

Abstract
A detailed understanding of the atomic configuration of the compound semiconductor surface, especially after reconstruction, is very important for the device fabrication and performance. While there have been numerous experimental studies using the scanning probe techniques, further theoretical studies on surface reconstruction are necessary to promote the clear understanding of the origins and development of such subtle surface structures. In this work, therefore, a pressure-temperature surface reconstruction diagram was constructed for the model case of the InAs (001) surface considering both the vibrational entropy and configurational entropy based on the density functional theory. Notably, the equilibrium fraction of various reconstructions was determined as a function of the pressure and temperature, not as a function of the chemical potential, which largely facilitated the direct comparison with the experiments. By taking into account the entropy effects, the coexistence of the multiple reconstructions and the fractional change of each reconstruction by the thermodynamic condition were predicted and were in agreement with the previous experimental observations. This work provides the community with a useful framework for such type of theoretical studies.
Introduction
The metal oxide semiconductor field effect transistor (MOSFET) is a key component in semiconductor devices that have been scaled down according to the well-known Moore’s law for the past half a century1. As the simple linear scaling of the physical gate length (L) of MOSFET has approached its physical limit, there were recently two major modifications of the material and device structure: the adoption of the high-k gate dielectric/metal gate in 20072 and the three-dimensional channel structure, such as FinFET, in 20143. Even with these innovations, L will keep decreasing, and at the L of 5 nm node, the presently prevailing Si channel will have to be replaced with a higher mobility channel material, such as InGaAs and Ge, for the n- and p-type MOSFETs, respectively. Nonetheless, these channel materials will not be used in a bulk single crystalline configuration but with a local heterogeneous epitaxial form on a Si substrate. Molecular beam epitaxy (MBE) or atomic layer epitaxy (ALE) must be the appropriate process for growing such layers, and numerous researches have been performed on this subject4–6.
Apart from the Si (or even Ge) single crystal surface, the compound semiconductors have inherently complicated surface structures, especially in terms of surface reconstruction. Surface reconstruction occurs to minimize the surface energy (γ) of a specific crystallographic plane by altering some of its bonding configurations, which will have a crucial influence on the dielectric film growth on top, and thus, on the device performance7–10. The film growth itself by either MBE or ALE can be influenced by the surface reconstruction, or conversely, the surface reconstruction can be affected by the film growth conditions11–14. Therefore, understanding these reconstruction behaviors under different environments is very important not only for the device fabrication and characterization but also for the theoretical understanding of the surface properties.
In this work, InAs was used as the prototypical compound semiconductor for the n-type MOSFET. Among the low index surfaces of InAs, there has been much interest on the (001) surface because a variety of surface reconstructions on (001) provide diverse controllability and because it is desirable to be grown on the commercial Si (001) wafer4–6. Many experimental studies confirmed that the dominant reconstruction of InAs (001) changes from c(4 × 4) to (2 × 4) and finally to (4 × 2), or vice versa, depending on the thermodynamic conditions13–15.
The density functional theory (DFT) calculations also examined the reconstructions of the InAs (001) surface11, 16–19. One of the inherent limitations of the conventional DFT calculations, however, is that they cannot deal with all the possible reconstructions using the limited-size supercell. For example, the DFT calculations failed to confirm any stable (4 × 2) reconstruction11 despite the experimental observation of the InAs (001) (4 × 2) reconstruction in the In-rich condition13, 14, 20–22. One of the plausible reasons for this is that certain unknown (4 × 2) reconstructions may be stable. In fact, several new InAs (001) (4 × 2) reconstructions were suggested, such as β3′(4 × 2)20, ζ(4 × 2)17, 18, and ζa(4 × 2)19, 21, 22, whose feasibilities are still being debated on. Another limitation of the conventional DFT calculations is that they cannot predict the coexistence of multiple reconstructions, although the coexistence of some reconstructions on InAs (001) was experimentally identified, such as the mixture of c(4 × 4) with (2 × 4)15, that of β2(2 × 4) with α2(2 × 4)11–13, and that of (2 × 4) with (4 × 2)14. The shortcoming of not being able to predict the coexistence of multiple reconstructions was overcome by considering the configurational entropy23, 24. These earlier theoretical works, however, showed the equilibrium fraction of each reconstruction as a function of the temperature (T) at a fixed chemical potential (μ), or as a function of μ at a fixed T. These calculation results are not highly comparable with those of the experimental procedure because μ can hardly be controlled directly but must be dealt with by changing to the pressure (P) and T. Therefore, predicting the surface reconstruction as a function of P and T is a much more preferred approach.
In this study, the equilibrium fraction of various reconstructions for a given P and T condition was predicted by taking into account both the vibrational entropy and configurational entropy using density functional thermodynamic calculations. The InAs (001) surface was studied as a model case. All the calculations were performed according to the following conceptual flow: the surface energy at 0K was calculated as a function of the chemical potential of As; then, the surface energy at 0K was converted to the function of P and T under the assumption of the thermodynamic equilibrium between the surface atom and the surrounding reservoir; then, the surface energy at non-0K was obtained as a function of P and T by considering the vibrational entropy; finally, the coexistence of the reconstructions was estimated by taking the configurational entropy into account. Unlike the previous calculations on the oxides with oxygen gas as the surrounding reservoir25, 26, the surrounding phase of InAs can be either an As solid or gaseous mixture composed of As2 and As4 molecules. Therefore, the procedure for calculating the (P-T) surface reconstruction diagram in InAs is more complicated. The calculated fraction of reconstructions and the (P-T) transition boundary were found to match those in the experimental reports on InAs (001)13–15. The methodology applied in this study can also be used for other surfaces and other materials to extend the understanding of surface reconstruction.
Results and Discussions
The surfaces were represented by slabs consisting of eight or nine atomic layers with an at-least-10 Å vacuum layer. To eliminate the effect of the dipole moment along the vacuum, dipole correction27 was used. A preliminary calculation was carried out for a slab with both surfaces passivated by pseudo-hydrogen atoms (ZH = 1.25), where only the hydrogen atoms were allowed to move and the total energy () was obtained. From this calculation, the sum of the hydrogen-passivated surface energy (), including the atomic energy of the pseudo-hydrogen and the bonding energy of the pseudo-hydrogen with In atoms on both ends (α), was obtained using the following equation:
| 1 |
where , , , , and are the chemical potentials of In and As in InAs, the numbers of In and As atoms, and the cross-sectional area of the slab, respectively.
