3 activation in monocytes, but this biology is independent of IL-6R signaling.
Figure 2. Plasma-derived soluble factors from patients with advanced PDAC induce STAT3 activation in human monocytes.
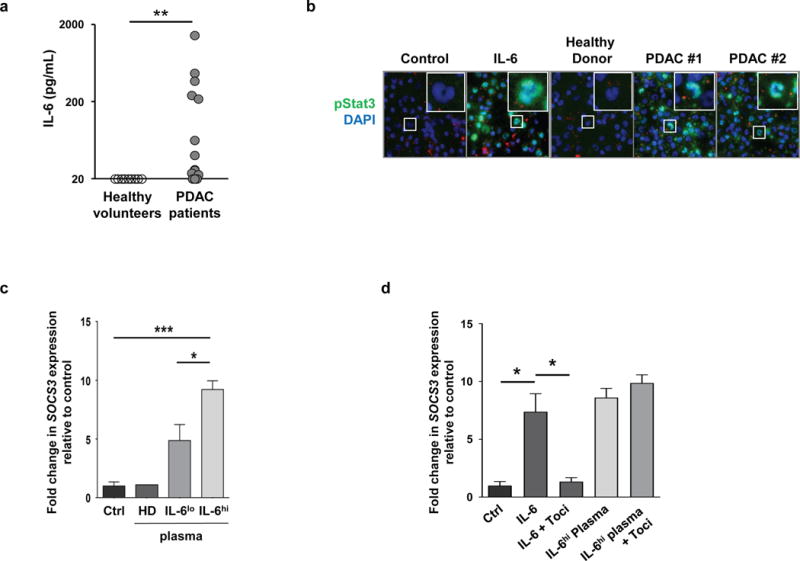
(A) Shown are protein levels of IL-6 (pg/mL) detected in the plasma of patients with newly diagnosed unresectable PDAC with comparison to healthy volunteers. **, p<0.01; Mann-Whitney test. (B) Representative images of immunofluorescence microscopy showing pSTAT3 expression in monocytes incubated with media alone (Ctrl), media with recombinant human IL-6 (50 ng/mL), or plasma obtained from healthy volunteers (HD) and two patients with PDAC with plasma IL-6 levels > 40 pg/mL. Expression of SOCS3 in human monocytes incubated with media alone (Ctrl) or plasma obtained from healthy volunteers (HD) and PDAC patients. Low (< 40 pg/mL, IL-6lo) and high (> 40 pg/mL, IL-6hi) levels of plasma IL-6 were defined based on the median level of IL-6 (40 pg/mL) detected in the plasma from 19 patients examined. SOCS3 expression was normalized to GAPDH, and is shown relative to Ctrl-treated monocytes; n=3 independent experiments. (C) Expression of SOCS3 in human monocytes incubated with media alone (Ctrl) or plasma obtained from healthy volunteers (HD) and PDAC patients. Low (< 40 pg/mL) and high (> 40 pg/mL) levels of plasma IL-6 were defined based on the median level of IL-6 (40 pg/mL) detected in the plasma from 19 patients examined. SOCS3 expression was normalized to GAPDH, and is shown relative to Ctrl-treated monocytes; n=3 independent experiments. (D) Shown is SOCS3 expression normalized to GAPDH in human monocytes pretreated with or without the IL-6Ralpha blocking antibody tocilizumab (Toci) and then incubated with media (Ctrl), IL-6 (100 ng/mL), or IL-6hi plasma from a PDAC patient. *, p<0.05; ***, p<0.001; two-tailed student’s t-test.
