

Abstract
The circulatory system distributes blood flow to each tissue and transports oxygen and nutrients. Peripheral circulation is required to maintain the physiological function in each tissue. Disturbance of circulation, therefore, decreases oxygen delivery, leading to tissue hypoxia which takes place in several cardiovascular disorders including atherosclerosis, pulmonary arterial hypertension and heart failure. While tissue hypoxia can be induced because of cardiovascular disorders, hypoxia signaling itself has a potential to modulate tissue remodeling processes or the severity of the cardiovascular disorders. Hypoxia inducible factor-1α (HIF-1α) and HIF-2α belongs to a group of transcription factors which mediate most of the cellular responses to hypoxia at a transcriptional level. We, and others, have reported that HIF-α signaling plays a critical role in the initiation or the regulation of inflammation. HIF-α signaling contributes to the tissue remodeling processes; thus it has a potential to become a therapeutic target. Elucidation of the molecular link, therefore, between hypoxia signaling and tissue remodeling will greatly help us to understand the pathophysiology of the cardiovascular disorders. The purpose of this review is to give a brief overview of the current understanding about the function HIF-α in inflammation processes especially by focusing on its roles in macrophages. In addition, the pathophysiological roles of hypoxia signaling for the development of cardiovascular disease will be discussed.
Keywords: Cardiovascular diseases, Inflammation, Hypoxia, HIF-1α
Introduction
While molecular oxygen is required to maintain the homeostasis in each cell, the oxygen consumption rate greatly varies depending on each tissue. For instance, the brain consumes approximately 3 ml O2/min/100g tissue. The oxygen consumption rate in the heart is even larger at a rate of 8–15 ml O2/min/100g tissue. It should be noted that it could rise up to 70 ml O2/min/100g during vigorous exercise period1, 2). In general, the oxygen concentration below the tissue-specific physiological level is called hypoxia.
In the hypoxic environment, each cell exhibits several types of responses at transcriptional, translational or post-translational levels. Most of the gene expressions are, at the transcriptional level, significantly suppressed in the hypoxic environment. In contrast, the expression of a group of genes is significantly enhanced in hypoxia. These genes are termed as hypoxia-inducible genes. Representatives of hypoxia-inducible genes are genes related to angiogenesis (vascular endothelial growth factor-a, Vegf-a)3), erythropoiesis (erythropoietin, Epo)4), cellular metabolism (pyruvate dehydrogenase kinase, isoform 1, Pdk 1, or lactate dehydrogenase-a, Ldh-a)5, 6) and inflammation (inducible nitric oxide synthase, iNOS)7). Hypoxia-inducible factor-1α (HIF-1α) and HIF-2α act as transcriptional mediators in their hypoxic induction8, 9). The activity of HIF-α is regulated at a post-translational level. In normoxic conditions, HIF-α protein is hydroxylated through an oxygen dependent process, and is degraded through a ubiquitin-proteasome system10). In contrast, the HIF-α protein is stabilized in hypoxic condition and translocated into the nucleus. After forming a heterodimer complex with HIF-1β, HIF-α binds to the hypoxia responsive elements (HRE) with a core consensus sequence (RCGTG), and activates the transcription of hypoxia-inducible genes11, 12)(Fig. 1).
Fig. 1.
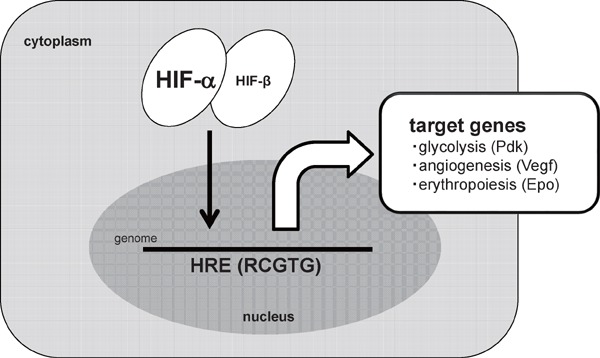
HIF-α mediated cellular responses to hypoxia
In a hypoxia environment, HIF-α protein is stabilized and binds to the hypoxia responsive element (HREs) of each target gene, including glucose metabolism (Pyruvate dehydrogenase kinase (Pdk)), angiogenesis (Vascular endothelial growth factor-a (Vegf-a)) and erythropoiesis (Erythropoietin (Epo)).
Sterile Inflammation in Cardiovascular Diseases
Most basic or clinical researches support the notion that an inflammatory process underlies the development of atherosclerotic processes. Monocytes are recruited in the subendothelial space of the arterial wall and differentiate into macrophage at the initial phase of atherosclerosis. Repeated innate immune responses, in turn, promote the deposition of lipid-loaded macrophages or foam cells, which secrete proinflammatory mediators and result in plaque destabilization13, 14). Consistent with these findings, the serum levels of inflammatory markers, including Creactive protein (CRP) and interleukin-6 (IL-6), are elevated in atherosclerotic patients15–17).
The inflammatory process also contributes to the development of vascular remodeling in systemic hypertension or pulmonary artery hypertension (PAH)18, 19). An elevated serum level of CRP precedes the new onset of systemic hypertension in an elderly healthy population20, 21). Similarly, serum level of proinflammatory cytokines including interleukin 6 (IL-6) is elevated in PAH patients, and the cytokine abundance has a significant impact on the subsequent prognosis of PAH patients22–24).
Heart failure is a condition in which the heart can't pump enough amount of blood to meet the body's needs. Heart failure with reduced ejection fraction (HFrEF; also known as systolic heart failure) develops after myocardial infarction or as one of the manifestation of cardiomyopathy. In contrast, heart failure with preserved ejection fraction (HFpEF) occurs in patients with systemic hypertension. It should be noted that the serum levels of inflammatory markers are increased in both HFrEF and HFpEF patients25–27). Importantly, the serum levels of CRP, IL-6 and tumor necrosis factor alpha (TNF-α) are associated with their prognosis28–33).