Through the thermodynamic equilibrium, and vary with the following constraints:
| 2 |
| 3 |
By combining the equations (2) and (3):
| 4 |
where, , , and are the chemical potentials in the bulk solid states. Note that, is constrained by the upper limit of and the lower limit of , respectively, and that the upper and lower limits are not constant, but decrease as T increases. The detailed calculations are explained in the on-line supplementary information (SI).
At 0K, the equation (4) becomes:
| 5 |
where, , , and are the energy of the bulk solid states at 0K. The μ for the bulk solid states at 0K was calculated using the tetragonal phase of In, the rhombohedral phase of As, and the zinc blende phase of InAs, respectively. Then, the equation (1) can be described as a function of through the substitution of equation (2):
| 6 |
For the calculation of the reconstruction, the passivating hydrogen atoms on the top surface were eliminated, and the atoms at the top five layers were relaxed, while the other bottom layers and passivating hydrogen atoms at the bottom surface were fixed until the remaining forces were less than 0.02 eV/Å. Then, at 0K, for each reconstruction was calculated as a function of using the following equation:
| 7 |
where, is the total energy of this slab at 0K. It should be pointed out that is the function only of with the slope of , and that the other values in equation (7) are all fixed for the given slab model.
Figure 1(a) shows the top view of the calculated reconstructions of InAs (001). For reference, the as-cleaved As-terminated surface structure was also included. The more the reconstructions included in the calculations, the more accurate the prediction of the fraction of each reconstruction. Therefore, all the previously suggested (4 × 2) reconstructions, such as β3′(4 × 2)20, ζ(4 × 2)17, 18, and ζa(4 × 2)19, 21, 22, were examined. In addition, for the c(4 × 4) reconstructions, not only the homodimers consisting of the six surface As atoms but also the heterodimer configurations were considered, as shown in Fig. 1(b). All the possible heterodimer configurations were constructed by replacing one to six As atoms at the top layer with In atoms, as summarized in Table 1. For example, ‘As5+In1’ corresponds to the topmost dimer layer composed of five As atoms and one In atom. The reconstructions in Fig. 1(b) show the structures with the lowest surface energy for a given surface composition.
Figure 1.

(a) Top view of the reconstructions of the InAs (001) surface compared with the cleaved surface (b) Top view of the c(4 × 4) heterodimers of various surface compositions. For example, ‘As5+In1’ means that the topmost layer is composed of five As atoms and one In atom. These configurations show the lowest surface energy for a given surface composition. All the surface areas correspond to their surface unit cell size except the cleaved surface whose surface unit cell size is indicated by a shaded square. The largest circles indicate the topmost layer.
Table 1.
Two-dimensional symmetry group (2SG), the number of symmetry operations (σ), and degeneracy factor (g) for the various reconstructions of InAs (001). For the c(4 × 4) heterodimers, the configuration showing the lowest surface energy for a given surface composition is in bold.
| Reconstruction | 2SG | σ | Reconstruction | 2SG | σ | ||
|---|---|---|---|---|---|---|---|
| α2(4 × 2) | p1 | 1 | 8 | ζa(4 × 2) | pmm | 4 | 2 |
| αα3(4 × 2) | pmm | 4 | 2 | α2(2 × 4) | p1 | 1 | 8 |
| β2(4 × 2) | pm | 2 | 4 | α3(2 × 4) | pmm | 4 | 2 |
| β3(4 × 2) | pm | 2 | 4 | β2(2 × 4) | pm | 2 | 4 |
| β3′(4 × 2) | p1 | 1 | 8 | β3(2 × 4) | pm | 2 | 4 |
| ζ(4 × 2) | pmm | 4 | 2 | c(4 × 4) | cmm | 8 | 2 |
| c(4 × 4) As6 | cmm | 8 | 2 | c(4 × 4) As3+In3(d) | p1 | 2 | 8 |
| c(4 × 4) As5+In1(a) | p1 | 2 | 8 | c(4 × 4) As3+In3(e) | p1 | 2 | 8 |
| c(4 × 4) As5+In1(b) | cm | 4 | 4 | c(4 × 4) As3+In3(f) | p1 | 2 | 8 |
| c(4 × 4) As4+In2(a) | p2 | 4 | 4 | c(4 × 4) As2+In4(a) | p2 | 4 | 4 |
| c(4 × 4) As4+In2(b) | p1 | 2 | 8 | c(4 × 4) As2+In4(b) | p1 | 2 | 8 |
| c(4 × 4) As4+In2(c) | cm | 4 | 4 | c(4 × 4) As2+In4(c) | cm | 4 | 4 |
| c(4 × 4) As4+In2(d) | p1 | 2 | 8 | c(4 × 4) As2+In4(d) | p1 | 2 | 8 |
| c(4 × 4) As4+In2(e) | cm | 4 | 4 | c(4 × 4) As2+In4(e) | cm | 4 | 4 |
| c(4 × 4) As4+In2(f) | cmm | 8 | 2 | c(4 × 4) As2+In4(f) | cmm | 8 | 2 |
| c(4 × 4) As3+In3(a) | cm | 4 | 4 | c(4 × 4) As1+In5(a) | p1 | 2 | 8 |
| c(4 × 4) As3+In3(b) | p1 | 2 | 8 | c(4 × 4) As1+In5(b) | cm | 4 | 4 |
| c(4 × 4) As3+In3(c) | cm | 4 | 4 | c(4 × 4) In6 | cmm | 8 | 2 |
Figure 2(a) shows the calculated of the InAs (001) reconstructions shown in Fig. 1(a), as a function of both (top-axis) and that of (bottom-axis), respectively. The top-axis representation is conventional as in the previous reports11, 16–19, while the bottom-axis representation was added for ease of conversion from to the corresponding (P-T) in the latter part of this study. Note that both ranges satisfy equation (5) and the slope in γ as a function of is proportional to the excess number of In atoms compared to the As atoms in the slab, as represented in equation (7). Therefore, the fact that the slope of the most stable reconstruction decreases as increases shows a consensus with the general conjecture that the reconstruction with more excess As atoms is more stable as increases. In the middle of the regime, the (2 × 4) reconstructions, such as α3(2 × 4), α2(2 × 4), and β2(2 × 4), are the most stable, and their positive, zero, and negative slopes correspond to their deficient, identical, and excess number of As atoms compared to the In atoms, respectively. In the In-rich condition (i.e., the low regime), ζa(4 × 2) with the steepest slope was identified as the reconstruction with the lowest surface energy. On the contrary, in the As-rich condition (i.e., the high regime), c(4 × 4) with a negative slope is the most stable.