IL-6 receptor blocking antibodies target inflammatory monocytes and enhance the therapeutic efficacy of cytotoxic chemotherapy
STAT3 signaling has recently been shown to be a mechanism of chemoresistance in mouse models of PDAC (17, 34). However, the translation of STAT3 inhibitors to the clinic has been challenging and thus, this approach is not an immediate therapeutic option. However, we postulated that inhibition of the IL-6/STAT3 pathway, by disrupting the interaction of IL-6 with IL-6Ralpha, may also improve the sensitivity of PDAC to cytotoxic chemotherapy. Blockade of IL-6 signaling using IL6R blocking antibodies has been used to rapidly reverse symptoms of cytokine release syndrome (35) and is approved for the treatment of polyarticular juvenile idiopathic arthritis, rheumatoid arthritis, and systemic juvenile idiopathic arthritis (36, 37). However, the therapeutic potential of IL6R blockade in cancer remains ill-defined. We found in KPC mice and control littermates, as seen in humans, that IL-6Ralpha was expressed on Ly6Chi F4/80+ inflammatory monocytes as well as CD3+ T cells (Figure 3A, Supplemental Figure 4). Consistent with this expression pattern, systemic administration of an IL-6Ralpha blocking antibody selectively targeted inflammatory monocytes and T cells within the peripheral blood (Figure 3B). Because inflammatory monocytes are recognized mediators of chemoresistance, we next examined the binding kinetics of IL-6R blocking antibodies to this subset of monocytes in vivo. We administered anti-IL6Ralpha mAb (clone 15A7) to mice weighing 20–30 grams at a dose of 0.2 mg via intraperitoneal injection. This dose is compatible with doses of human anti-IL6R blocking antibodies (4–8 mg/kg) used in the treatment of rheumatologic diseases (36, 37). We found that unbound anti-IL6Ralpha monoclonal antibody (mAb) was short-lived within the peripheral blood lasting only hours after injection compared to isotype control (Figure 3C). In contrast, we detected membrane bound anti-IL6Ralpha mAb on inflammatory monocytes for up to 4 days in the peripheral blood (Figure 3D). Based on this pharmacokinetic profile, we adopted a twice weekly treatment schedule for administration of anti-IL6Ralpha mAb to provide continuous IL-6R blockade in vivo in mouse models of PDAC.
Figure 3. IL-6Ralpha blocking antibody 15A7 binds a subset of peripheral blood monocytes in vivo.
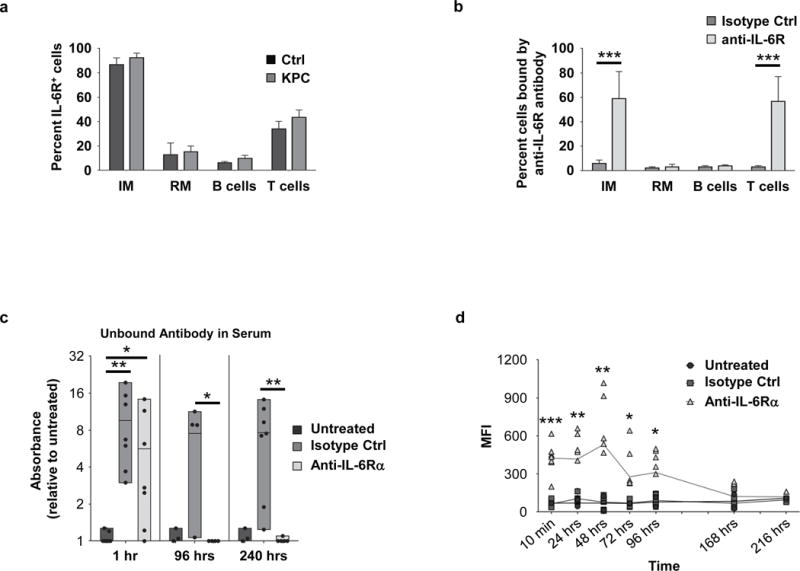
(A) IL-6Ralpha expression on peripheral blood mononuclear cells from control littermate mice and tumor-bearing KPC mice, n=3 independent experiments. (B) Shown is the percent of peripheral blood cells with surface bound anti-IL-6Ralpha antibody detected at 10 minutes post injection; n=8 mice combined from two independent experiments. (C) Detection of unbound control or anti-IL6Ralpha antibody in sera at defined time points after injection; n=3–7 mice per time point. (D) Shown is mean fluorescence intensity (MFI) of rat IgG bound antibody detected on the cell surface of inflammatory monocytes at defined time points after anti-IL-6Ralpha or isotype control antibody injection with comparison to untreated mice. n=3–11 mice per time point for control or anti-IL-6Ralpha antibody treated mice; *, p<0.05; **, p<0.01; ***, p<0.001; two-tailed student’s t-test.
We next examined the therapeutic impact of administering anti-IL6Ralpha mAb with or without chemotherapy on PDAC growth. We first tested this approach in syngeneic B6 mice implanted subcutaneously with 152.PDA, a cell line derived from a PDAC tumor arising spontaneously in KPC mice (10). We found that the combination of gemcitabine chemotherapy and anti-IL6Ralpha mAb delayed tumor outgrowth and improved overall survival compared to either treatment alone (Figure 4A, B). We then tested this therapeutic strategy in KPC mice with spontaneously arising ultrasound-confirmed PDAC tumors and detected a significant increase in tumor regressions measured by ultrasonography at 14 days after gemcitabine/anti-IL6Ralpha mAb treatment compared to baseline (Figure 4C). In contrast, we detected no statistically significant effect on tumor growth with gemcitabine alone. In addition, while a modest slowing of tumor outgrowth in a subset of mice treated with anti-IL6Ralpha mAb alone was observed, this effect was not statistically significant compared to control treated mice.