These observations clearly indicate that an inflammatory signal plays a critical role during the processes of cardiovascular remodeling. It still remains unclear, however, why sterile inflammation develops in the vessel wall or in the cardiac tissue. Moreover, it should also be elucidated whether these inflammatory processes play an adaptive or mal-adaptive function.
HIF-α Switching in M1 / M2 Macrophage
Previously, it has been formerly considered that macrophages consist of a single cell population, and simply activate inflammatory processes in response to the tissue injury or infection34). A number of recent studies have revealed clearly that macrophages are composed of heterogeneous cell populations. Currently, several types of classification exist in defining each macrophage population based on its origin, location or the function. Tissue macrophages may exist as a resident macrophage or may differentiate from monocyte populations19). One macrophage population activates inflammation (pro-inflammatory), but the other suppresses or resolves the inflammatory processes (anti-inflammatory)35–37). A number of definitions still exist regarding the pro- or anti-inflammatory macrophage population. Among them, it has been widely used for easy understanding of macrophage heterogeneity including M1 (pro-inflammatory) and M2 polarization (anti-inflammatory).
In in vitro experiments, M1 is usually induced by a combination of Th1 cytokine, interferon-γ (IFN-γ) and a ligand for toll-like receptor 4, lipopolysaccharide (LPS). IFN-γ and LPS robustly induce the expression of pro-inflammatory genes including inducible nitric oxide (NO) synthase (iNOS), and elicit the production NO. NO is one of the critical mediators in inflammation. Thus, the iNOS gene is considered as a classical M1 marker gene. On the other hand, M2 macrophage is induced by Th2 cytokines such as interleukin-4 (IL-4) or IL-13. Arginase 1 (Arg1) or the mannose receptor expression is highly expressed in M2 macrophages, thus these genes are known as M2 marker genes38).
It should be noted that both iNOS and ARG1 enzymes catalyze and compete for the same metabolic substrate, l-arginine. While iNOS in M1 macrophage produces NO from l-arginine and strikingly promotes the inflammatory processes, ARG1 in M2 macrophage suppresses NO production39). The antagonistic activity, therefore, between iNOS and Arg1 crucially regulates the production of NO.
The roles of HIF-1α in M1 macrophage activation have been extensively investigated. LPS or IFN-γ mediated HIF-1α induction is required for iNOS gene expression in M1 macrophages7, 40, 41). In agreement with this, the severity of septic shock was significantly attenuated in myeloid-specific HIF-1α deficient (LysM-cre;HIF-1αfl/fl) mice42, 43). Moreover, chemically induced cutaneous inflammation or experimental arthritis was also attenuated in HIF-1α deficient mice44). These results indicate that HIF-1α plays a critical role in M1 macrophage activation.
In contrast to the pro-inflammatory processes those are activated in M1 macrophages, little is known about its resolution process. We examined the expression of HIF-1α and HIF-2α in murine macrophages, and found that HIF-1α and HIF-2α are specifically expressed in M1 and M2 macrophages, respectively43). While LPS or IFN-γ significantly upregulated HIF-1α protein abundance, LPS and IFN-γ strikingly suppressed HIF-2α gene expression. In contrast, IL-4 or IL-13 significantly increased HIF-2α protein abundance in hypoxia. Importantly, we revealed that HIF-2α induces Arg1 gene expression in M2 macrophages. While both iNOS and Arg1 gene expression increase in hypoxia, iNOS and Arg1 utilizes distinct isoform of HIF-α in its hypoxic induction. Through a loss-of-function approach, we identified that HIF-1α potentiates, but HIF-2α suppresses NO production in vivo. These results revealed that the balance between HIF-1α and HIF-2α, named as HIF-α switching, critically determine the on/off regulation of NO production (Fig. 2).
Fig. 2.
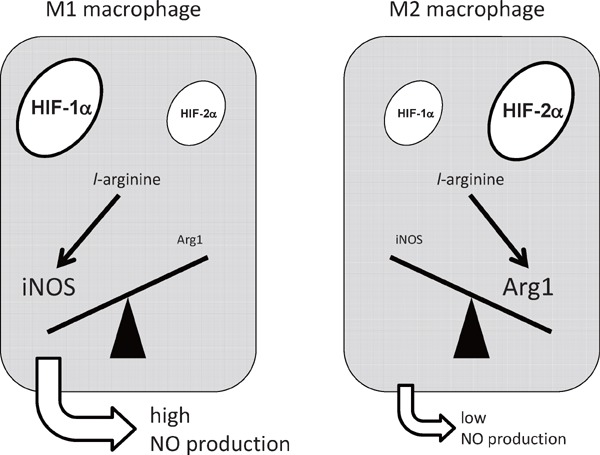
HIF-α switching regulates nitric oxide (NO) production from macrophages.
HIF-1α activates iNOS gene expression, resulting in the increase of NO synthesis. In contrast, HIF-2α induces the expression of Arg1, which suppresses the NO production. The balance between HIF-1α and HIF-2α, namely HIF-α switching, regulates macrophage NO production.
The Role of Active Glycolysis in Macrophage Migration
In addition to its activation, migration is another important function of macrophages initiating inflammation42, 45). The processes of cell migration consist of a series of dynamic remodeling in actin cytoskeleton. While lamellipodia is an actin projection on the leading edge of the migrating cell, filopodia is a slender projection that extends beyond the lamellipodia46). During cell migration, dynamic recycling of actin polymerization and depolymerization takes place through an ATP dependent manner. It should be noted, therefore, that the cell migration process consumes abundant ATP in the cytosol47, 48).
ATP can be synthesized through at least two metabolic pathways, including glycolysis in the cytoplasm and tricarboxylic cycle (TCA cycle) in the mitochondria. In glycolysis, two molecules of ATP are synthesized from one glucose molecule, accompanying the generation of lactate. Glucose can also be metabolized through a TCA cycle in the mitochondria via oxidative phosphorylation. Mitochondrial electron transport chain produces 36 molecules of ATP from glucose through an oxygen dependent manner.
