

Abstract
There is an urgent need for the improvement of drought-tolerant bread and durum wheat. The huge and complex genome of bread wheat (BBAADD genome) stands as a vital obstruction for understanding the molecular mechanism underlying drought tolerance. However, tetraploid wheat (Triticum turgidum ssp., BBAA genome) is an ancestor of modern bread wheat and offers an important model for studying the drought response due to its less complex genome. Additionally, several wild relatives of tetraploid wheat have already shown a significant drought tolerance. We sequenced root transcriptome of three tetraploid wheat varieties with varying stress tolerance profiles, and built differential expression library of their transcripts under control and drought conditions. More than 5,000 differentially expressed transcripts were identified from each genotype. Functional characterization of transcripts specific to drought-tolerant genotype, revealed their association with osmolytes production and secondary metabolite pathways. Comparative analysis of differentially expressed genes and their non-coding RNA partners, long noncoding RNAs and microRNAs, provided valuable insight to gene expression regulation in response to drought stress. LncRNAs as well as coding transcripts share similar structural features in different tetraploid species; yet, lncRNAs slightly differ from coding transcripts. Several miRNA-lncRNA target pairs were detected as differentially expressed in drought stress. Overall, this study suggested an important pool of transcripts where their manipulations confer a better performance of wheat varieties under drought stress.
Introduction
Wheat (Triticum ssp.) is one of the major sources of continuously increasing food demand, ranking third in crop production worldwide1. Domestication and cultivation efforts of agricultural practices resulted in an increased yield2 with the approximate global production of 700 million tons per year distributed over 200 million hectares1. Despite this spread distribution, obtained rate of yield is not sufficient to meet world food demand since production rate is significantly limited by biotic and abiotic stress factors3. Drought is one of the major abiotic stress factors worldwide, causing decrease in grain quality and yield loss through all cereals, including wheat4. Recent studies have suggested a substantial increase in drought caused by climate change and global warming5. In order to maintain sufficient amount of yield with an improved nutritional quality, development of new wheat varieties with an increased drought tolerance is urgently needed toward future challenges.
Wild plants evolved sophisticated stress tolerance and adaptation mechanisms to drought where the domestication of modern wheat varieties has led to the loss of these valuable genes in the process of domestication6. Introgression of the valuable elements from wild relatives has been an attractive approach for agronomical improvement of modern wheat varieties for decades7, due to their rich gene pool for the resistance to many different stress factors. Tetraploid emmer wheat (T. turdigum ssp. dicoccoides, 2n = 28, AABB) is the wild progenitor of the allohexaploid bread wheat (T. aestivum, 6n = 42, AABBDD) and the domesticated tetraploid durum wheat (T. turgidum ssp. durum, 4n = 24, AABB)2. Recent studies on tetraploid wheat varieties revealed contrasting drought tolerance in tetraploid wild emmer wheat varieties and domesticated tetraploid durum wheat8, 9. Ergen and colleagues surveyed drought response of several genotypes of wild and domesticated tetraploid wheat varieties; they were able to show that wild emmer wheat, genotype TR39477, exhibits the highest drought tolerance while TTD-22 genotype has the lowest tolerance under drought stress. On the other hand, durum wheat variety Kiziltan showed a moderate tolerance in response to slow drought imposition9; however, complete mechanism of these drought responses remains elusive. A better understanding of the genomic background and the molecular mechanisms of drought responses in wild progenitors of wheat might reveal such favorable regulatory elements lost during domestication and cultivation processes.
