

Abstract
p53 encodes a transcription factor that transactivates downstream target genes involved in tumour suppression. Although osteosarcoma frequently has p53 mutations, the role of p53 in osteosarcomagenesis is not fully understood. To explore p53-target genes comprehensively in calvarial bone and find out novel druggable p53 target genes for osteosarcoma, we performed RNA sequencing using the calvarial bone and 23 other tissues from p53 +/+ and p53 −/− mice after radiation exposure. Of 23,813 genes, 69 genes were induced more than two-fold in irradiated p53 +/+ calvarial bone, and 127 genes were repressed. Pathway analysis of the p53-induced genes showed that genes associated with cytokine-cytokine receptor interactions were enriched. Three genes, CD137L, CDC42 binding protein kinase gamma and Follistatin, were identified as novel direct p53 target genes that exhibited growth-suppressive effects on osteosarcoma cell lines. Of the three genes, costimulatory molecule Cd137l was induced only in calvarial bone among the 24 tissues tested. CD137L-expressing cells exhibited growth-suppressive effects in vivo. In addition, recombinant Fc-fusion Cd137l protein activated the immune response in vitro and suppressed osteosarcoma cell growth in vivo. We clarified the role of CD137L in osteosarcomagenesis and its potential therapeutic application. Our transcriptome analysis also indicated the regulation of the immune response through p53.
Introduction
Among the cancer-related genes that have been identified, p53 is the most frequently mutated gene in human cancers1. p53 encodes a transcription factor that transactivates downstream target genes involved in various cellular functions, including apoptosis1, cell cycle arrest2, metabolism3, stem cell maintenance4, and metastasis5, Through the regulation of these cellular functions, p53 plays a pivotal role in tumour suppression. In addition to these functions, emerging evidence has illuminated a new role of p53 in the regulation of the immune system6.
Osteosarcoma is the most common primary malignant bone tumour7. Comprehensive genome analyses of osteosarcoma have revealed that the most frequent mutation is that of the p53 gene (up to 80% of cases)8–12. The association between p53 inactivation and osteosarcomagenesis is also observed in patients with Li-Fraumeni syndrome, an autosomal dominant disorder characterized by a germline mutation in p53, who have a higher risk of developing osteosarcoma13. Moreover, deletion of p53 in mouse osteoblast has been reported to result in the development of osteosarcoma, and osteoblast or osteoblast precursor in bone is considered to be cells of origin in osteosarcoma7, 14. Thus, p53 behaves as a core tumour suppressor in osteosarcoma. However, the roles of p53 in the pathogenesis of osteosarcoma are not fully understood.
Recent genome-wide profiling of p53 binding and transcriptional activity has shown that the precise cellular responses triggered by p53 are cell-type dependent15. Moreover, various malignant tumours occur in patients with Li-Fraumeni syndrome or p53 −/− mice, and the incidence of these tumours differs13, 16. These results suggest that the tumour suppressive roles of p53 are organ- or cell-type dependent. In these contexts, unravelling the comprehensive p53 functions specific to bone or osteoblasts is crucial to elucidate the roles of p53 in osteosarcomagenesis.
Current therapies for osteosarcoma incorporate surgical resection and combination chemotherapy (doxorubicin, cisplatin and methotrexate), which cures approximately 70% of patients17. However, survival for patients with metastatic or relapsed osteosarcoma has remained virtually unchanged over the past 30 years, with an overall 5-year survival rate of approximately 20%18. Consequently, new therapies are needed. Because the restoration of wild-type p53 function in osteosarcoma cells has not succeeded clinically owing to the complexity of p53 signalling1, identification of druggable p53 downstream molecules or pathways could be key to attacking p53-deficient tumours such as osteosarcoma.
Therefore, we aimed to clarify p53 target genes comprehensively in calvarial bone which mainly consists of osteoblasts using RNA sequencing and to find novel p53 target genes, especially for bone-specific p53 targets. In addition, the purpose of this study was to reveal the roles of p53 in osteosarcomagenesis and to identify novel druggable p53 targets.
Results
p53 target genes in calvarial bone and enriched pathways
We conducted RNA sequencing of 280 samples from 24 tissues including calvarial bone of p53 +/+ and p53 −/− mice19. p53 +/+ and p53 −/− mice were sacrificed for organ sampling 24 h after 10 Gy of whole-body X-ray irradiation. Samples were categorized into 4 groups: (K) p53 −/− mice without irradiation, (W) p53 +/+ mice without irradiation, (KX) p53 −/− mice with irradiation, and (WX) p53 +/+ mice with irradiation (n = 3 per group). First, we analyzed mRNA expression profiles of 23,813 transcripts in 12 calvarial bone samples (K, W, KX, WX, n = 3 per group) (Fig. 1a).
Figure 1.

Regulation of CD137L, CDC42BPG and FST by p53. (a) Outline of the screening process. The expression profiles of 23813 genes in calvarial bone were detected by RNA sequencing, and 69 genes were selected by the indicated criteria as p53-induced genes. A second screening revealed three novel direct p53 targets. Of the 3 genes, one gene (CD137L) was up-regulated specifically in bone among 24 tissues. (b) At 24 h after transfection of each siRNA, U2OS cells were treated with ADR (2 μg/ml for 2 h). At 36 h after treatment, qPCR was performed. siRNA against EGFP was used as a control. β-actin was used for the normalization of expression levels. Error bars represent SD (n = 2). (c) qPCR was performed for the same calvaria sample as the RNA sequencing. β-actin was used for normalization of the expression levels. Error bars represent SD (n = 3). **P < 0.001, Student’s t-test. (d) qPCR was performed 36 h after treatment with ADR (2 μg/ml for 2 h) in p53 +/+ or p53 −/− calvarial osteoblasts. β-actin was used for normalization of the expression levels. Error bars represent SD (n = 3). **P < 0.001, Student’s t-test.
We screened genes whose expression level was induced or repressed only in the WX group. Of 23813 genes, 69 genes were induced more than two-fold in p53 +/+ mice after radiation exposure compared with the other groups (Supplementary Table 1a). Representative p53 target genes including p21, Bax and Fas were induced by radiation in p53 +/+ mice (Supplementary Fig. 1a). Pathway analysis for the 69 induced genes showed that genes related to the p53 signalling pathway, cytokine-cytokine receptor interactions, the TGF-β signalling pathway, and apoptosis were enriched (Table 1). On the other hand, the expression of 127 genes was more than two-fold repressed (Supplementary Table 1b). Enriched pathway of p53-repressed genes included the cell cycle pathway. p53-binding peak was detected in 49 genes (71%) among 69 induced genes in CHIP sequence data20, while p53-binding peak was detected in only 8 genes (6%) among 127 repressed genes. Moreover, 51 of 127 genes were shown to be regulated by p53-p21-DREAM pathway in the previous report21. Therefore, many of down-regulated genes are likely to be regulated through indirect mechanisms.