Figure 4. IL-6Ralpha antibodies enhance the efficacy of gemcitabine chemotherapy in mice with PDAC.
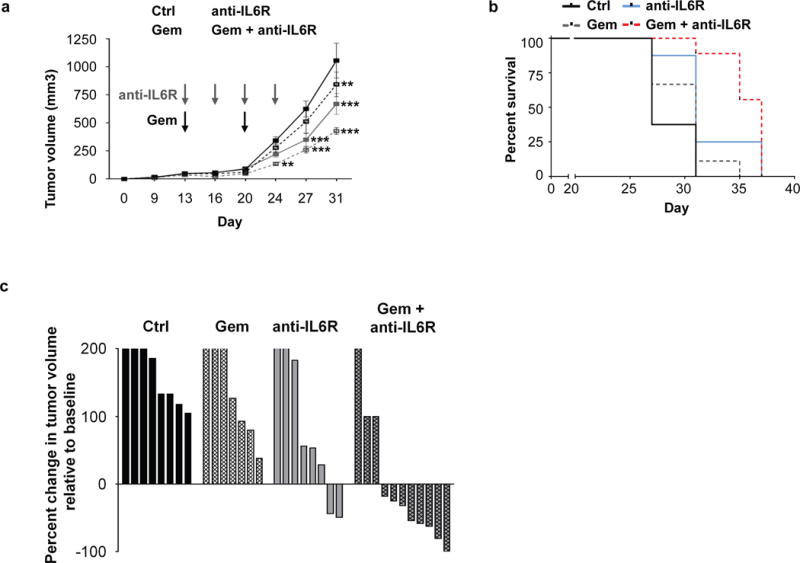
B6 mice were implanted with PDAC cells and two weeks later treated as indicated. (A) Shown are tumor growth curves (n=10 mice per group). Statistical significance determined using two-way ANOVA with comparison to control treated mice. *, p<0.05; **, p<0.01; ***, p<0.001. (B) Overall survival curves of the 4 experimental groups (n=10 mice per group). Ctrl vs anti-IL-6Ralpha, p=0.0011; Gem vs anti-IL-6Ralpha/Gem, p=0.0042; anti-IL-6Ralpha vs anti-IL-6Ralpha/Gem, p=0.0425; Gehan-Breslow-Wilcoxon test. (C) KPC mice with ultrasound confirmed tumors were treated weekly with PBS or gemcitabine (Gem) with or without twice weekly anti-IL-6Ralpha or control antibody treatment. Shown is a waterfall plot of percent change in primary pancreatic tumor volume at 14 days after treatment relative to baseline with each bar representing an individual lesion (n=7–11 per group). Anti-IL6Ralpha/Gem vs Ctrl, p<0.05, one-way ANOVA.
IL-6R blockade inhibits intratumoral Stat3 phosphorylation, decreases tumor cell proliferation, and sensitizes tumors to chemotherapy-induced cell death
We next investigated the mechanism of anti-tumor activity produced by treatment with anti-IL6Ralpha mAb in combination with chemotherapy. At one day after treatment, we detected anti-IL6Ralpha antibodies in the stromal tissue that surrounds malignant cells in PDAC tumors (Supplementary Figure 5). We also found that systemic administration of anti-IL6Ralpha mAb inhibited STAT3 phosphorylation, as seen by immunohistochemistry, in PDAC tumors arising in the KPC model (Figure 5A, B). This decrease in pSTAT3 expression was seen mainly in malignant cells. Similarly, we found a decrease in pSTAT3 expression in malignant cells when PDAC tumors were grown in syngeneic IL-6 deficient (IL-6−/−) mice supporting a role for host-derived IL-6 (Figure 5C, D). While some tumor cells have been reported to express IL-6Ralpha, we found that IL-6Ralpha expression on PDAC tumor cells was undetectable by flow cytometry (Supplemental Figure 6). Further, IL-6Ralpha expression was limited to a subset of CD45+ leukocytes within PDAC tumor tissue (Supplemental Figure 7).
Figure 5. IL-6R antibodies inhibit STAT3 activation, decrease tumor cell proliferation, and sensitize tumors to chemotherapy-induced apoptosis.