Figure 2.

(a) Calculated surface energy at 0K for the InAs (001) reconstructions shown in Fig. 1(a) and (b) c(4 × 4) composed of heterodimers as well as homodimers (=As6). (c) Low values in (a) and (b).
Figure 2(b) shows the calculated of the c(4 × 4) As6, which is reproduced from Fig. 2(a) and indicated as As6, and the other c(4 × 4) reconstructions with As-In heterodimers as a function of both and , respectively. The several lines for a certain slope denote the multiple configurations for a certain surface composition summarized in Table 1 and the lowest and thick line for a given slope correspond to the configuration in Fig. 1(b). In general, as increases, the c(4 × 4) dimers with more As atoms become more stable, as can be expected, from ‘In6’ to ‘As3+In3’, then to ‘As4 + In2’. Only the structure of ‘In6’ shows a positive slope corresponding to its excess In atoms in the slab, while that of ‘As1+In5’ shows zero slope, indicating its overall stoichiometry. All the others’ negative slopes represent the excess As atoms in the slab. Note that in most of the regime, the c(4 × 4) heterodimers remarkably reduced values compared to the c(4 × 4) homodimer.
In Fig. 2(c), only the reconstructions with low values in Fig. 2(a) and (b) are shown. The critical values at which the values of the two stable reconstructions are identical were set as μI, μII, μIII, μIV, and μV respectively. For example, at μI, the of the ζa(4 × 2) is the same as that of the α3(2 × 4). Overall, the c(4 × 4) heterodimers with low values expand the stable regime of c(4 × 4) compared to the c(4 × 4) homodimers.
Although the calculation results shown in Fig. 2 are extensive, their usefulness compared with the results of the experiments is limited because was given as a function of , not as a function of P and T. To draw the (P-T) surface reconstruction diagram of the InAs (001) surface, should be calculated as a function of P and T, which is certainly achievable from the assumption of the thermodynamic equilibrium between the surface atom and the surrounding reservoir. The bulk states of As can be regarded as the surrounding reservoir and present either in a solid state with the rhombohedral crystalline structure or in two forms of gaseous states: As2 and As4. Liquid phase As was ruled out because the triple point of As appears at 35.8 atm and 1090K while the growth conditions of the thin film in the MBE chamber are a high vacuum and a temperature of around 750K11–13, 28. On the other hand, the pressure of In gas has been known to be ~1 order lower than that of As gas in the MBE chamber29 and hence can be ignored.
The relative stability of the surrounding reservoir was determined by calculating the Gibbs free energy of each phase. The Gibbs free energy of the gas phase was calculated considering the partial pressure of each gas under equilibrium. In the gas phase, the calculated equilibrium fraction of As2 increases as T increases or as Ptotal decreases, as shown in Fig. 3(a). Figure 3(b) shows the fractions of PAs2 and PAs4 as a function of T when Ptotal is fixed at 4 × 10−9 atm, which is comparable to the usual vacuum level for the MBE chamber13, 14, 28. In Fig. 3(c), both in the solid phase and that in the gas phase were drawn as a function of Ptotal and T. For the solid phase, decreases as T increases. On the contrary, it does not depend on P due to the assumption of the constant volume, which is a reasonable assumption because the volume change in solid is negligible compared to that in gas upon the variation of P. The phase boundary between the solid and gas phases in the (P-T) phase diagram of bulk As in Fig. 3(d) was obtained from the iso-chemical potential line between the solid and gas phases shown in Fig. 3(c).
Figure 3.

(a) Equilibrium fraction of PAs2 as a function of T and Ptotal(=PAs2 + PAs4). (b) Fractions of PAs2 and PAs4 as a function of T when the Ptotal is fixed at 4 × 10−9 atm. (c) Chemical potentials of arsenic solid (dark) and gas (light). (d) (P-T) phase diagram of pure As phase.
By combining the reconstruction with the lowest as a function of in Fig. 2(c) and as a function of P and T in Fig. 3(c), Fig. 2(c) could be converted to the (P-T) region, as shown in Fig. 4(a). The conversion was done under the assumption that (i) the bulk As forms the surrounding reservoir of the InAs surface, (ii) the surrounding As is in thermodynamic equilibrium with the surface As in InAs (i.e., ) and finally (iii) the at non-0K, is almost the same as the or critical values of the is not significantly different from those of the . By doing so, the dependence of on is converted to the dependence on , and finally to that on (P-T) owing to the dependence of on the (P-T) as shown in Fig. 3(c). Note that Fig. 4(a) is the direct conversion of Fig. 2(c).
Figure 4.