Table 1.
Pathway analysis for the p53-induced and p53-repressed genes.
| Term | Genes | −log (p value) | Fold enrichment | |
|---|---|---|---|---|
| p53-induced genes | p53 signalling pathway | P21, Bbc3, Z3, Bax, Rprm, Mdm2, Pmaip1, Fas, Perp, Sesn2, Ccng1 | 12.27 | 30.49 |
| Cytokine-cytokine receptor interaction | Inhbb, Tnfrsf10b, Gdf5, Eda2r, Fas, Cd137l | 2.16 | 4.7 | |
| TGF-beta signalling pathway | Inhbb, Gdf5, Fst | 1.15 | 6.6 | |
| Apoptosis | Tnfrsf10b, Bax, Fas | 1.15 | 6.6 | |
| p53-repressed genes | Cell cycle | Cdk1, E2f2, Cdc6, Ttk, Chek1, Mcm2, Mcm3, Mcm5, Mcm6, Ccne2, Cdc45, Ccnb2, Plk1, Bub1, Bub1b, Ccna2 | 10.99 | 21.1 |
| DNA replication | Prim1, Dna2, Pole, Mcm2, Mcm3, Mcm5, Mcm6 | 5.70 | 33.75 | |
| Oocyte meiosis | Ccne2, Cdk1, Ccnb2, Plk1, SgoL1, Bub1, Fbxo5 | 2.82 | 10.27 | |
| p53 signalling pathway | Ccne2, Cdk1, Ccnb2, Rrm2, Chek1 | 2.44 | 12.23 | |
| Progesterone-mediated oocyte maturation | Cdk1, Ccnb2, Plk1, Bub1, Ccna2 | 1.81 | 9.93 | |
| Pyrimidine metabolism | Prim1, Rrm2, Pole, Tk1 | 1.54 | 7.03 |
Screening of novel p53-target genes in calvarial bone by RNA sequencing
Of the 69 p53-induced genes, 28 have already been reported as direct p53 targets. With the exception of 10 genes that did not have a human homolog, we identified 31 candidate p53 novel target genes (Supplementary Table 1a). For the second screening to identify novel p53 direct target genes from the 31 candidate genes, we explored genes whose expression levels were induced by adriamycin (ADR) and changed in a p53-dependent manner using a human osteosarcoma cell line (Fig. 1a). At 24 h after transfection with small interfering RNA (siRNA) targeting p53 (sip53), U2OS osteosarcoma cells were treated with 2 µg/ml ADR for 2 h. Expression of p21 (positive control) was induced by ADR treatment but supressed in sip53-treated cells compared with that of control cells (Supplementary Fig. 1b). We assessed the expression of all 31 candidates by quantitative real-time PCR (qPCR) and identified CD137L, CDC42 binding protein kinase gamma (CDC42BPG) and Follistatin (FST) as novel p53-regulated genes (Fig. 1b). We found that ADR induced the expression of the three genes in a dose-dependent manner in U2OS cells (Supplementary Fig. 1c). We also confirmed the expression of the three genes in mouse calvarial bone tissues by qPCR and observed their induction by X-ray irradiation only in the calvarial bone of p53 +/+ mice, concordant with our RNA sequencing analysis (Fig. 1c, Supplementary Fig. 1d).
Then, we evaluated the expression of these genes in calvaria-derived primary osteoblasts. We found that ADR treatment induced the mRNA expression of the Cd137l, Cdc42bpg and Fst genes in primary osteoblasts (Supplementary Fig. 1e). Moreover, Cd137l, Cdc42bpg and Fst mRNA levels were significantly increased in p53 +/+ primary osteoblasts with ADR than those of other groups (Fig. 1d). These findings clearly indicated that p53 regulates the expression of Cd137l, Cdc42bpg and Fst in response to DNA damage.
Identification of CD137L as a novel bone-specific p53 target gene
Subsequently, we surveyed the genomic sequences of the human and mouse CD137L, CDC42BPG and FST to detect p53-binding sequences (p53BS). The human and mouse CD137L, CDC42BPG and FST genes had potential p53BS within the first intron or promoter region (<5000 bp upstream from the transcription start site) (Supplementary Fig. 2a–c). We then subcloned a human or mouse CD137L DNA fragment, which included two putative p53BSs, into a pGL4.24 promoter vector (pGL4.24/CD137L-BS). We found that the co-transfection of pGL4.24/CD137L-BS with the wild-type p53 expression plasmid enhanced the luciferase activity (Fig. 2a). For the other 2 genes, co-transfection of pGL4.24/CDC42BPG-BS or pGL4.24/FST-BS also enhanced the luciferase activity (Fig. 2b,c). To examine the possible binding of p53 to these DNA segments, we carried out a chromatin immunoprecipitation (ChIP) assay using SaOS2 cells (p53-null) that were infected with either Ad-p53 or Ad-LacZ. qPCR of immunoprecipitated DNA indicated that the p53 protein bound to the genomic fragment containing the p53BSs (Fig. 2d–f). We analyzed published CHIP sequence data22 and found p53-binding peaks at 3.5-kb 3′ flanking region of the FST gene (FST-BS2). We cloned potential p53 binding sequence of FST gene (FST-BS2) and performed luciferase assay. As a result, p53 induced luciferase activity through this sequence (Fig. 2c). ChIP assay using SaOS2 cells also indicated the binding of p53 to this genomic fragment (Fig. 2f). These findings implied that p53 directly regulated the expression of the three genes through multiple p53BSs.
Figure 2.

Identification of CD137L, CDC42BPG and FST as p53 direct target genes. (a–c) Luciferase assay of the p53BS in human (left) or mouse (right) CD137L (a), CDC42BPG (b) or FST (c) using SaOS2 cells. Luciferase activity is indicated relative to the activity of the mock vector with SD (n = 3). *P < 0.05, *P < 0.001, Student’s t-test. (d–f) CHIP assay for CD137L (d), CDC42BPG (BS-C) (e) or FST (f) was performed using SaOS2 cells that were infected with Ad-LacZ (lane 1) or Ad-p53 (lane 2-4). DNA-protein complexes were immunoprecipitated with an anti-p53 antibody (lanes 1 and 2) followed by qPCR. Input chromatin represents a small portion (1%) of the sonicated chromatin collected prior to immunoprecipitation. Immunoprecipitates with an anti-IgG antibody (lane 3) or in the absence of an antibody (lane 4) were used as negative controls. Columns, mean; error bars, SD (n = 3).