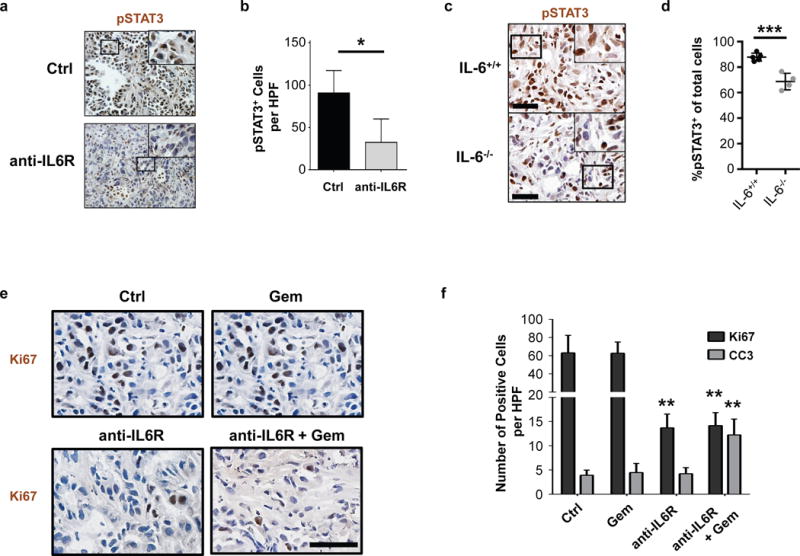
(A) Representative images showing immunohistochemical staining for pSTAT3 expression in tumor tissue from KPC mice treated with anti-IL6Ralpha blocking antibodies or isotype control. Scale bar = 100μm. (B) Quantification of pSTAT3+ cells detected in (A) by immunohistochemistry. n=3–4 mice per group, 4 high-powered fields per mouse; *, p<0.05; two-tailed student’s t-test. (C) Representative images showing immunohistochemical staining for pSTAT3 expression in tumor tissue from PDAC cell line implanted orthotopically into syngeneic wild-type (IL-6+/+) or IL-6 deficient (IL-6−/−) mice. (D) Quantification of the percentage of pSTAT3+ cells per total nucleated cells detected in (C) by immunohistochemistry. n=4–5 mice per group, 5 high-powered fields per mouse. ***, p<0.001; two-tailed student’s t-test. (E) Tumor-bearing mice were treated with or without anti-IL-6Ralpha for three days. Shown are representative images of Ki67 expression in PDAC tumors isolated one day after subsequent treatment with or without gemcitabine chemotherapy. Scale bar = 50μm. (F) Quantification of Ki67 and cleaved caspase 3 (CC3) detected in PDAC tumors isolated one day after gemcitabine chemotherapy treatment with or without three-day pretreatment with anti-IL-6Rα antibodies. n=4–5 mice per group, 5–8 high-powered fields per mouse. Statistical significance determined using two-tailed student’s t-test. Comparisons were made to Ctrl. **, p<0.01.
To understand the capacity of anti-IL6Ralpha mAb to enhance the sensitivity of tumors to gemcitabine, we next examined the impact of treatment on tumor proliferation (Ki67) and cell death (cleaved caspase 3). We detected a decrease in Ki67 expression among tumors treated with anti-IL-6Ralpha mAb with or without gemcitabine chemotherapy compared to control (Figure 5E, F). However, only the combination of anti-IL6Ralpha mAb and gemcitabine produced a significant increase in cleaved caspase 3 expression (Figure 5F, Supplemental Figure 8). Thus, our findings support a role for IL-6Ralpha blockade as a therapeutic approach to inhibit IL-6/STAT3 signaling for enhancing the efficacy of cytotoxic chemotherapy in PDAC.
DISCUSSION
The tumor microenvironment is a major therapeutic barrier in PDAC. Previous studies have identified poor vascularity and dense fibrosis as physical barriers that may limit the delivery of therapeutics to the tumor bed (2–4). In this study, we show that IL-6/STAT3 activation is a key determinant of the sensitivity of PDAC to chemotherapy. Using a clinically-relevant genetic mouse model of PDAC, we found that the tumor microenvironment was marked by preferential and hyperactivation of the STAT3 and STAT5 signaling pathways. Whereas STAT5 activation was seen within stromal cells, STAT3 activation was diffusely detected in myeloid cells, fibroblasts, and malignant epithelial cells. In addition, soluble factors present in the peripheral blood of PDAC patients induced STAT3 activation and SOCS3 expression in peripheral blood monocytes indicating systemic activation of the STAT3/SOCS3 pathway. As STAT3 signaling is a major mediator of cancer inflammation and can inhibit the efficacy of cytotoxic therapies, we hypothesized that IL-6-induced STAT3 activation would be a therapeutic target for improving chemotherapy efficacy in PDAC. We found that administration of IL-6R blocking antibodies to inhibit IL-6 signaling blocked STAT3 activation in the tumor microenvironment and enhanced the sensitivity of malignant cells to cytotoxic chemotherapy. Given the availability of clinical grade IL-6R antagonists, our findings have direct and immediate implications for translating IL-6R blocking antibodies as a strategy to improve the efficacy of chemotherapy in PDAC.