While the oxygen concentration inside the blood vessel remains high, its concentration is strikingly decreased in the inflammatory area49). During the migration processes of monocyte derived macrophages, the oxygen concentration gradually decreases as macrophages migrate from the blood stream into the inflammatory area. It has been also well documented that mitochondrial activity is significantly suppressed in hypoxic condition, which is termed as the Pasteur effect or classic glycolysis50). ATP production in hypoxic condition should also be decreased. Given that macrophage migration consumes abundant ATP, how is it that macrophage can migrate under hypoxic environment?
We initially investigated which energy substrate is required for macrophage migration in hypoxia. Using Boyden chamber assay, we identified that glucose, but not glutamine, is critically required for macrophage mobilization in hypoxia51). Intriguingly, dichloroacetate (DCA), a chemical inhibitor of pyruvate dehydrogenase kinase (PDK) significantly suppressed macrophage migration capacity. These results indicated that glycolysis, but not mitochondrial respiration plays a critical role in maintaining macrophage migration capacity under hypoxic environment.
To further investigate the link between glycolytic metabolism and macrophage migration, we examined the intracellular localization of glycolytic enzymes in macrophages. Pyruvate kinase muscle isozyme (PKM2) belongs to glycolytic enzymes, and is responsible for glycolytic ATP synthesis in the cytosol. Intriguingly, PKM2 co-localizes with F-actin in filopodia and lamellipodia in primary macrophages. These results implied that local production of ATP in the cytosol where it is rapidly consumed during cell migration processes may be beneficial to accommodate the demand of ATP during cell migration processes52, 53)(Fig. 3).
Fig. 3.
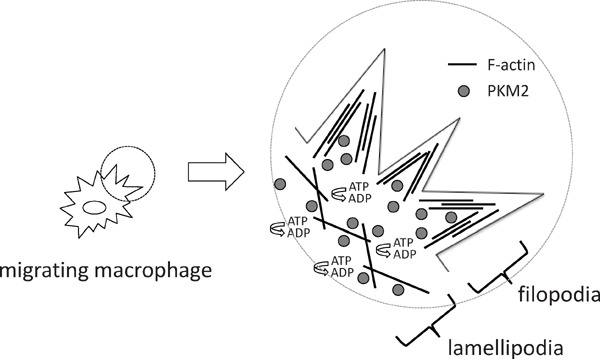
PKM2 co-localizes with F-actin in macrophages
ATP is consumed at filopodia or lamellipodia during cell migration processes. Pyruvate kinase, muscle (PKM2) co-localizes with F-actin in filopodia or lamellipodia.
We also examined the molecular processes by which hypoxia signaling suppresses glucose oxidation in primary macrophages. We established a novel experimental system in which we could measure the oxygen consumption rate in hypoxic environment. Based on these approaches, we identified a novel mode of glycolytic reprogramming which takes place in primary macrophages, termed as active glycolysis (Fig. 4). In active glycolysis, HIF-1α mediated Pdk1 induction actively elicits glycolytic reprogramming even in the presence of mitochondrial electron transport chain activity.
Fig. 4.
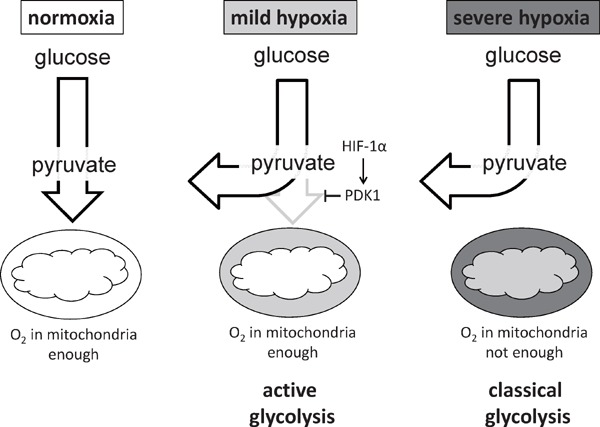
HIF-1α - PDK1 axis induces active glycolysis in hypoxic macrophage.
In severely hypoxic condition, mitochondrial activity is decreased, resulting in the glycolytic reprogramming (classical glycolysis). In mild hypoxia, HIF-1α - PDK1 axis actively accelerates glycolysis in the presence of sustained mitochondrial activity (active glycolysis).
Collectively, these results indicated that cytosolic distribution of intracellular ATP accelerates macrophage motility. Intriguingly, active glycolysis occurs not only in primary macrophages, but also in primary hepatocytes, indicating that this metabolic alteration may be one of the common features in non-malignant cells.
Hypoxia Signaling in Atherosclerosis
Chronic inflammation in the vessel walls underlies the development of atherosclerosis. Various types of the cells including endothelial cells, smooth muscle cells, fibroblasts, monocytes/macrophages and T lymphocytes are involved in atheroma plaque formation. Among them, monocyte/macrophages predominantly promote the progression of atheroma. Foam cells, one of the macrophages in atheroma which is surrounded by the lipid, elicits a fatty streak formation in the vessel walls54).
The cells of the blood vessel wall depend on the oxygen supply from the luminal blood or the adventitial vasa vasorum. In developed atherosclerosis, oxygen consumption rate increases in vessel wall, leading to the occurrence of tissue hypoxia at the plaque lesion. Pimonidazole immunohistochemistry, a hypoxia marker, in human atheroma patients revealed that a hypoxic area exists in the macrophage-rich center of the plaque lesion55). Intriguingly, HIF-1α is expressed in the macrophages of human atheroma plaques55–57).
The low-density lipoprotein receptor in deficient mice (Ldlr −/−) is commonly used as a model of atheroma progression. Using this animal model, the roles of macrophage HIF-1α in plaque progression were examined. Bone marrow transplantation of myeloid-specific HIF-1α deficient mice, but not of wild type mice, significantly decreased plaque burden in the aorta of Ldlr −/− mice. The expression of the genes related to the inflammation or M1 macrophage accumulation was strikingly suppressed in HIF-1α deficient mice58).