To compare the expression of these genes in mouse 24 tissues, we calculated the median fragments per kilobase of exon per million mapped fragments (FPKM) value of WX/maximum FPKM value of the median K, median KX or median W for 24 tissues in our RNA sequencing data. We compared the fold change in the calvarial bone with those in the 23 other tissues. Cd137l was more than two-fold induced only in the calvarial bone among the 24 tissues analysed (Fig. 3a). Cdc42bpg and Fst did not show bone-specific induction. We also found that CD137L protein was induced by DNA damage in U2OS cells, and sip53 remarkably reduced the CD137L protein level (Fig. 3b). In addition, immunohistochemistry indicated the induction of Cd137l protein by X-ray irradiation in mouse calvarial osteoblasts of p53 +/+ mice (Fig. 3c). Thus, we selected CD137L for further functional analysis.
Figure 3.

Identification of CD137L as a bone-specific p53 target gene. (a) Induction of Cd137l among 24 tissues. Samples were categorized into 4 groups: (K) p53 −/− mice without irradiation, (W) p53 +/+ mice without irradiation, (KX) p53 −/− mice with irradiation and (WX) p53 +/+ mice with irradiation (n = 3 per group). We calculated the median FPKM value of WX/maximum FPKM value of the median FPKM value in K, KX or W for 24 tissues. (b) At 24 h after transfection with each siRNA, U2OS cells were treated with ADR (2 μg/ml for 2 h). At 36 h after treatment, whole cell extracts were subjected to western blotting with an anti-CD137L, anti-p21, anti-p53, or anti-β-actin antibody. siRNA against EGFP was used as a control. β-actin was used for normalization of the expression levels. These images were cropped from full-length blots (Supplementary Fig. 9). (c) Immunohistochemical staining of Cd137l in mouse p53α or p53−/− calvaria with or without radiation exposure. Representative images from three tissues in each group are shown. Scale Bars (left) = 50 μm, Scale Bars (right) = 20 μm. Black arrowhead shows osteoblast.
Growth suppressive effect of p53 or CD137L
Because osteoblast precursors are considered the cells of origin in osteosarcoma7, we evaluated the proliferation of primary osteoblasts. Primary osteoblasts from p53 −/− mice showed increased proliferation compared with osteoblasts from p53 +/+ mice (Fig. 4a), as previously reported23. Ki67-positive osteoblasts were more frequently found in the p53 −/− calvarial bone compared with those in p53 +/+ mice (Fig. 4b). In addition, Ki67-positive osteoblasts were not observed in the p53 +/+ calvarial bone with radiation exposure, demonstrating the growth suppressive effect of p53 in osteoblasts.
Figure 4.

Growth suppressive effects of p53 or CD137L. (a) p53 +/+ calvarial osteoblasts or p53 −/− calvarial osteoblasts were cultured on plates. Cell proliferation was assessed at 48 h. Error bars represent SD (n = 3). *P < 0.05, Student’s t-test. (b) Immunohistochemical staining of Ki67 in mouse p53+/+ or p53−/− calvaria with or without radiation exposure. Representative images from three tissues in each group are shown. Scale Bars (left) = 50 μm, Scale Bars (right) = 20 μm. (c) A colony formation assay was performed. After ectopic expression of CD137L or mock, the number of SaOS2, U2OS or LM8 cells was determined. Whole cell extracts were subjected to western blotting with a human or mouse anti-CD137L antibody. Error bars represent SD (n = 3). **P < 0.001, Student’s t-test. β-actin (U2OS and SaOS2) and α-tubulin antibody (LM8) was used as loading control. (d) Cd137l (n = 3) or mock (n = 3) stable expression cell lines were inoculated into the left and right flanks of C3H mice; each group contains 2 mice. The tumour volume was calculated every 2 or 3 days. *P < 0.05, Wilcoxon rank-sum test.
To analyse the function of CD137L, we carried out colony formation assays using human osteosarcoma cell lines (U2OS and SaOS2) and a mouse osteosarcoma cell line (LM8) transfected with plasmids expressing CD137L or mock. Ectopic expression of CD137L reduced cell proliferation, suggesting a growth-suppressive effect of CD137L (Fig. 4c). Moreover, ectopic expression of CDC42BPG, FST344 (major isoform of FST) or FST311 (minor isoform of FST) reduced cell proliferation, which indicated that these genes also had growth suppressive effects in osteosarcoma cell lines (Supplementary Fig. 3a,b).
Then, we investigated the growth suppressive effect of Cd137l in vivo. LM8 cells stably expressing mock or Cd137l (n = 3 each, Supplementary Fig. 4a,b) were subcutaneously inoculated into CH3 mice. As a result, Cd137l-expressing LM8 cells showed reduced tumour growth compared with mock-expressing LM8 cells (Fig. 4d, Supplementary Fig. 4c).
Role of the CD137L reverse or direct signal in osteosarcoma
Although CD137L is a ligand of CD137, CD137L transmits a reverse signal through its interaction with CD137 and mediates diverse cellular responses, including proliferation and differentiation, in a variety of cells24, 25. We generated recombinant protein containing the extracellular domain of CD137 and the Fc region of human IgG protein (human CD137-Fc or mouse Cd137-Fc) or Cd137l and the Fc region of human IgG protein (mouse Cd137l-Fc) (Supplementary Fig. 5a,b). After transfection of HEK293 cells with plasmid expressing CD137L or mock, the cells were incubated with CD137-Fc or mock-Fc for 2 h. Interaction between CD137L and CD137-Fc was confirmed by immunocytochemistry using an anti-human IgG antibody (Supplementary Fig. 5c). We also confirmed the interaction between Cd137 and Cd137l-Fc.
To investigate the effects of the CD137L reverse signalling, SaOS2 cells or osteoblasts were seeded on plates precoated with CD137-Fc or mock-Fc. CD137-Fc significantly reduced the proliferation of SaOS2 cells or p53 +/+ osteoblasts compared with cells treated with mock-Fc (Fig. 5a,b). These results indicate that CD137L reverse signalling inhibits the proliferation of osteosarcoma cells and osteoblasts.