IL-6 has long been associated with advanced tumor stage, poor survival, and cachexia in PDAC (27–29). However, to our knowledge IL-6 has not been previously demonstrated to be a target for improving cytotoxic chemotherapy in this disease. IL-6 can activate STAT3 signaling in cells via two mechanisms. The first involves a classical signaling mechanism in which IL-6 binds to IL-6Ralpha and gp130 on target cells to induce STAT3 activation. However, only a few cell types, including myeloid cells, hepatocytes, and T cells, express the membrane-bound form of IL-6Ralpha. This is in contrast to membrane-bound gp130 which is ubiquitously expressed by cells. The second mechanism involves IL-6 binding to a soluble form of IL-6Ralpha (sIL6R) to form a complex that then interacts with membrane-bound gp130 to induce signaling in cells lacking IL-6Ralpha, a process termed IL-6 trans-signaling. sIL6R levels are naturally detected in the circulation (38), but are increased in patients with cancer which could lead to formation of IL-6/sIL6R complexes and subsequent IL-6 trans-signaling (39, 40). Previous work has implicated IL-6 trans-signaling as a critical mechanism for STAT3 activation in malignant cells that is necessary to promote PanIN progression and development of PDAC (15).
IL-6 is a cytokine that has been attributed to multi-drug resistance due to its ability to modulate the expression of several genes involved in regulating survival (e.g. anti-apoptotic proteins including Bcl-xL and Mcl-1), proliferation (e.g. Ras/Raf/MEK/MAPK, PI3K/AKT, and JAK/STAT pathways), and cell cycle progression (41). In PDAC, IL-6 has been implicated in the maintenance and progression of pancreatic cancer precursor lesions (42), but its role in defining the biology of invasive PDAC is ill-defined. Our findings using IL-6R blocking antibodies show that IL-6 is a critical factor for pancreatic cancer cell proliferation in vivo. This finding is consistent with recent studies showing a role for IL-6 produced by fibroblasts in supporting pancreatic cancer cell growth in tumor organoid models (43). Thus, in the absence of IL-6 as a pro-survival signal, our data suggest that pancreatic cancer cells become more susceptible to chemotherapy-induced apoptosis.
In our studies, we used anti-IL6Ralpha antibodies which block IL-6 binding to IL-6Ralpha and as a result, can be used to inhibit both classical and trans-signaling mechanisms of IL-6. Using this strategy, we found that IL-6Ralpha blocking antibodies enhanced the cytotoxic activity of chemotherapy and inhibited tumor outgrowth. However, we were unable to measure significant levels of IL-6Ralpha expression on malignant cells. As a result, anti-IL6Ralpha blocking antibodies may act to inhibit the binding of IL-6 to soluble IL-6Ralpha and in doing so, disrupt IL-6 trans-signaling in malignant cells – a mechanism that has been previously shown to be critical for progression of pancreatic intraepithelial neoplasia (15). Consistent with such a mechanism, we also found that IL-6Ralpha blocking antibodies could be detected in the stromal tissue surrounding malignant cell nests. A previous report investigating a non-selective JAK1/2 inhibitor for suppressing STAT3 signaling in tumors found that JAK1/2 inhibition increased microvessel density and enhanced drug delivery without impacting stromal fibrosis (17). However, we did not observe any significant changes with anti-IL6Ralpha treatment on vessel patency within tumors. Nonetheless, strategies designed to improve macromolecular permeability and drug delivery (4) could further enhance the therapeutic benefit achieved with IL-6Ralpha blockade. It is also possible, though, that anti-tumor activity seen with anti-IL-6Ralpha antibodies used in combination with cytotoxic chemotherapy is dependent on disrupting IL-6 signaling in non-malignant cells outside or adjacent to tumor tissue. Consistent with this hypothesis, IL-6 has been shown to stimulate monocyte chemotaxis (44). Thus, IL-6 blockade may regulate monocyte recruitment to tumors or even the phenotype of tumor-infiltrating monocytes, which we and others have previously shown to be key determinants of the efficacy of cytotoxic therapies, including chemotherapy and radiation (9, 10).
IL-6 is a key regulator of STAT3 activation and is detected in PDAC at elevated levels in the tumor microenvironment as well as the peripheral blood. A recent report by Ohlund et al. suggested that the cellular source of IL-6 in the tumor microenvironment of PDAC is mainly non-malignant cells, including cancer-associated fibroblasts and tumor-infiltrating leukocytes, rather than malignant epithelial cells (43). Our findings using IL-6 deficient mice support a role for host-derived IL-6 in regulating STAT3 activation in malignant cells. However, even in the absence of host-derived IL-6, we still detected STAT3 phosphorylation in some malignant cells and many non-malignant cells in PDAC tumors. This finding suggests a role for other factors beyond host-derived IL-6 in regulating STAT3 activation in the tumor microenvironment as well as a role for potentially distinct mechanisms that stimulate STAT3 signaling in malignant and non-malignant cells.