In recent years, technological advances have made road into the reduction in the cost of sequencing experiments and availability of several genomic and transcriptomic data from bread wheat and its relatives/progenitors10, 11. Particularly, transcriptomic studies shed light into the differential expression and regulation of several transcripts under biotic and abiotic stress conditions, which further provide insights about the molecular mechanism associated with stress tolerance4, 12, 13. Total transcriptome sequencing and annotation possess a potential for detection of protein coding transcripts, differentially regulated under stress conditions, together with their non-coding interacting partners which is associated with a large portion of transcriptomes14. The content and the amount of the non-coding RNAs (ncRNAs) in the genome show an increased correspondence with the genome complexity which further supports their regulatory roles15, 16. Over the last decades, extensive studies in both animals and plants have shed light into the functions and mechanisms of ncRNAs such as microRNAs (miRNAs) and small interfering RNAs (siRNAs) in the transcriptional and post-transcriptional regulation of gene expression17, 18. While the miRNAs and siRNAs are referred as small RNAs (sRNAs) based on their small length ranging between 18 to 24 nucleotides, another type of ncRNAs longer than 200 nucleotides has been recently defined as long non-coding RNAs (lncRNAs)19, 20. LncRNAs resemble messenger RNAs (mRNAs) in their structure and biogenesis process, i.e., they are mainly transcribed by RNA Pol II and poly-adenylated, as though mRNAs20. Additionally, they might possess multiple exons and are subjected to alternative splicing. The major factor distinguishing lncRNAs from mRNAs is the lack of discernable coding potential of lncRNAs21. Besides, lncRNAs are composed of ~3 exons on average as opposed to ~11 exons in mRNAs and exhibit a more tissue-specific expression pattern compared to mRNAs where their expression is also relatively less than mRNAs in a given tissue21.
Emerging evidence has suggested that lncRNAs have regulatory roles in the major biological processes such as development, vernalization, nodulation and environmental stress adaptation both in direct and indirect manner20. As an example, two lncRNAs, the long antisense intragenic RNA (COOLAIR) and the intronic noncoding RNA (COLDAIR), have been detected as mediating the flowering process in Arabidopsis through silencing and epigenetic repression of Flowering Locus C (FLC)22. Additionally, several studies evinced the functions of lncRNAs in the biogenesis and targeting process of small-noncoding RNAs by possessing miRNA-siRNA precursor potential and sRNA target mimicry19. RNA-dependent DNA Methylation (RdDM) in plants, for example, utilizes lncRNAs acting as precursors of siRNAs which later target lncRNAs acting as scaffold RNAs recruiting siRNA-AGO4 complex together with RDM1 (RNA-directed DNA Methylation 1) to a target genomic loci for DNA methylation-mediated silencing23. In another example, lncRNA IPS1 has been shown to inhibit miR399-mediated cleavage of PHO2 as a competitor for PHO2 mRNA24. LncRNAs have also been identified as differentially expressed under several stress conditions and their regulation on both mRNA and sRNA pool detected as critical for stress tolerance and maintenance of vitality25; however, not that much effort has been done in drought responsive lncRNAs and their association with coding and other non-coding RNA species, particularly in cereals. Here, we present a detailed analysis of drought responsive mRNAs and lncRNAs along with their particular interaction with each other in three different tetraploid wheat varieties. Results revealed the presence of more than 200 putative stress responsive lncRNAs per cultivar which provided insights about drought tolerance mechanism in ancestor of modern wheat. Additionally, this study presents a brief method for precise identification and detailed characterization of lncRNAs for plants lacking both an annotated genome and a reference genome.
Results
De novo assembly of transcriptomes
In our previous study, two wild emmer wheat genotypes, T. turgidum ssp. dicoccoides TR39477 and TTD-22 showed marked differences in tolerance to drought stress when compared to the modern durum wheat, T. turgidum ssp. durum var. Kiziltan. Upon slow drought treatment, Kiziltan exhibited a moderate reaction whereas TR39477 and TTD-22 exhibited the most and the least tolerance, respectively9. High-throughput sequencing of root samples from control and drought-stressed Kiziltan, TR39477 and TTD-22 led to more than 27,000,000 raw sequence reads8. In order to remove adaptor sequences and perform the quality trimming, Trimmomatic26 analysis evaluated and more than 95% of raw reads were cleaned after initial processing with Trimmomatic (Table 1). The clean reads were assembled using Trinity software27 yielding a total of 243,670, 211,709 and 203,230 transcripts for Kiziltan, TR39477 and TTD-22, respectively. The average contig lengths detected as altering between 666 and 779 nucleotides long where the total transcriptome size ranges between 99.7 to 146.6 Mb (Table 1).
Table 1.