Lectin like oxidized low density lipoprotein (LDL) receptor-1 (Lox-1) is the major receptor for oxidatively modified low density lipoproteins (OxLDLs)59), and strikingly promotes the progression of atherosclerosis60). While Lox-1 is expressed in macrophages, endothelial cells or smooth muscle cells61, 62), its expression is increased in the hypoxic environment58). In agreement with this, macrophage lipid content is significantly increased in hypoxia through a Lox-1 dependent manner. Importantly, HIF-1α in macrophage significantly contributes for the hypoxic induction of Lox-1 gene in hypoxia. These results demonstrate the tissue hypoxia in the plaque lesion is not just a consequence of increased plaque burden, but that in turn, HIF-1α signaling promotes M1 macrophage activation at the center of atheroma plaque.
The roles of HIF-1α signaling, however, in plaque formation seem to be more complicated than expected. CD11c is known as one of the surface markers in antigen-presenting cells (APCs). Conditional ablation of HIF-1α in APCs significantly augment the severity of atheroma formation in Ldlr−/− mice63). Accumulation of Th1 at the plaque area was significantly accelerated in APC specific HIF-1α deficient mice.
Collectively, the roles of HIF-1α signaling in each cell during atherogenesis has to be examined in more detail. It still remains unclear as to the molecular processes by which inflammation takes place in the vessel wall. Is it induced through an antigen-dependent or independent process? If there is a specific antigen, what are they? Additional experiments by focusing in the hypoxic signaling will help to elucidate the molecular link between sterile inflammation in atheroma formation.
The Roles of Hypoxia Signaling in Vascular Remodeling
We have shown that the balance between HIF-1α and HIF-2α, namely HIF-α switching, critically regulate the production of NO in primary macrophages. NO also acts as a powerful vasodilator in the circulatory system. NO mediated increase of cyclic guanosine monophosphate (cGMP) in smooth muscle cells regulates vascular tone64–66). NO could also elicit vasodilation through a cGMP-independent fashion including S-nitorosylation of target proteins, activation of sarco/endoplasmic reticulum calcium ATPase or production of cyclic inosine monophosphate67).
Skin is one of the largest organs, and critically regulates systemic vascular resistance through the production of NO. Vasodilation in the skin also works as a radiator and helps to maintain core body temperature. While its vascular tone has to be tightly regulated depending on the external environment, its molecular process still remains unclear. We therefore hypothesized that HIF-α switching regulates vascular tone in the skin, and tested the roles of keratinotye HIF-α signaling in vascular function. We generated keratinocyte-specific HIF-1α and HIF-2α deficient mice (K14-cre; HIF-1αfl/fl or HIF-2αfl/fl) and measured systemic blood pressure (BP). While systemic BP is elevated in HIF-1α deficient mice, it is decreased in HIF-2α deficient mice68). Cardiac fibrosis elicited by AngiotensinII infusion was also attenuated in HIF-2α deficient mice. These results indicate that HIF-α switching in the skin critically regulates vascular tone in the skin. Consistent with our hypothesis, the core body temperature in HIF-1α deficient mice was elevated when the mice were exposed to the warm environment. Intriguingly, decrease of HIF-1α, but elevation of HIF-2α expression was detected in hypertensive patients, indicating that HIF-α in the skin could contribute in the regulation of systemic BP.
Pulmonary arterial hypertension (PAH) is manifested by an increased BP in pulmonary artery, resulting in the right ventricular heart failure69–71). It is also known that a hypoxic environment elicits pulmonary vasoconstriction and arterial remodeling. While hypoxia signaling seems to play pivotal roles in PAH72, 73), the precise roles of HIF-αs in pulmonary arterial remodeling have been unclear. Hypoxic exposure is commonly used as a murine model of PAH. Recently, the roles of HIF-α switching were examined using hypoxia induced PAH model74). Pulmonary endothelial specific HIF-2α deficient mice (L1-cre;HIF-2αfl/fl) exhibited tolerance to hypoxia induced PAH compared to control mice or HIF-1α deficient mice. Notably, PA remodeling was significantly attenuated in HIF-2α deficient mice. As a molecular mechanism, HIF-2α mediated induction of arginase-1 critically regulates NO production in pulmonary vasculature.
These results clearly indicated that HIF-2α-Arginase1 axis could become a therapeutic target to improve the NO availability of the pulmonary arteries or systemic circulation. Recently, HIF-2α antagonist was synthesized by using a structure-based design approach, and is currently used to treat patients with renal cell carcinoma75, 76). It seems tempting, therefore, to test the therapeutic efficacy of HIF-2α antagonists in PAH. Elucidating the molecular process by which HIF-2α signal is activated in systemic hypertension or PAH will also help to understand the pathophysiology of vascular remodeling in more detail.
Hypoxia Signaling and Cardiac Remodeling
Hypoxia signaling, as described in the previous section, plays a critical role in the inflammatory process or intracellular metabolism. Importantly, both of them strikingly affect the cardiac function. While heart failure is predominantly a hemodynamic condition, inflammatory signal is strikingly activated in heart failure patients77, 78). Serum level of inflammatory cytokines including tumor necrosis factor (TNF)-α or interleukin-6 (IL-6) is significantly elevated in heart failure patients31). Importantly, the level of these inflammatory cytokines correlated with the severity of the heart failure79, 80). At a histological level, it has also been shown that inflammatory cells including macrophages accumulate to the cardiac tissues of human heart failure subjects81). The roles of inflammatory processes in myocardial infarction have been studied using in vivo murine model of myocardial infarction82–84). Two types of monocytes/macrophages, including Ly-6Chi and Ly-6Clo, sequentially accumulated in response to the myocardial death85–87). While Ly-6Chi monocytes/macrophages engulf the injured tissues, Ly-6Clo monocytes/macrophages promoted angiogenesis, and scar formation. These results demonstrate that each population of inflammatory cells exerts a distinct function in cardiac remodeling. These results raised the hypothesis that sterile inflammation underlies the development of heart failure. Two multicenter clinical trials, however, using anti-TNF-α antibody demonstrated that inhibition of TNF-α did not improve the clinical courses of heart failure patients88, 89). These results indicate that further study is required to fully elucidate and identify the role of each inflammatory cell in cardiac remodeling.