Figure 5.

The role of CD137L reverse or direct signalling in osteosarcoma. (a) SaOS2 cells were cultured with 10 or 20 μg/ml CD137-Fc or mock-Fc. Cell proliferation was assessed at 48 h. Error bars represent SD (n = 3). *P < 0.05, **P < 0.001, Student’s t-test. (b) p53 +/+ calvarial osteoblasts were cultured with 40 μg/ml Cd137-Fc or mock-Fc protein. Cell proliferation was assessed at 48 h. Error bars represent SD (n = 3). *P < 0.05, Student’s t-test. (c,d) IL2 production (c) and proliferation (d) of CD4+ or CD8+ T cells were assessed 48 h after stimulation with Cd137l-Fc. **P < 0.001, Student’s t-test. (e) C3H/HeJ mice (each group, n = 3) were subcutaneously injected into the left flank with 0.1 ml of PBS containing 1 × 106 LM8 cells. After tumours reached 0.5 cm in diameter, each of the mice was treated with Cd137l-Fc fusion protein or control mock-Fc by intraperitoneal injection for 3 days. The tumour volume was calculated every 2 or 3 days. Arrow shows the days of injection. *P < 0.05, Wilcoxon rank-sum test.
The CD137L receptor/ligand system is used by antigen-presenting cells to costimulate T cell activity together with the T cell receptor/CD3 signal26. To investigate the effect CD137L on T cell activation, IL-2 production and cell proliferation were assessed using CD8+ and CD4+ T cells isolated from C3H/HeJ mice spleens. CD4+ and CD8+ cells were seeded in 24-well culture plates precoated with an anti-CD3 antibody in the presence of Cd137l-Fc or mock-Fc. Cd137l-Fc significantly induced IL-2 production and increased proliferation of CD8+ and CD4+ T cells (Fig. 5c,d).
Finally, we explored the effect of Cd137l-Fc on the growth of LM8 cells in vivo. Six-week-old male C3H/HeJ mice were subcutaneously injected with 1 × 106 LM8 cells in the left flank. When tumours reached 0.5 cm in diameter, at approximately the 7th day after tumour implantation, mice (n = 3 each) were treated with 0.2 ml (50 µg) of Cd137l-Fc fusion proteins or control mock-Fc by intraperitoneal injection for 3 consecutive days. As a result, Cd137l-Fc injection significantly suppressed tumour growth at day19 compared with mock-Fc (Fig. 5e), demonstrating the growth suppressive effect of CD137/CD137L pathway.
Discussion
Here, we comprehensively investigated p53-regulated genes in calvarial bone. We found several p53-regulated pathways from comprehensive set of p53 targets and identified CD137L, CDC42BPG, and FST as novel direct p53 targets. Of these three genes, CD137L was considered a bone-specific p53 target that mediates growth suppression and immune response of osteosarcoma cells (Fig. 6a).
Figure 6.
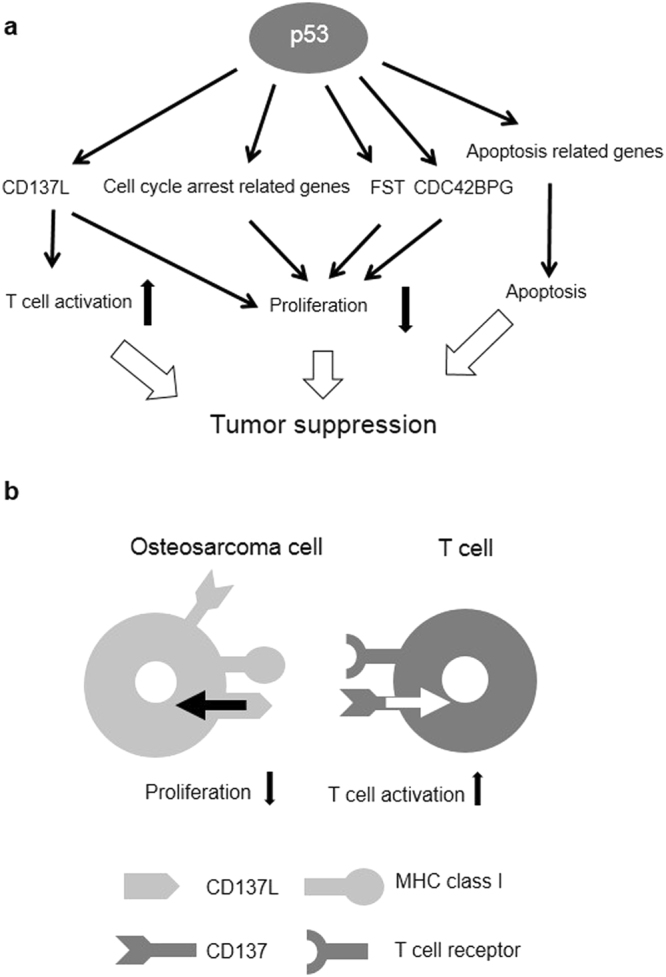
A schema of p53 or CD137L function. (a) A schema of the p53-regulated pathways or genes and the tumour suppressive roles. (b) A schema of CD137L function through direct or reverse signalling.
As we expected, pathway analysis showed that apoptosis-related genes were enriched in the p53-induced genes. Conversely, cell cycle-related genes were enriched in the p53-repressed genes. Numerous studies have revealed the role of canonical p53-mediated apoptosis and cell cycle arrest in tumour suppression1, 2. In bone or osteoblasts, p53-driven apoptotic response or the cell cycle arrest induced by acute DNA damage presumably contribute to the tumour-suppressive roles of p53 (Fig. 6a). Moreover, the cytokine-cytokine receptor interaction pathway may be related to the tumour-suppressive roles of p53 in bone according to our study. Inhbb, Tnfrsf10b, Gdf5, Eda2r, Fas and Cd137l were included in the cytokine-cytokine receptor interaction. Of these genes, Tnfrsf10b, Eda2r and Fas were reported to be p53 target genes and tumour-suppressor genes27–30.
CD137L was identified as a novel bone-specific p53 target gene. CD137L (also known as tumour necrosis factor superfamily 9 [TNFSF9] or 4-1BBL) is a member of the TNF superfamily, and the CD137L receptor/ligand system is used by antigen-presenting cells to costimulate T cell activity26. The activation of T cells plays a crucial role in the antitumour immune response, and cancer immunotherapy has now been clinically validated as an effective treatment28. To activate naive T cells, two key signals through major histocompatibility complex (MHC) class I/T cell receptor and costimulatory molecules are required26. The amplitude and quality of the T cell response are regulated by a balance between costimulatory and coinhibitory signals26, 31. p53 can regulate the T cell response by activating the expression of MHC class I molecules32 and suppressing the expression of coinhibitory molecules, such as PDL133. This is the first report that shows p53 regulates costimulatory molecules, and our results emphasize the importance of p53 in immune regulation.