Statistics on quality trimming and assembly construction for the transcriptome assemblies.
| Samples | Kiziltan Control | Kiziltan Drought | TR39477 Control | TR39477 Drought | TTD-22 Control | TTD-22 Drought |
|---|---|---|---|---|---|---|
| The quality trimming of samples | ||||||
| Before trimming | 35,463,556 | 36,944,980 | 35,212,424 | 30,670,932 | 27,505,294 | 32,690,630 |
| After trimming | 33,772,655 | 35,171,816 | 33,698,813 | 29,223,580 | 26,200,743 | 31,249,427 |
| Assembly statistics for the samples | ||||||
| Number of transcripts | 204128 | 18817 | 169762 | 159940 | 168314 | 155170 |
| Median contig length (b) | 482 | 516 | 478 | 483 | 468 | 494 |
| Average contig | 666.67 | 779.58 | 731.65 | 741.67 | 726.12 | 752.61 |
| Total length (Mb) | 99.78 | 146.69 | 129.32 | 125.24 | 122.22 | 116.78 |
| GC% | 49.72 | 49.45 | 49.98 | 49.92 | 50.63 | 50.25 |
| N50 | 1024 | 1082 | 1001 | 1108 | 1004 | 1056 |
Functional annotation of transcriptomes
Gene content of each transcriptome assembly was evaluated through four layers of analyses as described in Fig. 1. All transcripts were initially screened against known small non-coding RNA sequences and mitochondria/chloroplast originated sequences of Triticum families with Blast tool kit28. Overall, less than %1 of transcripts with significant hits in these screenings were considered as contaminants and excluded from the Kiziltan, TR39477 and TTD-22 transcriptome assemblies. Subsequent to contaminant analysis, open reading frame (ORF) content of the remaining transcripts was analyzed and transcripts possess ORFs longer than 80 aa were further evaluated for their coding potential through ab initio techniques; CPC29, CNCI30 and AUGUSTUS31. Totally, 60% of Kiziltan, 62% of TR39477 and 64% of TTD-22 transcriptome showed coding potential evidence, respectively. These transcripts were further evaluated for detection of their homology to known protein sequences and/or presence of functional protein domains. Ultimately, a total of 84,288, 75,996 and 78,456 putative protein-coding transcripts were identified from Kiziltan, TR39477 and TTD-22, which corresponded to 35%, 37% and 39% of the assemblies, respectively.
Figure 1.

The pipeline for the identification and annotation of both coding transcripts and lncRNAs. Before the identification of both coding transcripts and lncRNAs, the organellar transcripts and small RNAs were eliminated from the transcripts which were all together named as ‘contaminants’. The open reading frame (ORF) of transcripts were detected with two different softwares, Transdecoder and EMBOSS:getorf, since the presence of functional ORF is the most important criteria for identification of both coding transcripts and lncRNAs. Coding potential of transcripts was calculated with two different programs, CNCI and CPC, in addition to the online gene prediction tool, AUGUSTUS. Additionally, the homology between transcripts and known proteins sequences were inspected, together with the functional protein domains.
Although the assemblies were constructed with the utilization of data from pooled samples of three different biological replicate for each variety, assembled contigs might show none-to-very low expression level; thus, identification of actively-expressed transcripts is necessary for further characterization of transcriptomic data. To determine expression levels of transcripts, transcript abundances were quantified in terms of Fragment Per Kilobase Million mapped reads (FPKM) using RSEM package under Trinity software. Expression activity of protein-coding transcripts was evaluated based on the normalized FPKM. Percent of transcripts that failed to satisfy FPKM cutoffs in both control and drought stressed samples were plotted over a range of FPKM thresholds (Supplementary Fig. 1). Overall, 1% change observed between five cut-offs from 0.1 to 0.5 FPKM; however, a sudden 1% change occurred thereafter. The point, 0.5 FPKM, was chosen arbitrarily as this was the point where the slope of the curve changes, indicating the significance of this point, thereby suggesting it as a potential threshold. With this threshold, 95% of each transcriptome were found to be actively-expressed transcripts, indicating the quality of the transcriptome assemblies and good coverage of the sequencing. In total, 81,168 (96%), 73,465 (97%) and 75,861 (97%) actively-expressed protein-coding transcripts (called coding transcript from this point) were identified in Kiziltan, TR39477 and TTD-22, respectively (Supplementary File 1).