In physiological condition, the myocardium predominantly utilizes free fatty acids as its energy substrate, and acquires ATP through the oxidative phosphorylation. Molecular oxygen is required to maintain the activity of mitochondrial electron transport chain. Notably, cardiomyocytes change their energy substrates from fatty acids to glucose in response to the mechanical or ischemic stresses90–92). We previously reported that this metabolic alteration could become a helpful diagnostic tool in the evaluation of the cardiac function93).
It still remains unclear whether the metabolic reprograming in cardiomyocytes is an adaptive or maladaptive process in maintaining cardiac function. It has been shown that tissue hypoxia develops during cardiac remodeling processes94–96). Moreover, HIF-1α in murine cardiomyocytes plays an important role in modulating its intracellular metabolism by activating PPARgammma97). Therefore, elucidation of the hypoxia signaling in cardiomyocytes will help us to understand roles of metabolic alteration in cardiac function.
Conclusion
Tissue hypoxia seems to be one of the common features in cardiovascular disorders including atherosclerosis, vascular remodeling and heart failure. Notably, HIF-α signal has a potential to become a therapeutic target in the managing cardiovascular remodeling. While a number of questions remain unsolved as to the roles of inflammation or metabolic alteration in cardiovascular disorders, further study on the hypoxia signaling will help us to understand its pathological processes in more detail.
Acknowledgement
N.T. was supported by a grant from a Grant-in-Aid for Scientific Research on Innovative Areas “Oxygen Biology: a new criterion for integrated understanding of life” (No. 26111003) of MEXT, Japan. H.A. was supported by Grant-in-Aid for Young Scientists (B) (15K19369, 17K15990).
COI
N.T. has received grant supports from DAIICHI SANKYO COMPANY, LIMITED, and Bayer Yakuhin, Ltd.. H. S. has received grant supports from The Uehara Memorial Foundation.
References
- 1). West JB. Cardiac energetics and myocardial oxygen consumption. Physiologic basis of medical practice. Williams and Wilkins; Baltimore, Maryland, USA: 1991: 250-260 [Google Scholar]
- 2). Braunwald E. Coronary blood flow and myocardial ischemia. Heart disease: A textbook of cardiovascular medicine. W.B. Saunders Company; Philadelphia, Pennsylvania, USA: 2001: 1161-1183 [Google Scholar]
- 3). Tuder RM, Flook BE, Voelkel NF. Increased gene expression for vegf and the vegf receptors kdr/flk and flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest. 1995; 95: 1798-1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4). Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992; 12: 5447-5454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Kim JW, Tchernyshyov I, Semenza GL, Dang CV. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell metabolism. 2006; 3: 177-185 [DOI] [PubMed] [Google Scholar]
- 6). McClelland GB, Brooks GA. Changes in mct 1, mct 4, and ldh expression are tissue specific in rats after long-term hypobaric hypoxia. Journal of applied physiology (Bethesda, Md. : 1985). 2002; 92: 1573-1584 [DOI] [PubMed] [Google Scholar]
- 7). Melillo G, Musso T, Sica A, Taylor LS, Cox GW, Varesio L. A hypoxia-responsive element mediates a novel pathway of activation of the inducible nitric oxide synthase promoter. J Exp Med. 1995; 182: 1683-1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Tian H, McKnight SL, Russell DW. Endothelial pas domain protein 1 (epas1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997; 11: 72-82 [DOI] [PubMed] [Google Scholar]
- 9). Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993; 90: 4304-4308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein vhl targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999; 399: 271-275 [DOI] [PubMed] [Google Scholar]
- 11). Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (hif-1alpha) and hif-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003; 23: 9361-9374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of hif-binding sites by chip-seq. Blood. 2011; 117: e207-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Libby P. Inflammation in atherosclerosis. Nature. 2002; 420: 868-874 [DOI] [PubMed] [Google Scholar]
- 14). Gautier EL, Huby T, Witztum JL, Ouzilleau B, Miller ER, Saint-Charles F, Aucouturier P, Chapman MJ, Lesnik P. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation. 2009; 119: 1795-1804 [DOI] [PubMed] [Google Scholar]
- 15). Ridker PM. Clinical application of c-reactive protein for cardiovascular disease detection and prevention. Circulation. 2003; 107: 363-369 [DOI] [PubMed] [Google Scholar]
- 16). Biasucci LM, Liuzzo G, Fantuzzi G, Caligiuri G, Rebuzzi AG, Ginnetti F, Dinarello CA, Maseri A. Increasing levels of interleukin (il)-1ra and il-6 during the first 2 days of hospitalization in unstable angina are associated with increased risk of in-hospital coronary events. Circulation. 1999; 99: 2079-2084 [DOI] [PubMed] [Google Scholar]
- 17). Valgimigli M, Ceconi C, Malagutti P, Merli E, Soukhomovskaia O, Francolini G, Cicchitelli G, Olivares A, Parrinello G, Percoco G, Guardigli G, Mele D, Pirani R, Ferrari R. Tumor necrosis factor-alpha receptor 1 is a major predictor of mortality and new-onset heart failure in patients with acute myocardial infarction: The cytokine-activation and long-term prognosis in myocardial infarction (c-alpha) study. Circulation. 2005; 111: 863-870 [DOI] [PubMed] [Google Scholar]
- 18). Pullamsetti SS, Savai R. Macrophage regulation during vascular remodeling: Implications for pulmonary hypertension therapy. American journal of respiratory cell and molecular biology. 2017; 56: 556-558 [DOI] [PubMed] [Google Scholar]