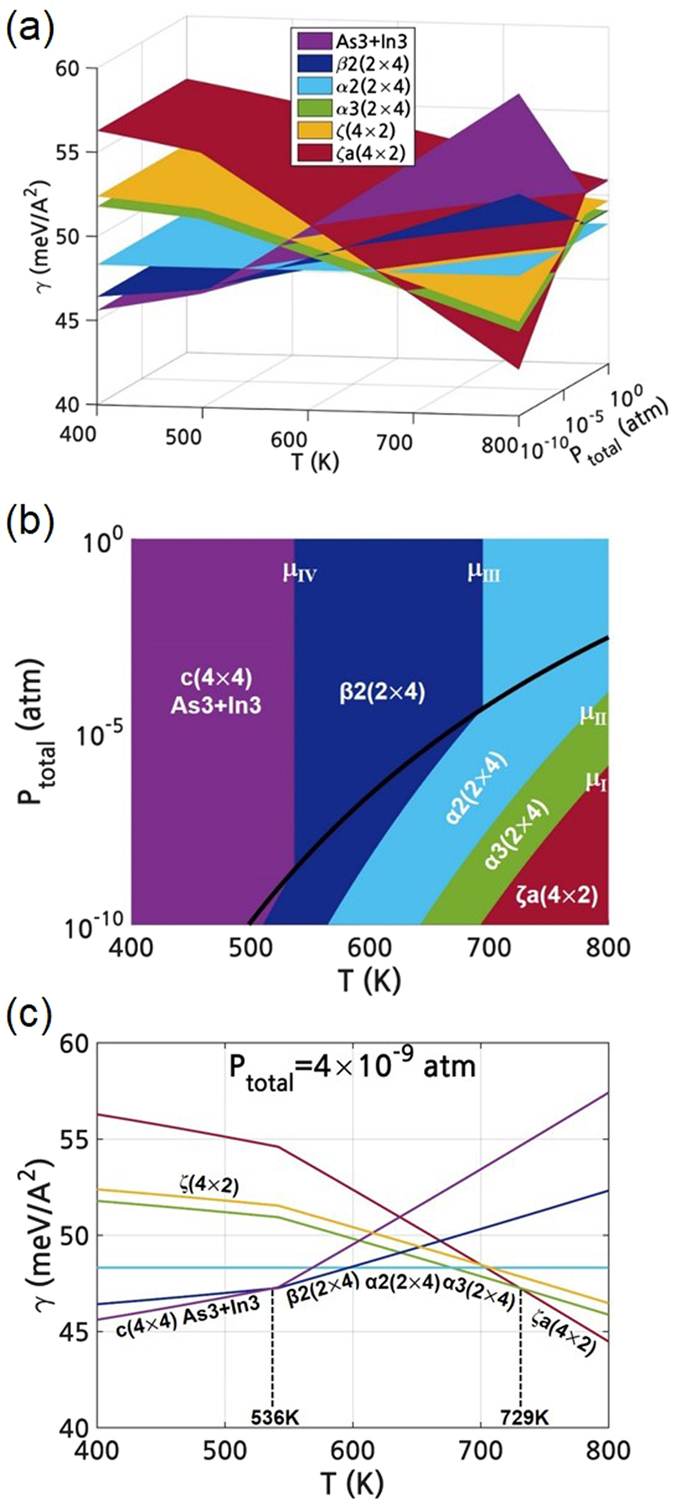
(a) Calculated surface energy of the InAs (001) reconstructions with low surface energy as a function of P and T. (b) (P-T) surface reconstruction diagram of the InAs (001) surface. (c) Calculated surface energy of the InAs (001) reconstructions with low surface energy as a function of T at fixed Ptotal of 4 × 10−9 atm. All figures were obtained without considering the surface phonons.
Figure 4(b) is the surface reconstruction phase diagram of InAs (001) and shows the reconstruction with the lowest surface energy at a given (P-T), which is the bottom view of Fig. 4(a). Transition lines between different reconstructions correspond to the critical values such as μI~ μIV in Fig. 2(c) where the reconstruction having the lowest surface energy changes. As the stable state of bulk As is either solid or gas depending on P and T, as shown in Fig. 3(d), the (P-T) region in Fig. 4(b) was also divided into two regions according to the surrounding phase of either solid or gas. The bold curve in Fig. 4(b) is the phase boundary line between the bulk solid and gas phases of As, which are identical in the phase boundary curve indicated in Fig. 3(d). Therefore, some lines such as μIII and μIV are partially not dependent on P where the solid As is the surrounding reservoir of the InAs surface due to the assumption of the constant volume of solid.
Figure 4(c) shows the transition of the surface reconstruction at Ptotal of 4 × 10−9 atm; c(4 × 4) ‘As3+In3’ → β2(2 × 4) → α2(2 × 4) → α3(2 × 4) → ζa(4 × 2) as T increases. These tendencies are consistent with the previous scanning tunneling microscopy (STM) observations showing the dependence of the InAs (001) reconstructions on the annealing temperature11–15. The transition temperature from c(4 × 4) to (2 × 4) and from (2 × 4) to (4 × 2) was calculated as 536K and 729K, respectively. These calculated transition temperatures are slightly lower but are in feasible agreement with the experimental values of 550K15 and 750K13, 14 in the UHV condition (~4 × 10−9 atm), respectively.
It is worth emphasizing that the low P and high T refer to the low (). Therefore, such a change in the dominant reconstruction as a function of P and T is in direct accordance with the changes in the lowest as a function of in Fig. 2(c) (from right to left). Moreover, the conversion of the variation of to the variation of P and T makes it practical to directly compare the calculation data with the experimental observations. The procedure explained so far was performed under the assumption that the is almost the same as the or the variations of the surface energy caused by the surface vibrational entropy for the various reconstructions are the same. In InAs, however, the differences in the bonding geometries and stoichiometry of the various surface reconstructions as well as the discrepancy in the mass between In and As are non-trivial, which might induce the different T dependence of various surface reconstructions. Therefore, the surface vibrational entropy was also taken into account. In fact, in some semiconductors, the vibrational entropy of the surface plays a critical role in the energetics and properties30, 31.
For the calculation of the surface vibration, the atoms of the top three layers were treated as coupled harmonic oscillators and these atoms were displaced from their equilibrium positions. Under the assumption that the vibration of the atoms below these three topmost layers are the same as that of the atoms in the bulk state, the for each reconstruction was calculated using the following equation:
| 8 |
where is the vibrational free energy of the three topmost layers, and are the numbers of In and As atoms on the three topmost layers, and are the vibrational free energy of the bulk InAs and bulk As, respectively. The detailed calculations of vibrational free energy and are explained in the computational methods and the on-line supplementary information (SI). The first term is the same as in the equation (7) and the other terms were added to consider the difference between the effects of surface phonons and those of the bulk phonons.