The actively-expressed coding transcripts were inspected for their expression patterns in control and drought treated samples. All three T. turgidum varieties represented a high portion of common transcripts between controlled and stressed samples where more than 70% of transcripts were detected as common (60,520; 57,012 and 56,164 transcripts for Kiziltan, TR39477 and TTD-22, respectively). Sample specific transcripts were most abundant in control samples, where 14,595, 10,865 and 14,204 transcripts expressed from solely controlled Kiziltan, TR39477 and TTD-22 varieties, respectively, as opposed to that of 7% of transcripts (6,053; 5,588 and 5,493 transcripts for Kiziltan, TR39477 and TTD-22, respectively) expressed only drought-stressed samples. Blast alignments of drought-specific transcripts revealed that 1,034 homologous transcripts (>80% of query identity and coverage) expressed in both tolerant and susceptible varieties. Drought-tolerant TR39477 revealed 36 different transcripts which does not have any similarity to transcripts from drought-susceptible TTD-22 while 4 of the TTD-22 transcripts were detected as TTD-specific (Supplementary Table 1). These transcripts were remarked as effective on the different drought stress tolerance and adaptation mechanism of these wheat varieties.
Functional annotation of all coding transcripts was conducted by Gene Ontology (GO) term assignment followed with KEGG pathway and COG analysis via Blast2GO software32. In total, 860,446 GO terms were assigned to 157,013 (68% of all coding transcripts) transcripts. Assigned GO annotations were clustered in three main categories; Molecular Function (MF), Cellular Component (CC) and Biological Process (BP), based on the blast hits from the NCBI non-redundant (nr) Viridiplantae protein database with an e-value cutoff of 1E-5. Across all GO annotations, ‘ATP binding’, ‘membrane’ and ‘protein phosphorylation’ were predominant in the MF, CC and BP categories, respectively. Since these included coding transcripts from both treated and untreated samples in the three varieties, the categories most represented by transcripts could be considered as representatives of housekeeping genes. These sequences were further inspected in terms of the blast hit distribution through different plants which revealed the homology pattern of coding transcripts of T. turgidum varieties (Supplementary Fig. 2). Blast hit distributions showed that T. turgidum coding sequences possess the highest homology with Aegilops tauschii sequences where 26% of transcript revealed as identical to proteins from this species, followed by Triticum Urartu (24%) and Hordeum vulgare ssp. vulgare (24%) (Supplementary Fig. 2A). Additionally, e-value distribution of blast top hits indicated the general quality of the assembled coding transcripts where more than 50% of the hits have e-values smaller than 1e-110 (Supplementary Fig. 2B). Following Blast2GO annotations, mapping against KEGG database were performed to retain relative biological pathways of the coding transcripts33. In total, 50,250 transcripts were assigned to a total of 133 pathways in KEGG database. KEGG pathways the most represented by transcripts were purine metabolism (7,799 transcripts, 15.5%), thiamine metabolism (6,302 transcripts, 12.5%) and biosynthesis of antibiotics (3,147 transcripts, 6.3%) across all transcripts. Additionally, Cluster of Orthologous Groups (COG) screenings were performed using EggNog database, under Blast2GO software34 and coding transcripts sharing similar functions were classified into 23 functional groups. The largest group represented by transcripts had functions defined as ‘unknown’ (5,935 transcripts, 23.5%) followed by ‘posttranslational modification, protein turnover and chaperones’ (2,545 transcripts, 10.1%), ‘signal transduction mechanisms’ (2,397 transcripts, 9.5%), ‘intracellular trafficking, secretion and vesicular transport’ (1,600 transcripts, 6.3%) and ‘translation, ribosomal structure and biogenesis’ (1,506 transcripts, 6%).
The functions of differentially expressed transcripts were further analyzed after the annotation of all transcripts. In all the three plants, ‘oxidation-reduction’ and ‘protein phosphorylation’ were the most represented BP terms in transcripts exhibited differential expression in response to drought stress (Fig. 2). Interestingly, drought-susceptible TTD-22 revealed an increased number of upregulated genes which were categorized in ‘response to stress’ group regarding to BP assessment of Blast2GO (from 17 to 37 transcripts) while this category represented less number of associate transcripts in Kiziltan (from 24 to 13 transcripts) and TR39477 (from 37 to 4 transcripts) under drought stress. The orthologous groups of drought specific transcripts were also analyzed to determine their functional importance. The most representative COG id by all the drought specific transcripts, KOG0987, was associated with a DNA helicase which is functional in cell cycle control and cell division. Another important one, COG0507, connected with ‘exodeoxyribonuclease v alpha’ protein involving in replication and recombination. In order to further understand the drought tolerance of TR39477, the unique transcripts which are specifically expressed under drought stress, 36 transcripts which do not show any resemblance to TTD-22 transcripts, were analyzed in regards to their associated KEGG pathway. Mostly, pathways associated with secondary metabolite synthesis were detected as enriched by these unique transcripts such as ‘Glutathione, Sphingolipid and Thiamine metabolism’. Also, ‘Glycosaminoglycan and glycan degradation pathways’ were enhanced by unique transcripts of TR39477. Accordingly, identification and annotation of all such transcripts provided insides regarding drought-responsive metabolomic changes in durum wheat together with their transcript partners.