- 19). Fujiu K, Wang J, Nagai R. Cardioprotective function of cardiac macrophages. Cardiovasc Res. 2014; 102: 232-239 [DOI] [PubMed] [Google Scholar]
- 20). Dauphinot V, Roche F, Kossovsky MP, Schott AM, Pichot V, Gaspoz JM, Gosse P, Barthelemy JC. C-reactive protein implications in new-onset hypertension in a healthy population initially aged 65 years: The proof study. Journal of hypertension. 2009; 27: 736-743 [DOI] [PubMed] [Google Scholar]
- 21). Mattace-Raso FU, Verwoert GC, Hofman A, Witteman JC. Inflammation and incident-isolated systolic hypertension in older adults: The rotterdam study. Journal of hypertension. 2010; 28: 892-895 [DOI] [PubMed] [Google Scholar]
- 22). Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. The European respiratory journal. 2003; 22: 358-363 [DOI] [PubMed] [Google Scholar]
- 23). Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. American journal of respiratory and critical care medicine. 1995; 151: 1628-1631 [DOI] [PubMed] [Google Scholar]
- 24). Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke-Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010; 122: 920-927 [DOI] [PubMed] [Google Scholar]
- 25). Raymond RJ, Dehmer GJ, Theoharides TC, Deliargyris EN. Elevated interleukin-6 levels in patients with asymptomatic left ventricular systolic dysfunction. Am Heart J. 2001; 141: 435-438 [DOI] [PubMed] [Google Scholar]
- 26). Kosmala W, Derzhko R, Przewlocka-Kosmala M, Orda A, Mazurek W. Plasma levels of tnf-alpha, il-6, and il-10 and their relationship with left ventricular diastolic function in patients with stable angina pectoris and preserved left ventricular systolic performance. Coronary artery disease. 2008; 19: 375-382 [DOI] [PubMed] [Google Scholar]
- 27). Williams ES, Shah SJ, Ali S, Na BY, Schiller NB, Whooley MA. C-reactive protein, diastolic dysfunction, and risk of heart failure in patients with coronary disease: Heart and soul study. European journal of heart failure. 2008; 10: 63-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, Mann DL. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: A report from the studies of left ventricular dysfunction (solvd). Journal of the American College of Cardiology. 1996; 27: 1201-1206 [DOI] [PubMed] [Google Scholar]
- 29). Tsutamoto T, Hisanaga T, Wada A, Maeda K, Ohnishi M, Fukai D, Mabuchi N, Sawaki M, Kinoshita M. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. Journal of the American College of Cardiology. 1998; 31: 391-398 [DOI] [PubMed] [Google Scholar]
- 30). Roig E, Orus J, Pare C, Azqueta M, Filella X, Perez-Villa F, Heras M, Sanz G. Serum interleukin-6 in congestive heart failure secondary to idiopathic dilated cardiomyopathy. Am J Cardiol. 1998; 82: 688-690, a688 [DOI] [PubMed] [Google Scholar]
- 31). Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990; 323: 236-241 [DOI] [PubMed] [Google Scholar]
- 32). Miettinen KH, Lassus J, Harjola VP, Siirila-Waris K, Melin J, Punnonen KR, Nieminen MS, Laakso M, Peuhkurinen KJ. Prognostic role of pro- and anti-inflammatory cytokines and their polymorphisms in acute decompensated heart failure. European journal of heart failure. 2008; 10: 396-403 [DOI] [PubMed] [Google Scholar]
- 33). Anand IS, Latini R, Florea VG, Kuskowski MA, Rector T, Masson S, Signorini S, Mocarelli P, Hester A, Glazer R, Cohn JN. C-reactive protein in heart failure: Prognostic value and the effect of valsartan. Circulation. 2005; 112: 1428-1434 [DOI] [PubMed] [Google Scholar]
- 34). Bonecini-Almeida MG, Chitale S, Boutsikakis I, Geng J, Doo H, He S, Ho JL. Induction of in vitro human macrophage anti-mycobacterium tuberculosis activity: Requirement for ifn-gamma and primed lymphocytes. J Immunol. 1998; 160: 4490-4499 [PubMed] [Google Scholar]
- 35). Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003; 3: 23-35 [DOI] [PubMed] [Google Scholar]
- 36). Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004; 25: 677-686 [DOI] [PubMed] [Google Scholar]
- 37). Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008; 8: 958-969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38). Komohara Y, Fujiwara Y, Ohnishi K, Shiraishi D, Takeya M. Contribution of macrophage polarization to metabolic diseases. Journal of atherosclerosis and thrombosis. 2016; 23: 10-17 [DOI] [PubMed] [Google Scholar]
- 39). El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, Konig T, Schleicher U, Koo MS, Kaplan G, Fitzgerald KA, Tuomanen EI, Orme IM, Kanneganti TD, Bogdan C, Wynn TA, Murray PJ. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nature immunology. 2008; 9: 1399-1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40). Bonello S, Zahringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Gorlach A. Reactive oxygen species activate the hif-1alpha promoter via a functional nfkappab site. Arteriosclerosis, thrombosis, and vascular biology. 2007; 27: 755-761 [DOI] [PubMed] [Google Scholar]
- 41). Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. Nfkappab links innate immunity to the hypoxic response through transcriptional regulation of hif-1alpha. Nature. 2008; 453: 807-811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42). Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol. 2007; 178: 7516-7519 [DOI] [PubMed] [Google Scholar]
- 43). Takeda N, O'Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of hif-{alpha} isoforms in macrophages are essential for no homeostasis. Genes Dev. 2010; 24: 491-501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. Hif-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003; 112: 645-657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45). MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995; 81: 641-650 [DOI] [PubMed] [Google Scholar]
- 46). Mattila PK, Lappalainen P. Filopodia: Molecular architecture and cellular functions. Nature reviews. Molecular cell biology. 2008; 9: 446-454 [DOI] [PubMed] [Google Scholar]