Tumor-infiltrating monocytes have been found to promote many of the hallmarks of cancer, including tumor survival, angiogenesis, metastasis and immune evasion (45). Moreover, the phenotype of circulating monocytes shows inherent plasticity and can be altered even prior to monocyte infiltration into tumor tissue (12). We found that soluble factors present within the peripheral blood of patients with advanced PDAC can activate STAT3/SOCS3 signaling in monocytes in an IL-6 independent manner. Multiple factors may contribute to STAT3/SOCS3 activation besides IL-6, including growth factors (e.g. VEGF) and cytokines (e.g. IL-10). In our studies, we found that the concentration of IL-6 detected in the peripheral blood was below the threshold needed to stimulate STAT3/SOCS3 activation. Nonetheless, we determined that anti-IL-6Ralpha antibodies bound rapidly to peripheral blood inflammatory monocytes which have been implicated in chemoresistance (8–10). While we also found that anti-IL6Ralpha antibodies bound to T cells within the peripheral blood, T cells remained largely excluded from the tumor microenvironment. This finding is consistent with recent work showing that anti-IL6Ralpha treatment does not impact T cell recruitment, even in immunogenic models of PDAC, unless combined with anti-PD-L1 checkpoint therapy (46).
IL-6 signaling can be inhibited using either IL-6R blocking antibodies or IL-6 neutralizing antibodies. IL-6 receptor blocking antibodies have been mainly used to treat rheumatic diseases, with the exception of cytokine release syndrome induced by chimeric antigen receptor T cell therapy in patients with hematological malignancies (35). In contrast, IL-6 neutralizing antibodies have been studied across a wide range of malignancies, including multiple myeloma, renal cell carcinoma, Castleman’s Disease, pancreatic cancer and prostate cancer (47, 48). As monotherapy, IL-6 neutralizing antibodies have not demonstrated significant clinical activity in solid tumors. However, in preclinical models of PDAC, IL-6 neutralizing antibodies have been shown to stimulate T cell immunosurveillance when used in combination with PD-L1 checkpoint blockade (46). In addition, IL-6 neutralizing antibodies have been found to enhance the effects of cytotoxic chemotherapy in several xenograft models (49). Our selection of a receptor blocking antibody versus a cytokine neutralizing antibody was based on the poor permeability of the PDAC microenvironment to macromolecules such as antibodies (4). Despite this, though, we found that IL-6R blocking antibodies could be detected in stromal tissue after systemic administration. This finding may reflect active diffusion of IL-6R antibodies into the tumor bed. However, it also remains possible that IL-6R blocking antibodies are passively delivered to stromal tissue by binding to tumor-infiltrating leukocytes prior to their entry into tumors. Consistent with this latter possibility, we have previously reported that an antibody directed against CD40 is delivered to the PDAC microenvironment by binding to CD40+ tumor-infiltrating myeloid cells (13). Thus, receptor blocking versus cytokine neutralizing antibodies may act distinctly to alter the therapeutic sensitivity of PDAC.
In summary, our findings show that disrupting IL-6 signaling using IL-6 receptor blocking antibodies can shift the tumor microenvironment of PDAC from chemoresistant with hyperactivation of the STAT3 pathway to chemosensitive with decreased STAT3 activation (Figure 6). Thus, we propose that the use of IL-6R blocking antibodies to “condition” tumors and inhibit STAT3 activation is a novel strategy for enhancing the sensitivity of PDAC to chemotherapy.
Figure 6. Conceptual model describing a role for IL6R blocking antibodies in enhancing the sensitivity of PDAC to chemotherapy.