Figure 2.
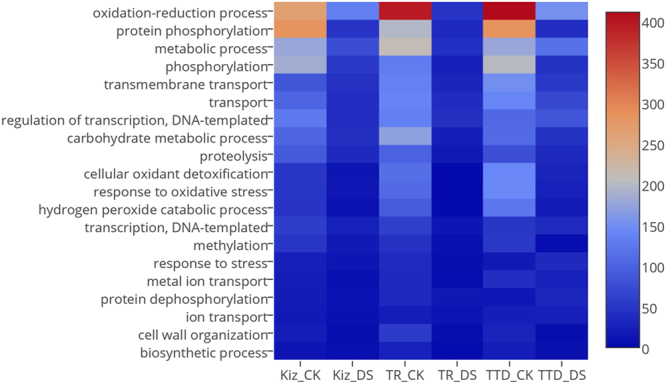
Heatmap for top 20 biological processes represented by stress-responsive coding transcripts in each sample. Several biological processed were detected and drought treated samples. (Graph legend: Kiz_CK: mRNAs from variety Kiziltan expressed under control conditions, Kiz_DS: mRNAs from variety Kiziltan expressed under drought stress, TR_CK: mRNAs from variety TR39477 expressed under control conditions, TR_DS: mRNAs from variety TR39477 expressed under drought stress, TTD_CK: mRNAs from variety TTD-22 expressed under control conditions, TTD_DS: mRNAs from variety TTD-22 expressed under drought stress.
Putative lncRNAs and their expression pattern under drought stress
Identification of long-noncoding RNAs was performed by following the pipeline illustrated in Fig. 1. Totally; 26% (63,773 transcripts), 29% (61,823 transcripts) and 22% (43,932 transcripts) of the transcriptome assemblies from Kiziltan, TR39477 and TTD-22 varieties, respectively, were associated with lncRNAs. Subsequent to identification, the actively-expressed putative lncRNAs were inspected based on normalized FPKM value obtained from transcripts abundance estimation analysis and total of 59,110 (93%), 57,944 (94%) and 40,858 (93%) putative lncRNAs were identified as actively-expressed putative lncRNAs (called lncRNAs from now on) from Kiziltan, TR39477 and TTD-22 varieties, respectively. The slightly lower ratio of active expression in lncRNAs (93–94%) compared to coding transcripts (96–97%) might arise from the tendency of lower expression of lncRNAs. Additionally, inspection of the expression patterns of transcripts in each control and drought-stressed samples revealed a similar distribution of the expressions of the lncRNA and coding transcripts between the three biological replicates only with little systematic biases (Fig. 3). Error plots showed lower expression levels of lncRNAs compared to coding transcripts across all three plants which is also supported by literature21. Moreover, overall expression pattern of lncRNAs were not altered by drought treatment, except a few transcripts.
Figure 3.

Expression pattern of coding transcripts and lncRNAs in three different T. turgidum samples. (A) The expression pattern of coding transcripts and lncRNAs from Kiziltan variety which exhibit moderate performance under drought conditions. (B) The expression pattern of coding transcripts and lncRNAs from drought tolerant TR39477 variety. (C) The expression pattern of coding transcripts and lncRNAs from drought susceptible TTD-22 variety.