CD137L seems to have two roles in the suppression of osteosarcomagenesis (Fig. 6b). First, reverse CD137L signalling showed a growth-suppressive effect on osteoblasts and SaOS2 cells. CD137L reverse signalling was reported to induce apoptosis via the intrinsic pathway and cell cycle arrest34, and this was considered as a major mechanism of CD137L mediated growth suppression in vitro. T cells were shown to express CD13726. We also found that DNA damage induced CD137 expression in osteoblasts or U2OS cells (Supplementary Fig. 6a,b). Thus, these cells may contribute to the induction of reverse CD137L signalling and suppress the tumour growth of osteosarcoma or osteosarcoma-initiating cells. However, many p53-regulated genes have a growth suppressive effect1, and the sole effect of CD137L on cell proliferation seems to be small.
Another function of CD137L in the suppression of osteosarcomagenesis is the activation of the T cell response. Recombinant Fc-fusion Cd137l protein activated the T-cells in vitro, and this data implies that direct CD137L signalling can mediate the osteosarcoma microenvironment and exert an anti-tumour immune effect through the activation of T cells. Mice inoculated with hepatocellular carcinoma cells expressing CD137L have been reported to develop a strong cytotoxic T lymphocyte response and long-term immunity against tumours35, and this data supports our hypothesis. Among 24 mouse tissues, induction of Cd137l was only found in bone, and these data indicates the importance of CD137L in osteosarcomagenesis. However, the mechanism how p53 induce Cd137l only in bone tissue is remained to be elucidated. Screening of Remap database indicates a binding of RUNX1 and RUNX3 that are essential to bone differentiation36, 37 to CD137L locus. Our RNA sequence data showed that Runx1 and Runx3 are highly expressed in the bone among 24 tissues19. In addition, RUNX family was shown to interact with p53 in DNA damage response38. Therefore, RUNX family would be co-factors for bone specific induction of Cd137l.
Our Microarray analysis using MCF10A and HCT116 cells indicated that CD137L mRNA expression was remarkably induced by ADR in MCF10A p53 +/+ and HCT116 p53 +/+ cells, although CD137L is also induced in MCF10A p53 −/− and HCT116 p53 +/+ cells (Supplementary Fig. 7). On the other hand, CD137L was not induced in H1299 lung cancer cells by ectopic expression of p53. These result suggested that human CD137L expression was regulated by DNA damage in various types of cells through both p53-dependent and p53-independent manner. Difference in induction pattern between human CD137L and mouse Cd137l might be partially explained by the fact that human CD137L has four p53-binding elements that are not conserved in mouse Cd137l (Supplementary Fig. 8). In our study, CD137L-Fc showed an anti-tumour effect in vivo. Although further studies are needed to reveal the mechanism of this effect, CD137L/CD137 signalling could potentially be an actionable novel therapeutic target for osteosarcoma and other malignancies. One possible explanation of this effect is the T cell activation by CD137L-Fc. The ability of CD137L to potentiate strong and durable immune effector responses has made CD137L a clinically viable target for cancer immunotherapy for several types of cancer39.
We found that CDC42BPG and FST were novel p53 targets that also had growth-suppressive effects on U2OS and SaOS2 cells. Aside from the growth suppressive effect, CDC42BPG may exert an anti-tumour effect by inhibiting CDC42, which activates migration and metastasis in osteosarcoma cell lines40, 41. p53 was shown to promote apoptosis through activation of Cdc42/JNK1 pathway42 and CDC42 also negatively regulate p53 through miR-2943. CDC42BPG may be related to the p53/CDC42 feedback loop by inhibiting CDC42. FST may have a tumour-suppressor role via the inhibition of BMP2 activity44. BMP2 contributes to osteosarcoma growth45.
In conclusion, our transcriptome analysis revealed comprehensive p53-regulated pathways and genes in bone. We also uncovered the role of CD137L in osteosarcomagenesis and its potential therapeutic application. This study provides new insights into the roles of p53 in osteosarcomagenesis and the regulation of the immune response thorough p53.
Materials and Methods
Cell culture and transfection
U2OS and SaOS2 cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). LM8 and HEK293 cell lines were purchased from Riken BRC (Tsukuba, Ibaraki, Japan). The FreeStyle 293-F cell line was purchased from Thermo Fisher Scientific (Waltham, MA, USA). U2OS and SaOS2 are human osteosarcoma cell lines. LM8 is a murine osteosarcoma cell line. Primary osteoblasts were isolated from the calvarial bone of newborn mice as previously described23.
Cells were transfected with siRNA oligonucleotides, commercially synthesized by Sigma Genosys (Woodlands, TX, USA), using Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA). The sequences of the siRNA oligonucleotides are shown in Supplementary Table 2. All methods were carried out accordance with the Guidelines of the Institute of Medical Science, University of Tokyo.
RNA sequencing
p53−/− mice were provided by the RIKEN BioResource Center. Genotypes were confirmed by PCR analysis. All mice were maintained under specific pathogen-free conditions. Institute of Medical Science, University of Tokyo committee approved the experimental protocols and all experiments were performed in accordance with the Guidelines for Animal Experiments of the Institute of Medical Science, University of Tokyo. We used 4 groups of mice: K, W, KX and WX (n = 3 per group). p53+/+ and p53−/− mice were X-ray-irradiated using a MBR-1520R-3 system (Hitachi). The calvarial bone and 23 additional tissues (thymus, heart, lung, kidney, spleen, liver, bladder, esophagus, stomach, colon, small intestine, testis, parorchis, seminal vesicle, muscle, bone marrow, tongue, eye ball, cerebrum, uterus, cartilage, ovary, mammary gland) were extracted at 24 h after exposure to 10 Gy radiation. One-week-old mice were used to obtain bone and cartilage tissues, and 10 week female mice were used for breast, uterus, and ovary. Six-week-old mice were used to obtain the other 19 tissues. Tissues were maintained in RNAlater stabilization solution at 4 °C until RNA collection. RNA was collected using an RNeasy Plus Universal Mini Kit (Qiagen, Valencia, CA, USA) after crushing the tissues with a Precellys homogenizer in QIAzol reagent. Quality was measured using a Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA, USA). One microgram of RNA was subjected to polyA-tailed cDNA library construction using a TruSeq RNA Sample Preparation Kit v2 (Illumina, San Diego, CA, USA). Sequencing was performed in a Riken on an Illumina HiSeq. 2500 system according to a standard paired-end 101 bp protocol.