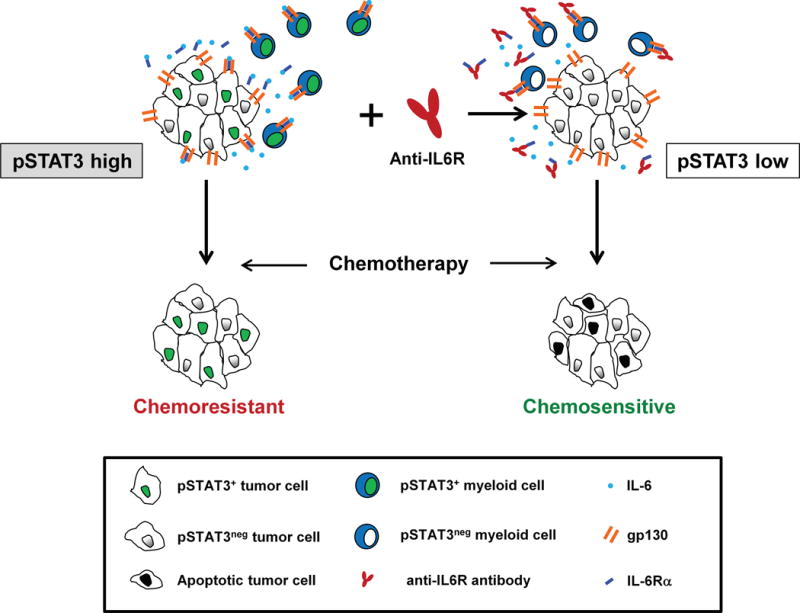
STAT3 signaling in malignant and non-malignant cells in the tumor microenvironment of PDAC is associated with chemoresistance. Treatment with anti-IL6R blocking antibodies disrupts IL-6/STAT3 signaling in both malignant and non-malignant cells which suppresses tumor proliferation and enhances the sensitivity of PDAC to chemotherapy-induced apoptosis.
Supplementary Material
Acknowledgments
The authors thank Michael Kalos and Erica Suppa for human cytokine analyses; Qian-Chun Yu, Hongwei Yu and Adam Bedenbaugh for advice and technical assistance with immunohistochemistry assays; and Weijing Sun for helpful discussions.
Financial support:
This work was supported by NIH grant K08 CA138907 (G.L. Beatty), NIH R01 CA197916 (G.L. Beatty), NIH National Institute of General Medical Sciences K12GM081295 (K.B. Long), NIH grant F30 CA196106 (J.W. Lee), a Molecular Biology and Molecular Pathology and Imaging Cores of the Penn Center for Molecular Studies in Digestive and Liver Diseases grant (P30 DK050306), the Damon Runyon Cancer Research Foundation grant DRR-15-12, for which G.L. Beatty is the Nadia’s Gift Foundation Innovator of the Damon Runyon-Rachleff Innovation Award, by Grant Number 15-20-25-BEAT from the 2015 Pancreatic Cancer Action Network-AACR Career Development Award supported by an anonymous foundation, and by grant 2013107 from the Doris Duke Charitable Foundation.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Research. 2014;74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–29. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–20. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014 doi: 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanford DE, Belt BA, Panni RZ, et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin Cancer Res. 2013;19:3404–15. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nywening TM, Wang-Gillam A, Sanford DE, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–62. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Research. 2013;73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalbasi A, Komar CA, Tooker GM, et al. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Research. 2014;74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016;6:400–13. doi: 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–69. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 16.Corcoran RB, Contino G, Deshpande V, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Research. 2011;71:5020–9. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagathihalli NS, Castellanos JA, Shi C, et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology. 2015;149:1932–43 e9. doi: 10.1053/j.gastro.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hurwitz HI, Uppal N, Wagner SA, et al. Randomized, Double-Blind, Phase II Study of Ruxolitinib or Placebo in Combination With Capecitabine in Patients With Metastatic Pancreatic Cancer for Whom Therapy With Gemcitabine Has Failed. J Clin Oncol. 2015;33:4039–47. doi: 10.1200/JCO.2015.61.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurwitz H, Van Custem E, Bendell JC, et al. Two randomized, placebo-controlled phase 3 studies (Rux) + capecitabine (C) in patients (pts) with advanced/metastatic pancreatic cancer (mPC) after failure/intolerance of first-line chemotherapy: JANUS (J1) and JANUS (J2) J Clin Oncol. 2017;35 abstract 343. [Google Scholar]
- 20.Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene. 2016;35:939–51. doi: 10.1038/onc.2015.150. [DOI] [PubMed] [Google Scholar]
- 21.Beatty GL, Shahda S, Beck JT, et al. A phase 1b/2 study of INCB039110 + nab-paclitaxel (N) and gemcitabine (G) in patients (pts) with advanced solid tumors and pancreatic cancer (PC) J Clin Oncol. 2017;35 abstract 362. [Google Scholar]
- 22.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 23.Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–20. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JW, Komar CA, Bengsch F, Graham K, Beatty GL. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+) ;LSL-Trp53(R172H/+) ;Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr Protoc Pharmacol. 2016;73:14 39 1–14 39 20. doi: 10.1002/cpph.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Untergasser A, Cutcutache I, Koressaar T, et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–91. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- 27.Ebrahimi B, Tucker SL, Li D, Abbruzzese JL, Kurzrock R. Cytokines in pancreatic carcinoma: correlation with phenotypic characteristics and prognosis. Cancer. 2004;101:2727–36. doi: 10.1002/cncr.20672. [DOI] [PubMed] [Google Scholar]