Figure 5(a) shows the as a function of P and T including the consideration of the surface vibration. The surface energy including the thermal effects, , was converted to the (P-T) region as in Fig. 4. Note that the ζa(4 × 2) reconstruction was excluded in Fig. 5 due to its instability. The detailed data on the imaginary frequencies in the surface phonon DOS of the ζa(4 × 2) as well as the surface phonon DOSs for various reconstructions are shown in the on-line supplementary information (SI). Compared to Fig. 4(a), the surface energies of the all the reconstructions were entirely lowered and the transition lines between different reconstructions in Fig. 5(a) are changed by the thermal effects. The surface reconstruction diagram in Fig. 5(b) shows that the stable (P-T) region of the c(4 × 4) ‘As3+In3’ was especially enlarged compared to Fig. 4(b). Therefore, at Ptotal of 4 × 10−9 atm, the transition temperature between the c(4 × 4) and (2 × 4) was raised from 536K in Fig. 4(c) to 555K in Fig. 5(c). This calculated transition temperature is closer to the experimental values of 550K15. However, the experimentally found transition from (2 × 4) to (4 × 2) was not shown due to the exclusion of unstable ζa(4 × 2). By considering the surface vibrational entropy, on the other hand, the surface energy of the ζ(4 × 2) become closer to that of the α3(2 × 4) as can be compared in Figs. 4(c) and 5(c).
Figure 5.
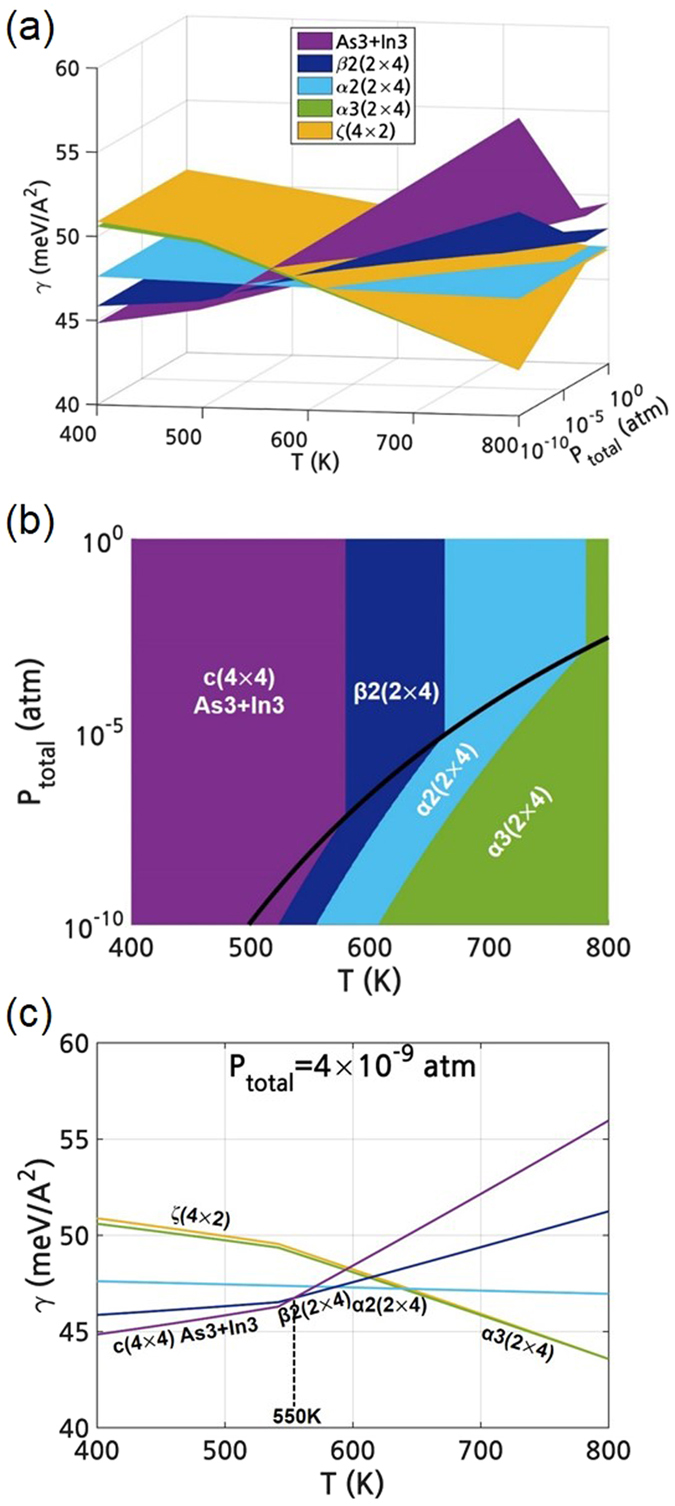
After considering the surface phonons, (a) calculated surface energy of the InAs (001) reconstructions with low surface energy as a function of P and T. (b) (P-T) surface reconstruction diagram of the InAs (001) surface. (c) Calculated surface energy of the InAs (001) reconstructions with low surface energy as a function of T at fixed Ptotal of 4 × 10−9 atm.
When some reconstructions show small energy difference, the reconstructions can coexist at the finite temperature due to the effects of the configurational entropy23, 24. The fraction of surface reconstruction j, , can be determined by using the partition function of each reconstruction j, ()24;
| 9 |
where j S (c(4 × 4), (2 × 4), (4 × 2), …; all calculated reconstructions)
| 10 |
| 11 |
where, is the surface energy of a certain reconstruction j, A is the surface area, is the Boltzmann constant, is the degeneracy factor related to the symmetry, and are the cell sizes of reconstruction j(n × m), and is the number of symmetry operations, as summarized in Table 1. It is noted that in this study, was calculated as a function of P and T in Fig. 5; hence, can also be calculated as a function of P and T. The calculated (P,T) enables the direct comparison with the experimental observations on the coexistence of multi-reconstructions as a function of P and T. It should be pointed out that this is a significant and more practical progress than the previous theoretical works23, 24 reporting as a function of T at a fixed μ, or as a function of μ at a fixed T.