- 47). Pantaloni D, Le Clainche C, Carlier MF. Mechanism of actin-based motility. Science. 2001; 292: 1502-1506 [DOI] [PubMed] [Google Scholar]
- 48). De Bock K, Georgiadou M, Carmeliet P. Role of endothelial cell metabolism in vessel sprouting. Cell metabolism. 2013; 18: 634-647 [DOI] [PubMed] [Google Scholar]
- 49). Leach RM, Treacher DF. Oxygen transport-2. Tissue hypoxia. BMJ (Clinical research ed.). 1998; 317: 1370-1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50). Webster KA. Evolution of the coordinate regulation of glycolytic enzyme genes by hypoxia. The Journal of experimental biology. 2003; 206: 2911-2922 [DOI] [PubMed] [Google Scholar]
- 51). Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, Miyazawa H, Yamaguchi Y, Miura M, Jenkins DM, Choi H, Kim JW, Asagiri M, Cowburn AS, Abe H, Soma K, Koyama K, Katoh M, Sayama K, Goda N, Johnson RS, Manabe I, Nagai R, Komuro I. Hif-1alpha-pdk1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016; 7: 11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52). De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P. Role of pfkfb3-driven glycolysis in vessel sprouting. Cell. 2013; 154: 651-663 [DOI] [PubMed] [Google Scholar]
- 53). Shiraishi T, Verdone JE, Huang J, Kahlert UD, Hernandez JR, Torga G, Zarif JC, Epstein T, Gatenby R, McCartney A, Elisseeff JH, Mooney SM, An SS, Pienta KJ. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget. 2015; 6: 130-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54). Bjornheden T, Bondjers G. Oxygen consumption in aortic tissue from rabbits with diet-induced atherosclerosis. Arteriosclerosis (Dallas, Tex.). 1987; 7: 238-247 [DOI] [PubMed] [Google Scholar]
- 55). Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD, Cleutjens JP, van den Akker LH, Corvol P, Wouters BG, Daemen MJ, Bijnens AP. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. Journal of the American College of Cardiology. 2008; 51: 1258-1265 [DOI] [PubMed] [Google Scholar]
- 56). Gao L, Chen Q, Zhou X, Fan L. The role of hypoxia-inducible factor 1 in atherosclerosis. Journal of clinical pathology. 2012; 65: 872-876 [DOI] [PubMed] [Google Scholar]
- 57). Hulten LM, Levin M. The role of hypoxia in atherosclerosis. Current opinion in lipidology. 2009; 20: 409-414 [DOI] [PubMed] [Google Scholar]
- 58). Crucet M, Wust SJ, Spielmann P, Luscher TF, Wenger RH, Matter CM. Hypoxia enhances lipid uptake in macrophages: Role of the scavenger receptors lox1, sra, and cd36. Atherosclerosis. 2013; 229: 110-117 [DOI] [PubMed] [Google Scholar]
- 59). Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989; 320: 915-924 [DOI] [PubMed] [Google Scholar]
- 60). Chen M, Masaki T, Sawamura T. Lox-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacology & therapeutics. 2002; 95: 89-100 [DOI] [PubMed] [Google Scholar]
- 61). Draude G, Hrboticky N, Lorenz RL. The expression of the lectin-like oxidized low-density lipoprotein receptor (lox-1) on human vascular smooth muscle cells and monocytes and its down-regulation by lovastatin. Biochemical pharmacology. 1999; 57: 383-386 [DOI] [PubMed] [Google Scholar]
- 62). Yoshida H, Kondratenko N, Green S, Steinberg D, Quehenberger O. Identification of the lectin-like receptor for oxidized low-density lipoprotein in human macrophages and its potential role as a scavenger receptor. The Biochemical journal. 1998; 334 (Pt 1): 9-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63). Chaudhari SM, Sluimer JC, Koch M, Theelen TL, Manthey HD, Busch M, Caballero-Franco C, Vogel F, Cochain C, Pelisek J, Daemen MJ, Lutz MB, Gorlach A, Kissler S, Hermanns HM, Zernecke A. Deficiency of hif1alpha in antigen-presenting cells aggravates atherosclerosis and type 1 t-helper cell responses in mice. Arteriosclerosis, thrombosis, and vascular biology. 2015; 35: 2316-2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64). Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3': 5'-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci U S A. 1977; 74: 3203-3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65). Denninger JW, Marletta MA. Guanylate cyclase and the No/cgmp signaling pathway. Biochimica et biophysica acta. 1999; 1411: 334-350 [DOI] [PubMed] [Google Scholar]
- 66). Hussain MB, Hobbs AJ, MacAllister RJ. Autoregulation of nitric oxide-soluble guanylate cyclase-cyclic gmp signalling in mouse thoracic aorta. British journal of pharmacology. 1999; 128: 1082-1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67). Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: Beyond enos. Journal of pharmacological sciences. 2015; 129: 83-94 [DOI] [PubMed] [Google Scholar]
- 68). Cowburn AS, Takeda N, Boutin AT, Kim JW, Sterling JC, Nakasaki M, Southwood M, Goldrath AW, Jamora C, Nizet V, Chilvers ER, Johnson RS. Hif isoforms in the skin differentially regulate systemic arterial pressure. Proc Natl Acad Sci U S A. 2013; 110: 17570-17575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69). Stenmark KR, Abman SH. Lung vascular development: Implications for the pathogenesis of bronchopulmonary dysplasia. Annual review of physiology. 2005; 67: 623-661 [DOI] [PubMed] [Google Scholar]
- 70). Carbone R, Bossone E, Bottino G, Monselise A, Rubenfire M. Secondary pulmonary hypertension--diagnosis and management. European review for medical and pharmacological sciences. 2005; 9: 331-342 [PubMed] [Google Scholar]
- 71). Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society. 2005; 2: 20-22 [DOI] [PubMed] [Google Scholar]
- 72). Gilroy J, Cahalan JL, Berman R, Newman M. Cardiac and pulmonary complications in duchenne's progressive muscular dystrophy. Circulation. 1963; 27: 484-493 [DOI] [PubMed] [Google Scholar]
- 73). Arias-Stella J, Saldana M. The terminal portion of the pulmonary arterial tree in people native to high altitudes. Circulation. 1963; 28: 915-925 [DOI] [PubMed] [Google Scholar]