RNA sequencing data analysis
We processed raw RNA sequencing data using a TopHat-Cufflinks pipeline. Before data processing, we checked the quality of data with FastQC. To quantify the expression levels of the genes and transcripts for all samples, 101-bp paired-end reads were aligned to the mouse reference genome mm9/GRCm37 by TopHat (v2.0.9). The mapping parameters follow the default setting in TopHat. After the read mapping, transcript and gene expression levels, which were represented by FPKM values, were calculated by Cufflinks (v2.2.1). Before we analysed the FPKM values in each of the tissues, 0.0001 was added to all the data to erase 0.
The criteria for p53-induced genes were as follows: (1) median FPKM value of WX/maximum FPKM value in median K, median KX or median W > 2, (2) t-test (WX vs. K, KX and W), P < 0.05, and (3) minimum FPKM value of WX > 1. The criteria for p53-repressed genes were (1) median FPKM value of WX/minimum FPKM value of median K, median KX or median W < 1/2, (2) t-test (WX vs. K, KX and W), P < 0.05, and (3) minimum FPKM value of W, K or KX > 1.
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) was used for pathway analysis46, 47.
cDNA microarray and its data processing
Gene expression analysis was performed using a SurePrint G3 Human GE 8 × 60 K microarray (Agilent, Santa Clara) according to the manufacturer’s protocol. Briefly, MCF10A p53 +/+, MCF10A p53 −/−, HCT p53 +/+ and HCT p53 −/−cells were treated with 2 µg/ml of adriamycin for 2 h and incubated at 37 °C until harvest. At 0 h, 12 h, 24 h and 48 h after ADR treatment, total RNA was isolated from the cells using standard protocols. Each RNA sample was labeled and hybridized to array slides. These data were shown in our previous studies48, 49. The MCF10A microarray data is available from the NCBI GEO database (GSE98727).
Quantitative real-time PCR
qPCR was conducted using SYBR Green I Master on a Light Cycler 480 (Roche, Basel, Switzerland). Primers sequences are indicated in Supplementary Table 2.
Gene reporter assay
DNA fragments, including the potential p53BS of each gene, were amplified and subcloned into pGL4.24 promoter vectors (Promega, Fitchburg, WI, USA). A reporter assay was performed using a Dual-Luciferase assay system (Promega) as previously described50. We used SaOS2 cells (p53 null) for a reporter assay.
Chromatin immunoprecipitation assay
ChIP assay was performed using an EZ-Magna ChIP G Chromatin Immunoprecipitation kit (Merck Millipore, Darmstadt, Germany) following the manufacturer’s protocol. Before immunoprecipitation, 1% of the supernatant was removed as “input”. Column-purified DNA was quantified by qPCR.
Western blotting
Western blotting was performed according to standard protocols. Anti-β-actin monoclonal antibody (clone AC15) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-p53 monoclonal antibody (clone DO-7) and anti-p21 monoclonal antibody (clone EA10) were purchased from Merck Millipore. Anti-HA monoclonal antibody (clone 3F10) was purchased from Roche. Human anti-CD137L monoclonal antibody (EPR1172Y) was purchased from GeneTex (Irvine, CA, USA). Mouse anti-Cd137l monoclonal antibody (sc-11819) was purchased from Santa Cruz (Dallas, TX, USA). α-Tubulin antibody (11H10) was purchased from Cell Signaling Technology (Danvers, MA, USA).
Immunohistochemistry
The calvarial bone was removed from 1-week-old p53 +/+ or p53 −/− mice. We used four groups of mice: K, W, KX and WX. Each group contained 3 mice. Goat anti-Cd137l antibody (sc-11819, Santa Cruz) and rat anti-Ki67 antibody (clone MIB-5, Agilent Technologies) were added to each slide after blocking of the endogenous peroxidase and proteins. Staining was evaluated by two independent investigators.
Colony formation assay
A colony formation assay was performed in six-well culture plates. Cells were transfected with pCAGGS/CD137L, pCAGGS/CDC42BPG, pCAGGS/FST or mock plasmid using FuGENE6 (Roche). Cells were cultured in the presence of Geneticin (1.0 mg/ml, 1.0 mg/ml or 1.2 mg/ml for U2OS, SaOS2 or LM8 cells, respectively) (Thermo Fisher Scientific) for 1-2 weeks. Colonies were stained with crystal violet (Sigma-Aldrich) and quantified using ImageJ software.
Cell proliferation assays for osteosarcoma cell lines and osteoblasts
Cell proliferation was assessed using a CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s protocol. Briefly, SaOS2 cells or osteoblasts were seeded at 4.0 × 103 or 7.5 × 103 cells/100 μl of medium/well, respectively, in 96-well culture plates that had been coated with CD137-Fc or mock-Fc. After culture for 48 h, the absorbance values were measured using a microplate reader (Biotek Instruments, Winooski, VT, USA).
Construction of stable cell lines and in vivo studies
LM8 cells were transfected using Lipofectamine 2000 reagent (Invitrogen) with the mock or Cd137l-pCAGGS expression vector, both of which had the neo gene to resist Geneticin. These clones were placed for 3 weeks in culture medium containing 1.0 mg/ml Geneticin to select for resistant cells. For each vector, 3 clones were selected, and we quantified Cd137l expression using western blotting and immunohistochemistry.
Six-week-old male C3H/HeJ mice were subcutaneously injected in the left and right flanks with a 0.1 ml phosphate buffered saline (PBS) containing 1 × 106 LM8 cells expressing Cd137l (Cd137l-1, Cd137l-2, Cd137l-3) or mock (mock 1, mock2, mock3). Each group contained 2 mice. The tumour volume was measured every 2 or 3 days and was calculated using the following formula: (long diameter) × (short diameter)2 × 0.52. The results are expressed as the mean ± standard deviation (SD). Statistical significance was determined by the Wilcoxon rank-sum test. Institute of Medical Science, University of Tokyo committee approved the experimental protocols and all experiments were performed in accordance with the Guidelines for Animal Experiments of the Institute of Medical Science, University of Tokyo.