Figure 6(a) shows the equilibrium fraction of the dominant reconstructions on InAs (001) as a function of P and T. The ranges of P and T are from 10−10 to 10−7 atm and from 500 to 800K, respectively. The fraction of the c(4 × 4) includes that of pure As-dimers and all the heterodimers. The fraction of the other surfaces was not represented due to their negligible fraction. Figure 6(a) clearly shows the coexistence of the multiple reconstructions and the variation of the dominant reconstruction depending on P and T. As P decreases and T increases, the dominant reconstruction changed: c(4 × 4) → β2(2 × 4) → α2(2 × 4) → α3(2 × 4) → ζ(4 × 2) and the fraction of the ζ(4 × 2) is comparable to that of the α3(2 × 4).
Figure 6.

After considering both the surface phonons and configurational entropy, (a) the fraction of the dominant InAs (001) reconstructions. Fraction of the dominant InAs (001) reconstructions at a fixed P of 4 × 10−9 atm (b) without including the c(4 × 4) heterodimers, and (c) including all the c(4 × 4) heterodimers.
Figure 6(b) and (c) show the fraction of reconstructions of InAs (001) as a function of T at a fixed P of 4 × 10−9 atm. For Fig. 6(b), the partition functions of only the reconstructions represented in Fig. 2(a) were included for the summation in equation (10), without considering the c(4 × 4) heterodimers. On the other hand, Fig. 6(c) was obtained by including all the reconstructions including all the c(4 × 4) heterodimers. These clearly demonstrate that the c(4 × 4) heterodimers boost the fraction of c(4 × 4) at the low T region and shows the transition of the dominant surface structures: c(4 × 4) → mixture of c(4 × 4) with (2 × 4) → (2 × 4) → mixture of (2 × 4) with (4 × 2) as T increases. The changes in the dominant reconstruction by T are in agreement with the previous experimental report that the transition from c(4 × 4) to (2 × 4)15 and from (2 × 4) to (4 × 2)13, 14 starts at around 550 and 750K at ~4 × 10−9 atm, respectively. When the configurational entropy are not considered, the direct transition from (2 × 4) to (4 × 2) does not appear in Fig. 5(c). However, the coexistence of the (2 × 4) and (4 × 2) is shown in Fig. 6(b) and (c) and the fractions of (2 × 4) and (4 × 2) become similar at 750K, which reflects the experimental transition T of 750K13, 14. In this study, by combining both the configurational entropy and vibrational entropy with the density functional thermodynamic calculations, the coexistence of the various reconstructions and the fractional change of each reconstruction in the coexistence region were successfully confirmed. Although the methodology of drawing the (P-T) surface reconstruction diagram was applied only to InAs (001) in this work, the same procedure can be readily applied to other surfaces and other materials to understand the surface reconstruction and to provide a guideline on the process window for the controlled thin film growth.
Conclusion
For the InAs (001) surface, a pressure-temperature (P-T) surface reconstruction diagram was constructed by density functional calculations. The fundamental strategy is calculating the surface energy of all the probable surface reconstruction configurations as a function of the chemical potential of As, whose value is further estimated as a function of P and T based on the assumption of the thermodynamic equilibrium between the surface atom and the surrounding reservoir. The calculation also takes into account the multiple phases of As as the surrounding reservoir. In addition, the equilibrium fraction of various reconstructions was determined as a function of P and T by considering both the vibrational entropy and configurational entropy with the density functional thermodynamic calculations. At a total pressure of 4 × 10−9 atm, which is comparable to the typical molecular beam epitaxy (MBE) condition for InAs film growth, the c(4 × 4) reconstruction was the most favorable for T < ~550K, and (4 × 2) became dominant as T increased to over 750K. The prediction of the transition boundary and the fractional change during the transition using this diagram provides a direct method of comparing the calculation results with the experimental conditions, and showed good agreement with the previous experimental reports. This methodology can be applied to other surfaces and other materials to understand the surface reconstruction and to provide a guideline on the process window for controlled thin film growth.
Computational Methods
Surface energy at 0K
All the calculations were performed using Vienna Ab-Initio Simulation Package (VASP)32–35. The project augmented wave method36, 37 within the local density approximation (LDA) parameterized by Ceperley et al.38, 39 was used with a cutoff energy of 300 eV. The 4d, 5 s, and 5p orbitals for In, and the 4 s and 4p orbitals for As, were treated as the valence electrons. Slab geometries consisting of eight or nine atomic layers with an at-least-10 Å vacuum layer were relaxed until the forces were less than 0.02 eV/Å.
Chemical potential of solid:
The μ of the bulk solids at non-0K was obtained from the Helmholtz free energy (F = E−TSvib) because the PV term in the Gibbs free energy (G = F + PV) is negligible due to the almost constant volume of solid, which is the general condition for the thin film growth of InAs. Harmonic oscillator approximation was used to calculate the vibration of crystal solid and the phonon density of states of solid was calculated using the finite displacement method implemented in PHONOPY40, 41.
Chemical potential of gas:
The μ of the gas phase was calculated by considering the partition functions of the translational, rotational, vibrational, and electronic motions, respectively, as the following equation:
| 12 |
| 13 |
| 14 |
where P is the partial pressure of i(gas), is the total partition function of i(gas) composed of N indistinguishable molecules, and , , and are the translational, rotational, vibrational, and electronic partition functions of one i molecule. By substituting the partition function, the translation contribution to the chemical potential is given by:
| 15 |
where, m is the molecular mass, and h is the Planck constant, respectively. The rotation contribution is given by:
| 16 |
where, , , and are the principal moments of inertia, σ is the symmetry number of the molecule, and ħ is . The vibration contribution is given by:
| 17 |
where, M is the number of the vibrational normal modes (3 N–5 for linear molecules, and 3 N–6 for non-linear molecules, respectively) and is the corresponding frequency. Note that term is the zero point energy of i(gas), . Finally, the electron contribution is given by:
| 18 |
where, is the total energy of the i(gas) and is the electronic spin degeneracy of the ground state, respectively.