Several lncRNAs from three different plants showed differential expression under drought treatment. These lncRNAs were identified with edgeR software with a p-value smaller than 0,001 and log2 (fold change) greater than 235. Based on these cut-offs, 200 (3% of all lncRNAs), 424 (6% of all lncRNAs) and 277 (4% of all lncRNAs) were detected as ‘drought-responsive’ from Kiziltan, TR39477 and TTD-22, respectively. Differentially expressed lncRNAs were further evaluated for their sample specific expressions. Most of the differentially expressed lncRNAs showed tendency to exhibit sample specific expressions, indicating distinct molecular functions they might perform. Intriguingly, 66, 52 and 77% of differentially expressed lncRNAs exhibited sample specific expressions in Kiziltan, TR39477 and TTD-22 samples, respectively (Fig. 4). From 64 to 202 differentially expressed lncRNAs were detected as common between control and drought treated samples of the three T. turgidum plants. In Kiziltan, 35% of stress-responsive lncRNAs were common whereas 48% and 23% stress-responsive lncRNAs were common between control and drought treated samples of TR39477 and TTD-22, respectively. These results indicate that common transcripts were more abundant in differentially-expressed lncRNAs from TR39477, with the most tolerant profile, than Kiziltan, with moderate reaction, and TTD-22, with the least tolerance. However, further characterizations are required for a complete understanding of the lncRNAs functions under drought stress.
Figure 4.

Common and drought specific lncRNAs from different T. turgidum varieties. Venn diagrams show the number of common and specific differentially expressed lncRNAs between control (CK) and drought-stressed (DS) samples of Kiziltan, TR39477 and TTD-22.
Differential expression of mRNAs and lncRNAs were also confirmed with Quantitative Real Time (qRT-PCR) experiment. Common mRNA and lncRNAs transcripts; defined with %80 identity and query coverage across whole samples; were analyzed and a group of differentially expressed ‘common’ transcripts was chosen for experimental conformation. From this pool, expression of randomly chosen 2 mRNA and 2 lncRNA transcripts were quantified followed by 4 hours of shock drought treatment with 2-week-old root and whole seedling tissues of Kiziltan genotype. The quantification results with QRT-PCR experiment showed accordance with RNA sequencing differential expression data analysis both for lncRNAs and mRNAs (Fig. 5). In addition, experimental results showed harmony between root and whole seedling tissues for lncRNAs and mRNAs.
Figure 5.

Relative normalized expression analysis results for common differentially expressed mRNA and lncRNAs samples. The quantification of transcript expression was performed with both root and whole-seedling tissue. The error bars were constructed based on standard deviation across three replicates of each sample.
Characteristics of actively expressed lncRNAs
All actively expressed lncRNAs were blasted against lncRNAs in A. thaliana from NONCODE database36. We identified 32, 24 and 15 lncRNAs that were homologous to those lncRNAs in A. thaliana, suggesting the weak conservation of lncRNAs between A. thaliana and T. turgidum species. Actively expressed lncRNAs were further analyzed for their structural features in wild relatives of wheat. To that end, the length distribution and GC content of expressed lncRNAs and coding transcripts were analyzed and compared. The average length of T. turgidum lncRNAs was 327 nucleotides long whereas that of coding transcripts was 1,198 nucleotides. Lengths of those lncRNAs ranged from 201 to 2,686 nt, 2,857 nt and 2,540 nt in Kiziltan, TR39477 and TTD-22, respectively. In general, the majority of lncRNAs were relatively short while almost half (47–50%) of coding transcripts were longer than 1,000 nt in all the T. turgidum varieties (Supplementary Fig. 3). The average GC content for lncRNAs was ranging from 43% to 45% and across all three varieties, the highest ratio of GC content was 82%. Interestingly, highest GC content of lncRNAs was detected in shorter lncRNAs, generally shorter than 1,000 nt which suggest an association between GC content and lncRNAs length (Supplementary Fig. 4). On the other hand, the average GC content of coding transcripts was detected as 52%, relatively higher than lncRNAs in all three varieties. Connection between GC content and length distribution was also observed in coding transcripts (Supplementary Fig. 4). While the length of the coding transcripts is increasing, the GC content was detected as narrowing around 50%. Overall, these results indicate that lncRNAs as well as coding transcripts share similar structural features in different T. turgidum species; yet, lncRNAs slightly differ from coding transcripts in gene structure in terms of structural characteristics.