Construction of human CD137-Fc, mouse Cd137-Fc and mouse Cd137l-Fc
The extracellular coding sequences of CD137 (human, amino acids 24–186; mouse, amino acids 24–187) or Cd137l (mouse, amino acids 104–304) and the Fc region of human IgG (amino acids 100–329) were amplified and subcloned into a pCAGGS expression plasmid according to previous reports51, 52. FreeStyle 293-F cells were transfected using FuGENE6 according to the manufacturer’s protocol. Recombinant proteins (human CD137-Fc, mouse Cd137-Fc and mouse Cd137l-Fc) were extracted from the culture media by affinity purification using protein A-Sepharose (Invitrogen), and it was purified using a dialysis cassette (0.1–0.5 ml, 10 K Molecular weight cut off, Thermo scientific). Immunocytochemistry was performed using a human IgG antibody (American Qualex International, San Clemente, CA, USA).
In vitro activity assay of mouse Cd137l-Fc for T cells
The effects of Cd137l-Fc on T cells were investigated by sandwich ELISA measurement of IL-2 production and a cell proliferation assay. T cells were isolated from splenocytes which were removed from 6-week-old male C3H/HeJ mice (CLEA Japan, Tokyo, Japan). CD4+ and CD8+ cells were sorted using FACS Aria (BD Biosciences, San Jose, CA, USA). CD4+ and CD8+ cells were seeded at 5.0 × 105 or 2.5 × 105 cells/500 μl medium/well, respectively, in 24-well culture plates precoated with 5.0 μg/ml anti-CD3 antibody (clone 145-11 C, Biolegend) in the presence of 20 μg/ml Cd137l-Fc or 20 μg/ml mock-Fc. Anti-CD28 antibody (clone 37.51, eBioscience, San Diego, CA, USA, 1.5 μg/ml) was used as a positive control. After 48 h, IL-2 production was determined by ELISA (mouse IL2 DuoSet ELISA, R and D systems, Minneapolis, MN, USA) for the above culture supernatants. All assays were performed in triplicate.
Effect of Cd137-Fc on tumour growth in vivo
Six-week-old male C3H/HeJ mice were subcutaneously injected in the left flank with 0.1 ml of PBS containing 1 × 106 LM8 cells. When tumours reached 0.5 cm in diameter, at approximately the 7th day after tumour implantation, each group of mice (n = 3) was treated with 0.2 ml (50 µg) of Cd137l-Fc fusion protein or control mock-Fc by intraperitoneal injection for 3 days. Statistical significance was determined by the Wilcoxon rank-sum test. Institute of Medical Science, University of Tokyo committee approved the experimental protocols and all experiments were performed in accordance with the Guidelines for Animal Experiments of the Institute of Medical Science, University of Tokyo.
Electronic supplementary material
Supplementary Figure 1-9, Supplementary Table 1 and 2
Acknowledgements
We thank Yuki Funauchi, Jinichi Mori, Asuka Terashima and Satomi Takahash for sample preparation and technical assistance. This work was supported by Ministry of Education, Culture, Sports, Science and Technology of Japan [25134707, 16H02676, and 16H01566 to K.M., 15K14377 to C.T., 15H05912 to S.M.] and Japan Agency for Medical Research and Development.
Author Contributions
Y.T., C.T., H.K., S.T. and K.M. initiated the project. Y.T. conducted most of the experiments and data analysis. M.H., T.M. and Y.V. assisted sample preparation and experiments. H.T. supported immunological study. Y.Z.Z., S.I., R.Y., S.M. and H.N. contributed to the analyses of RNA sequencing data. Y.T. and K.M. prepared the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-11208-x
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bieging KT, Mello SS, Attardi LD. Unraveling mechanisms of p53-mediated tumor suppression. Nat. Rev. Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brady CA, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maddocks OD, Vousden KH. Metabolic regulation by p53. J. Mol. Med. 2011;89:237–245. doi: 10.1007/s00109-011-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marión RM, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elyada E, et al. CKIα ablation highlights a critical role for p53 in invasiveness control. Nature. 2011;470:409–413. doi: 10.1038/nature09673. [DOI] [PubMed] [Google Scholar]
- 6.Muñoz-Fontela C, Mandinova A, Aaronson SA, Lee SW. Emerging roles of p53 and other tumor-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016;16:741–750. doi: 10.1038/nri.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat. Rev. Cancer. 2014;14:722–735. doi: 10.1038/nrc3838. [DOI] [PubMed] [Google Scholar]
- 8.Chen X, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;10:104–112. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perry JA, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA. 2014;111:5564–5573. doi: 10.1073/pnas.1419260111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bousquet M, et al. Whole exome sequencing in osteosarcoma reveals important heterogeneity of genetic alterations. Ann. Oncol. 2016;27:738–744. doi: 10.1093/annonc/mdw009. [DOI] [PubMed] [Google Scholar]
- 11.Kovac M, et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015;6 doi: 10.1038/ncomms9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorenz S, et al. Unscrambling the genomic chaos of osteosarcoma reveals extensive transcript fusion, recurrent rearrangements and frequent novel TP53 aberrations. Oncotarget. 2015;7:5273–5288. doi: 10.18632/oncotarget.6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germline transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348:747–749. doi: 10.1038/348747a0. [DOI] [PubMed] [Google Scholar]
- 14.Walkley CR, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes. Dev. 2008;22:1662–1676. doi: 10.1101/gad.1656808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikulenkov F, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 17.Collins M, et al. Benefits and adverse events in younger versus older patients receiving neoadjuvant chemotherapy for osteosarcoma: findings from a meta-analysis. J. Clin. Oncol. 2013;31:2303–2312. doi: 10.1200/JCO.2012.43.8598. [DOI] [PubMed] [Google Scholar]
- 18.Meyers PA, et al. Addition of pamidronate to chemotherapy for the treatment of osteosarcoma. Cancer. 2011;117:1736–1744. doi: 10.1002/cncr.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanikawa, C. et al. The Transcriptional Landscape of p53 Signalling Pathway. EBioMedicine 109–119, doi:10.1016/j.ebiom (2017). [DOI] [PMC free article] [PubMed]