- 74). Cowburn AS, Crosby A, Macias D, Branco C, Colaco RD, Southwood M, Toshner M, Crotty Alexander LE, Morrell NW, Chilvers ER, Johnson RS. Hif2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci U S A. 2016; 113: 8801-8806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75). Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, Carvo I, Signoretti S, Bruick RK, Josey JA, Wallace EM, Kaelin WG. On-target efficacy of a hif-2alpha antagonist in preclinical kidney cancer models. Nature. 2016; 539: 107-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76). Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, Hao G, Yousuf Q, Joyce A, Pedrosa I, Geiger H, Zhang H, Chang J, Gardner KH, Bruick RK, Reeves C, Hwang TH, Courtney K, Frenkel E, Sun X, Zojwalla N, Wong T, Rizzi JP, Wallace EM, Josey JA, Xie Y, Xie XJ, Kapur P, McKay RM, Brugarolas J. Targeting renal cell carcinoma with a hif-2 antagonist. Nature. 2016; 539: 112-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77). Yndestad A, Damas JK, Oie E, Ueland T, Gullestad L, Aukrust P. Role of inflammation in the progression of heart failure. Current cardiology reports. 2007; 9: 236-241 [DOI] [PubMed] [Google Scholar]
- 78). Anker SD, von Haehling S. Inflammatory mediators in chronic heart failure: An overview. Heart (British Cardiac Society). 2004; 90: 464-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79). Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, Niebauer J, Hooper J, Volk HD, Coats AJ, Anker SD. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000; 102: 3060-3067 [DOI] [PubMed] [Google Scholar]
- 80). Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: The cytokine hypothesis. Journal of cardiac failure. 1996; 2: 243-249 [DOI] [PubMed] [Google Scholar]
- 81). Azzawi M, Kan SW, Hillier V, Yonan N, Hutchinson IV, Hasleton PS. The distribution of cardiac macrophages in myocardial ischaemia and cardiomyopathy. Histopathology. 2005; 46: 314-319 [DOI] [PubMed] [Google Scholar]
- 82). Frangogiannis NG. The immune system and cardiac repair. Pharmacological research. 2008; 58: 88-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83). Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nature reviews. Cardiology. 2014; 11: 255-265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84). Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling--concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an international forum on cardiac remodeling. Journal of the American College of Cardiology. 2000; 35: 569-582 [DOI] [PubMed] [Google Scholar]
- 85). Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Ly-6chigh monocytes depend on nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014; 114: 1611-1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86). Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007; 204: 3037-3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87). Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: Protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010; 121: 2437-2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88). Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, Krum H, Liu P, Nieminen M, Tavazzi L, van Veldhuisen DJ, Waldenstrom A, Warren M, Westheim A, Zannad F, Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: Results of the randomized etanercept worldwide evaluation (renewal). Circulation. 2004; 109: 1594-1602 [DOI] [PubMed] [Google Scholar]
- 89). Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Investigators A-TTACHF Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-tnf therapy against congestive heart failure (attach) trial. Circulation. 2003; 107: 3133-3140 [DOI] [PubMed] [Google Scholar]
- 90). Knapp FJ, Goodman M, Callahan A, Kirsch G. Radio-iodinated 15-(p-iodophenyl)-3,3-dimethylpentadecanoic acid: A useful new agent to evaluate myocardial fatty acid uptake. J Nucl Med. 1986; 27: 521-531 [PubMed] [Google Scholar]
- 91). Stanley W, Lopaschuk G, Hall J, McCormack J. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. 1997; 33: 243-257 [DOI] [PubMed] [Google Scholar]
- 92). Dwivedi G, Al-Shehri H, deKemp RA, Ali I, Alghamdi AA, Klein R, Scullion A, Ruddy TD, Beanlands RS, Chow BJ. Scar imaging using multislice computed tomography versus metabolic imaging by f-18 fdg positron emission tomography: A pilot study. Int J Cardiol. 2013; 168: 739-745 [DOI] [PubMed] [Google Scholar]
- 93). Abe H, Iguchi N, Utanohara Y, Inoue K, Takamisawa I, Seki A, Tanizaki K, Takeda N, Tohbaru T, Asano R, Nagayama M, Takayama M, Umemura J, Sumiyoshi T, Tomoike H. Non-invasive diagnosis of coronary artery disease by 123i-bmipp/201tlcl dual myocardial spect in patients with heart failure. Int J Cardiol. 2014; 176: 969-974 [DOI] [PubMed] [Google Scholar]
- 94). Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. P53-induced inhibition of hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007; 446: 444-448 [DOI] [PubMed] [Google Scholar]
- 95). Shyu KG, Liou JY, Wang BW, Fang WJ, Chang H. Carvedilol prevents cardiac hypertrophy and overexpression of hypoxia-inducible factor-1alpha and vascular endothelial growth factor in pressure-overloaded rat heart. J Biomed Sci. 2005; 12: 409-420 [DOI] [PubMed] [Google Scholar]
- 96). Wei H, Bedja D, Koitabashi N, Xing D, Chen J, Fox-Talbot K, Rouf R, Chen S, Steenbergen C, Harmon JW, Dietz HC, Gabrielson KL, Kass DA, Semenza GL. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of tgf-beta signaling. Proc Natl Acad Sci U S A. 2012; 109: E841-850 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 97). Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, Perriard JC, Larsen T, Pedrazzini T, Krek W. Activation of a hif1alpha-ppargamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell metabolism. 2009; 9: 512-524 [DOI] [PubMed] [Google Scholar]