For in the equation (14), only the translation contribution expressed by the equation (15) is dependent on both P and T. The other contributions expressed by the equations (16), (17), and (18) are the functions only of T. By inserting the standard pressure, (=1 atm), into the equation (15) to separate the dependency on P, the chemical potential of gas becomes:
| 19 |
The first three terms in the right side of the equation (19) are defined as , then
| 20 |
where, and are the partial pressure and the standard chemical potential of i(gas), respectively. is a function of T and was obtained from the DFT calculations on an isolated molecule in a 17.5 × 17.5 × 17.5 Å3 box. To the authors’ knowledge, of As2 and As4 has not been reported experimentally. The calculated of O2, however, using the same scheme, was well matched with the thermodynamic data42.
Then, the two partial pressure, and , can be calculated by solving:
| 21 |
| 22 |
| 23 |
at a given total pressure of Ptotal and T.
Surface phonon
The surface phonons were calculated to predict the effects of the vibrational entropy by the frozen phonon approach. Starting from the relaxed surface reconstructions, the atoms of the three topmost layers were displaced from their equilibrium positions and the electronic energy was calculated until the energy difference between consecutive steps is less than 10−6 eV within the reconstructed supercell (~8.5 × 17 Å2). The resulting Hellmann-Feynman forces were used to determine the force constants by the harmonic approximation. The dynamical matrix was obtained at the 21 × 21 sampled k-points on the surface two-dimensional Brillouin zone. From the calculated frequencies at each k-points, , the surface vibration contribution to the energy was calculated by following equation:
| 24 |
where, is the number of k-points in the two-dimensional Brillouin zone.
Electronic supplementary material
Acknowledgements
This work was supported by the Future Semiconductor Device Technology Development Program [10048490] funded by MOTIE (Ministry of Trade, Industry & Energy) and KSRC (Korea Semiconductor Research Consortium), and by the Institutional Research Program of KIST [2E27160]. The authors would also like to acknowledge the support from KISTI Supercomputing Center through the Strategic Support Program for Supercomputing Application Research [KSC-2015-C2-006, KSC-2016-C2-0045].
Author Contributions
I.W.Y. performed all the calculations, including the analyses. J.P. helped with all the calculations and discussed the technical contexts. G.H. provided the idea of considering the configurational entropy and helped identify the symmetry group for each reconstruction. C.S.H. supervised and helped prepare the manuscript from the viewpoint of the experimentalist. J.-H.C. arranged and supervised all the calculations and took charge of the manuscript preparation. All the authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-10881-2
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ferain I, Colinge CA, Colinge J-P. Multigate transistors as the future of classical metal-oxide-semiconductor field-effect transistors. Nature. 2011;479:310–316. doi: 10.1038/nature10676. [DOI] [PubMed] [Google Scholar]
- 2.Bohr MT, Chau RS, Ghani T, Mistry K. The high-k solution. IEEE spectrum. 2007;44:29–35. doi: 10.1109/MSPEC.2007.4337663. [DOI] [Google Scholar]
- 3.Natarajan, S. et al. In Electron Devices Meeting (IEDM), 2014 IEEE International. 3.7. 1–3.7. 3 (IEEE).
- 4.Geum, D.-M. et al. Ultra-high-throughput production of III-V/Si wafer for electronic and photonic applications. Scientific reports6 (2016). [DOI] [PMC free article] [PubMed]
- 5.Bolkhovityanov YB, Pchelyakov OP. GaAs epitaxy on Si substrates: modern status of research and engineering. Physics-Uspekhi. 2008;51:437–456. doi: 10.1070/PU2008v051n05ABEH006529. [DOI] [Google Scholar]
- 6.Hu W, et al. Epitaxy of In0.01Ga0.99As on Ge/offcut Si (001) virtual substrate. Thin Solid Films. 2012;520:5361–5366. doi: 10.1016/j.tsf.2012.04.027. [DOI] [Google Scholar]
- 7.Chang Y, et al. Effective reduction of interfacial traps in Al2O3/GaAs (001) gate stacks using surface engineering and thermal annealing. Applied physics letters. 2010;97 doi: 10.1063/1.3488813. [DOI] [Google Scholar]
- 8.Chang Y, et al. Inversion-channel GaAs (100) metal-oxide-semiconductor field-effect-transistors using molecular beam deposited Al2O3 as a gate dielectric on different reconstructed surfaces. Applied Physics Letters. 2013;102 doi: 10.1063/1.4793433. [DOI] [Google Scholar]
- 9.Lin H-Y, et al. Influences of surface reconstruction on the atomic-layer-deposited HfO2/Al2O3/n-InAs metal-oxide-semiconductor capacitors. Applied Physics Letters. 2011;98 doi: 10.1063/1.3571293. [DOI] [Google Scholar]
- 10.Lin Y-H, et al. Low interfacial trap density and high-temperature thermal stability in atomic layer deposited single crystal Y2O3/n-GaAs (001) Applied Physics Express. 2016;9 doi: 10.7567/APEX.9.081501. [DOI] [Google Scholar]
- 11.Ratsch C, Barvosa-Carter W, Grosse F, Owen J, Zinck J. Surface reconstructions for InAs (001) studied with density-functional theory and STM. Physical Review B. 2000;62 doi: 10.1103/PhysRevB.62.R7719. [DOI] [Google Scholar]
- 12.Barvosa-Carter W, et al. Atomic scale structure of InAs (001)-(2 × 4) steady-state surfaces determined by scanning tunneling microscopy and density functional theory. Surface science. 2002;499:L129–L134. doi: 10.1016/S0039-6028(01)01638-7. [DOI] [Google Scholar]
- 13.Guo X, et al. Critical surface phase of α2(2 × 4) reconstructed zig-zag chains on InAs (001) Thin Solid Films. 2014;562:326–330. doi: 10.1016/j.tsf.2014.02.116. [DOI] [Google Scholar]