- 20.Kenzelmann BD, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013;27:1016–1031. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer M, Grossmann P, Padi M, DeCaprio JA. Integration of TP53, DREAM, MMB-FOXM1 and RB E2F target geneanalyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016;44:6070–6086. doi: 10.1093/nar/gkw523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menendez D, et al. Diverse stresses dramatically alter genome wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013;41:7286–72301. doi: 10.1093/nar/gkt504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J. Cell Biol. 2006;172:115–125. doi: 10.1083/jcb.200507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lippert U, et al. CD137 ligand reverse signaling has multiple functions in human dendritic cells during an adaptive immune response. Eur. J. Immunol. 2008;38:1024–1032. doi: 10.1002/eji.200737800. [DOI] [PubMed] [Google Scholar]
- 25.Ju S, et al. A novel approach to induce human DCs from monocytes by triggering 4-1BBL reverse signaling. Int. Immunol. 2009;21:1135–1144. doi: 10.1093/intimm/dxp077. [DOI] [PubMed] [Google Scholar]
- 26.Wen T, Bukczynski J, Watts TH. 4-1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. J. Immunol. 2002;168:4897–4906. doi: 10.4049/jimmunol.168.10.4897. [DOI] [PubMed] [Google Scholar]
- 27.Nagata S. Fas-induced apoptosis, and diseases caused by its abnormality. Genes Cells. 1996;1:873–879. doi: 10.1046/j.1365-2443.1996.d01-214.x. [DOI] [PubMed] [Google Scholar]
- 28.Wu GS, Kim K, el-Deiry WS. KILLER/DR5, a novel DNA-damage inducible death receptor gene, links the p53-tumor suppressor to caspase activation and apoptotic death. Adv. Exp. Med. Bio. 2000;465:143–151. doi: 10.1007/0-306-46817-4_13. [DOI] [PubMed] [Google Scholar]
- 29.Tanikawa C, et al. XEDAR as a putative colorectal tumor suppressor that mediates p53-regulated anoikis pathway. Oncogene. 2009;28:3081–3092. doi: 10.1038/onc.2009.154. [DOI] [PubMed] [Google Scholar]
- 30.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008;8:467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 32.Wang B, Niu D, Lai L, Ren EC. p53 increases MHC class I expression by upregulating the endoplasmic reticulum aminopeptidase ERAP1. Nat. Commun. 2013;4 doi: 10.1038/ncomms3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cortez, M. A. et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 108, doi:10.1093/jnci/djv303 (2016). [DOI] [PMC free article] [PubMed]
- 34.Qian Y, et al. CD137 ligand-mediated reverse signaling inhibits proliferation and induces apoptosis in non-small cell lung cancer. Med. Oncol. 2015;32 doi: 10.1007/s12032-015-0499-9. [DOI] [PubMed] [Google Scholar]
- 35.Li G, et al. Triple expression of B7-1, B7-2 and 4-1BBL enhanced antitumor immune response against mouse H22 hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2011;137:695–703. doi: 10.1007/s00432-010-0905-9. [DOI] [PubMed] [Google Scholar]
- 36.Smith N, et al. Overlapping Expression of Runx1(Cbfa2) and Runx2(Cbfa1) Transcription Factors Supports Cooperative Induction of Skeletal Development. J Cell Physiol. 2005;203:133–143. doi: 10.1002/jcp.20210. [DOI] [PubMed] [Google Scholar]
- 37.Bauer O, et al. Loss of Osteoblast Runx3 Produces Severe Congenital Osteopenia. Mol Cell Biol. 2015;35:1097–1109. doi: 10.1128/MCB.01106-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozaki, T., Nakagawara,A., Nagase, H. RUNX Family Participates in the Regulation of p53-Dependent DNA Damage Response. Int J Genomics. 271347, doi:10.1155/2013/271347. (2013). [DOI] [PMC free article] [PubMed]
- 39.Melero I, et al. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer. 2015;15:457–472. doi: 10.1038/nrc3973. [DOI] [PubMed] [Google Scholar]
- 40.Yui Y, et al. Mesenchymal mode of migration participates in pulmonary metastasis of mouse osteosarcoma LM8. Clin. Exp. Metastasis. 2010;27:619–630. doi: 10.1007/s10585-010-9352-x. [DOI] [PubMed] [Google Scholar]
- 41.Ng Y, Tan I, Lim L, Leung T. Expression of the human myotonic dystrophy kinase-related Cdc42-binding kinase gamma is regulated by promoter DNA methylation and Sp1 binding. J. Biol. Chem. 2004;279:34156–34164. doi: 10.1074/jbc.M405252200. [DOI] [PubMed] [Google Scholar]
- 42.Thomas A, Giesler T, White E. p53 mediates Bcl-2 phosphorylation and apoptosis via activation of the Cdc42/JNK1 pathway. Oncogene. 2000;19:5259–5269. doi: 10.1038/sj.onc.1203895. [DOI] [PubMed] [Google Scholar]
- 43.Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activates p53 by targeting p85a and CDC42. Nat Struct Mol Biol. 2009;16:23–29. doi: 10.1038/nsmb.1533. [DOI] [PubMed] [Google Scholar]
- 44.Umulis D, O’Connor MB, Blair SS. The extracellular regulation of bone morphogenetic protein signaling. Development. 2009;136:3715–3728. doi: 10.1242/dev.031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo X, et al. Osteogenic BMPs promote tumor growth of human osteosarcomas that harbor differentiation defects. Lab. Investig. J. Tech. Methods Pathol. 2008;88:1264–1277. doi: 10.1038/labinvest.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 48.Yodsurang V et al. Identification of a novel p53 target, COL17A1, that inhibits breast cancer cell migration and invasion. Oncotarget. 2017 Jun 9, doi:10.18632/oncotarget.18433. [DOI] [PMC free article] [PubMed]
- 49.Mori J. et al. Cystatin C as a p53-inducible apoptotic mediator that regulates cathepsin L activity. Cancer Sci. 2016 Mar, 107(3):298–306. [DOI] [PMC free article] [PubMed]
- 50.Tanikawa C, Matsuda K, Fukuda S, Nakamura Y, Arakawa H. p53RDL1 regulates p53-dependent apoptosis. Nat. Cell Biol. 2003;5:216–223. doi: 10.1038/ncb943. [DOI] [PubMed] [Google Scholar]
- 51.Zhang N, et al. Targeted and untargeted CD137L fusion proteins for the immunotherapy of experimental solid tumors. Clin. Cancer Res. 2007;13:2758–2767. doi: 10.1158/1078-0432.CCR-06-2343. [DOI] [PubMed] [Google Scholar]
- 52.Madireddi S, et al. Galectin-9 controls the therapeutic activity of 4-1BB-targeting antibodies. J. Exp. Med. 2014;211:1433–1448. doi: 10.1084/jem.20132687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1-9, Supplementary Table 1 and 2