- 28.Nixon AB, Pang H, Starr MD, et al. Prognostic and predictive blood-based biomarkers in patients with advanced pancreatic cancer: results from CALGB80303 (Alliance) Clin Cancer Res. 2013;19:6957–66. doi: 10.1158/1078-0432.CCR-13-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martignoni ME, Kunze P, Hildebrandt W, et al. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer-related cachexia. Clin Cancer Res. 2005;11:5802–8. doi: 10.1158/1078-0432.CCR-05-0185. [DOI] [PubMed] [Google Scholar]
- 30.Farren MR, Mace TA, Geyer S, et al. Systemic Immune Activity Predicts Overall Survival in Treatment-Naive Patients with Metastatic Pancreatic Cancer. Clin Cancer Res. 2016;22:2565–74. doi: 10.1158/1078-0432.CCR-15-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand RE, Nolen BM, Zeh HJ, et al. Serum biomarker panels for the detection of pancreatic cancer. Clin Cancer Res. 2011;17:805–16. doi: 10.1158/1078-0432.CCR-10-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mroczko B, Groblewska M, Gryko M, Kedra B, Szmitkowski M. Diagnostic usefulness of serum interleukin 6 (IL-6) and C-reactive protein (CRP) in the differentiation between pancreatic cancer and chronic pancreatitis. Journal of clinical laboratory analysis. 2010;24:256–61. doi: 10.1002/jcla.20395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mauer J, Denson JL, Bruning JC. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015;36:92–101. doi: 10.1016/j.it.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Greten FR, Weber CK, Greten TF, et al. Stat3 and NF-kappaB activation prevents apoptosis in pancreatic carcinogenesis. Gastroenterology. 2002;123:2052–63. doi: 10.1053/gast.2002.37075. [DOI] [PubMed] [Google Scholar]
- 35.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–22. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2385–95. doi: 10.1056/NEJMoa1112802. [DOI] [PubMed] [Google Scholar]
- 37.Smolen JS, Beaulieu A, Rubbert-Roth A, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–97. doi: 10.1016/S0140-6736(08)60453-5. [DOI] [PubMed] [Google Scholar]
- 38.Narazaki M, Yasukawa K, Saito T, et al. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood. 1993;82:1120–6. [PubMed] [Google Scholar]
- 39.Pulkki K, Pelliniemi TT, Rajamaki A, Tienhaara A, Laakso M, Lahtinen R. Soluble interleukin-6 receptor as a prognostic factor in multiple myeloma. Finnish Leukaemia Group British Journal of Haematology. 1996;92:370–4. doi: 10.1046/j.1365-2141.1996.d01-1470.x. [DOI] [PubMed] [Google Scholar]
- 40.Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology. 2001;58:1008–15. doi: 10.1016/s0090-4295(01)01405-4. [DOI] [PubMed] [Google Scholar]
- 41.Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Seminars in Immunology. 2014;26:38–47. doi: 10.1016/j.smim.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Yan W, Collins MA, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Research. 2013;73:6359–74. doi: 10.1158/0008-5472.CAN-13-1558-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. The Journal of Experimental Medicine. 2017;214:579–96. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clahsen T, Schaper F. Interleukin-6 acts in the fashion of a classical chemokine on monocytic cells by inducing integrin activation, cell adhesion, actin polymerization, chemotaxis, and transmigration. J Leukoc Biol. 2008;84:1521–9. doi: 10.1189/jlb.0308178. [DOI] [PubMed] [Google Scholar]
- 45.Long KB, Beatty GL. Harnessing the antitumor potential of macrophages for cancer immunotherapy. Oncoimmunology. 2013;2:e26860. doi: 10.4161/onci.26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mace TA, Shakya R, Pitarresi JR, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2016 doi: 10.1136/gutjnl-2016-311585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossi JF, Lu ZY, Jourdan M, Klein B. Interleukin-6 as a therapeutic target. Clin Cancer Res. 2015;21:1248–57. doi: 10.1158/1078-0432.CCR-14-2291. [DOI] [PubMed] [Google Scholar]
- 48.Deisseroth A, Ko CW, Nie L, et al. FDA approval: siltuximab for the treatment of patients with multicentric Castleman disease. Clin Cancer Res. 2015;21:950–4. doi: 10.1158/1078-0432.CCR-14-1678. [DOI] [PubMed] [Google Scholar]
- 49.Zhong H, Davis A, Ouzounova M, et al. A Novel IL6 Antibody Sensitizes Multiple Tumor Types to Chemotherapy Including Trastuzumab-Resistant Tumors. Cancer Research. 2016;76:480–90. doi: 10.1158/0008-5472.CAN-15-0883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.