- 14.Yamaguchi H, Horikoshi Y. Surface structure transitions on InAs and GaAs (001) surfaces. Physical Review B. 1995;51 doi: 10.1103/PhysRevB.51.9836. [DOI] [PubMed] [Google Scholar]
- 15.Bell G, Belk J, McConville C, Jones T. Species intermixing and phase transitions on the reconstructed (001) surfaces of GaAs and InAs. Physical Review B. 1999;59 doi: 10.1103/PhysRevB.59.2947. [DOI] [Google Scholar]
- 16.Moll N, Scheffler M, Pehlke E. Influence of surface stress on the equilibrium shape of strained quantum dots. Physical Review B. 1998;58 doi: 10.1103/PhysRevB.58.4566. [DOI] [Google Scholar]
- 17.Schmidt W. III-V compound semiconductor (001) surfaces. Applied Physics A: Materials Science & Processing. 2002;75:89–99. doi: 10.1007/s003390101058. [DOI] [Google Scholar]
- 18.Miwa R, Miotto R, Ferraz A. In-rich (4 × 2) and (2 × 4) reconstructions of the InAs (001) surface. Surface science. 2003;542:101–111. doi: 10.1016/S0039-6028(03)00955-5. [DOI] [Google Scholar]
- 19.Lång J, et al. Ab initio and scanning tunneling microscopy study of an indium-terminated GaAs (100) surface: An indium-induced surface reconstruction change in the c(8 × 2) structure. Physical Review B. 2010;81 doi: 10.1103/PhysRevB.81.245305. [DOI] [Google Scholar]
- 20.Feldwinn DL, et al. Anomalous hybridization in the In-rich InAs (001) reconstruction. Surface Science. 2009;603:3321–3328. doi: 10.1016/j.susc.2009.09.026. [DOI] [Google Scholar]
- 21.Goryl G, Toton D, Goryl M, Tomaszewska N, Kolodziej J. Structure of the In-rich InAs (001) surface. Surface Science. 2011;605:2073–2081. doi: 10.1016/j.susc.2011.08.008. [DOI] [Google Scholar]
- 22.Kumpf C, et al. Structure of metal-rich (001) surfaces of III-V compound semiconductors. Physical Review B. 2001;64 doi: 10.1103/PhysRevB.64.075307. [DOI] [Google Scholar]
- 23.Penev E, Kratzer P, Scheffler M. Atomic Structure of the GaAs (001) − c(4 × 4) Surface: First-Principles Evidence For Diversity of Heterodimer Motifs. Physical review letters. 2004;93 doi: 10.1103/PhysRevLett.93.146102. [DOI] [PubMed] [Google Scholar]
- 24.Romanyuk O, Grosse F, Proessdorf A, Braun W, Riechert H. Stabilization of semiconductor surface reconstructions by configurational entropy. Physical Review B. 2010;82 doi: 10.1103/PhysRevB.82.125315. [DOI] [Google Scholar]
- 25.Reuter K, Scheffler M. Composition, structure, and stability of RuO2 (110) as a function of oxygen pressure. Physical Review B. 2001;65 doi: 10.1103/PhysRevB.65.035406. [DOI] [Google Scholar]
- 26.Reuter K, Scheffler M. Composition and structure of the RuO2 (110) surface in an O2 and CO environment: Implications for the catalytic formation of CO2. Physical Review B. 2003;68 doi: 10.1103/PhysRevB.68.045407. [DOI] [Google Scholar]
- 27.Neugebauer J, Scheffler M. Adsorbate-substrate and adsorbate-adsorbate interactions of Na and K adlayers on Al (111) Physical Review B. 1992;46 doi: 10.1103/PhysRevB.46.16067. [DOI] [PubMed] [Google Scholar]
- 28.Lim JY, Song JD, Yang HS. Effect of thin intermediate-layer of InAs quantum dots on the physical properties of InSb films grown on (001) GaAs. Thin Solid Films. 2012;520:6589–6594. doi: 10.1016/j.tsf.2012.06.077. [DOI] [Google Scholar]
- 29.Bastiman F, Cullis A, Hopkinson M. InAs/GaAs (001) wetting layer formation observed in situ by concurrent MBE and STM. Surface Science. 2009;603:3439–3444. doi: 10.1016/j.susc.2009.10.009. [DOI] [Google Scholar]
- 30.Valtiner M, Todorova M, Grundmeier G, Neugebauer J. Temperature stabilized surface reconstructions at polar ZnO (0001) Physical review letters. 2009;103 doi: 10.1103/PhysRevLett.103.065502. [DOI] [PubMed] [Google Scholar]
- 31.Wippermann S, Schmidt WG. Entropy explains metal-insulator transition of the Si (111)-In nanowire array. Physical review letters. 2010;105 doi: 10.1103/PhysRevLett.105.126102. [DOI] [PubMed] [Google Scholar]
- 32.Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science. 1996;6:15–50. doi: 10.1016/0927-0256(96)00008-0. [DOI] [Google Scholar]
- 33.Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical review B. 1996;54 doi: 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- 34.Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Physical Review B. 1993;47 doi: 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- 35.Kresse G, Hafner J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Physical Review B. 1994;49 doi: 10.1103/PhysRevB.49.14251. [DOI] [PubMed] [Google Scholar]
- 36.Blöchl PE. Projector augmented-wave method. Physical review B. 1994;50 doi: 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- 37.Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Physical Review B. 1999;59 doi: 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- 38.Ceperley DM, Alder B. Ground state of the electron gas by a stochastic method. Physical Review Letters. 1980;45 doi: 10.1103/PhysRevLett.45.566. [DOI] [Google Scholar]
- 39.Perdew JP, Zunger A. Self-interaction correction to density-functional approximations for many-electron systems. Physical Review B. 1981;23 doi: 10.1103/PhysRevB.23.5048. [DOI] [Google Scholar]
- 40.Togo A, Oba F, Tanaka I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Physical Review B. 2008;78 doi: 10.1103/PhysRevB.78.134106. [DOI] [Google Scholar]
- 41.Togo A, Chaput L, Tanaka I, Hug G. First-principles phonon calculations of thermal expansion in Ti3SiC2, Ti3AlC2, and Ti3GeC2. Physical Review B. 2010;81 doi: 10.1103/PhysRevB.81.174301. [DOI] [Google Scholar]
- 42.Chase, M. W. NIST-JANAF Thermochemical Tables 4th edn (ed. Chase, M.) Journal of Physical and Chemical Reference Data, Monograph No. 9 (American Chemical Society and American Institute of Physics, 1998).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.