Tetraploid durum wheat and wild emmer wheat genomes are derived from hybridization of A and B sub-genomes, each contributing to the composition of coding and lncRNA transcripts equally. Since reference genomes for Kiziltan, TR39477 and TTD-22 varieties are not available yet, we analyzed composition of coding and lncRNA transcripts from the recently published assembly of Zavitan (T. dicoccoides variety) genome37. Using GMAP, we were able to map 89% of the lncRNA transcripts and 97% of coding transcripts to Zavitan genome. As Zavitan is a different cultivar from our three genotypes, we can expect cultivar dependent lncRNAs, resulting in the slight lower ratio of mapped transcripts of lncRNAs. Among these alignments, 2% of coding and 3% of lncRNA transcripts were mapped to uncharacterized scaffolds. On average, 50 and 48% of coding transcripts and 47 and 50% of lncRNA transcripts were mapped to A and B sub-genomes, respectively. 48, 53 and 49% of coding transcripts and 46, 50 and 45% of lncRNA transcripts were mapped to A sub-genome whereas 50, 45 and 50% of coding transcripts and 51, 48 and 52% of lncRNA transcripts were mapped to B sub-genome in Kiziltan, TR39477 and TTD-22 varieties, respectively. The results showed enrichment of both coding and lncRNA transcripts in A sub-genome in TR39477 varieties and in B sub-genomes in Kiziltan and TTD-22 varieties. Both coding and lncRNA transcripts were similarly distributed over each chromosome at frequencies varied between 6 to 9%. These transcripts were most abundant at 2A chromosome for TR39477 and at 2B chromosome for Kiziltan and TTD-22 varieties. The results showed each sub-genome and chromosome of tetraploid wheat genome contributed in composition of lncRNAs as in case of coding transcripts.
One transcript can be derived from different loci and from opposite directions on the genome. The results showed similar distribution of coding and lncRNA transcripts on sense and antisense strands. Nearly 24% of all transcripts were shown to be transcribed from both directions whereas remaining alignments were distributed equally on sense and antisense strands. Consistent with previous studies on plants38, most of the lncRNAs (80%) were single-exon transcripts and 6% of lncRNAs could be transcribed as both single-exon and multi-exons transcripts from different loci. On the other hand, coding transcripts tended to have more exons where 76% of coding transcripts transcribed with multi-exons. lncRNA transcripts showed smaller number of exons where maximum exon number can reach up to 16 in a lncRNA transcript as opposed to that of 68 for coding transcripts.
Similar to protein coding transcripts, lncRNAs are also exposed to alternative splicing with a lower rate compared to mRNAs39. Trinity-constructed isoforms of each gene were accounted for the spliced isoforms and were used to determine alternative splicing ratios of lncRNAs. In Kiziltan, 18% (10,369) of actively expressed lncRNAs were exposed to alternatively splicing where this ratio was detected as 64% (51,634) for that of coding transcripts. Similarly, alternatively spliced lncRNAs were counted as 18% (10,611) and 16% (6,906) of total lncRNAs where that of 60% (45,138) and 59% (44,221) of coding transcripts were identified as alternatively spliced, in TR39477 and TTD-22, respectively. Furthermore, alternative splicing (AS) events were identified from all mapped transcripts to Zavitan genome. AS events occurred in ~14% of all actively expressed transcripts in each genome. Among the AS events, intron retention with 38% of the events is the predominant over remaining splicing events, followed by 29% to other events, 15% to alternative acceptor, 10% to alternative donor and 8% to exon skipping. Among the transcripts, 25, 22 and 21% of coding transcripts and 5, 5, 4% of lncRNA transcripts were involved in an AS event in Kiziltan, TR39477 and TTD-22 varieties, respectively. Consistent alternative splicing patterns in different T. turgidum varieties suggested that alternative splicing is not as prevalent in lncRNAs as it is in coding transcripts.
Among the lncRNAs of trinity-constructed isoforms, the ones with the most abundant splicing events were further inspected. For example, the two lncRNAs, Kiz_both_c65078_g3_i12 and Kiz_both_c65078_g3_i4, were detected as the isoforms of the same gene, Kiz_c65078_g3 which possessed 23 alternatively spliced isoforms in Kiziltan transcriptome. It was also noted that 8 isoforms of this gene showed sample specific expression, where one (Kiz_CK_c65078_g3_i21) of them identified as ‘coding transcript’ in the control sample. In TR39477 transcriptome, maximum number of gene isoforms was observed as 27 for the gene TR_c63034_g2. Among these isoforms, six of them were detected as actively expressed lncRNAs where only two of these lncRNAs showed differential expression during drought treatment. For TTD-22 transcriptome, the gene TTD_c62818_g1 had the most splicing events with 24 isoforms where six and three of them were identified as lncRNAs and coding transcripts, respectively. None of these isoforms showed differential expression during drought treatment. Yet, all lncRNA isoforms exhibited sample specific expressions where none of them were in common between control and drought treated samples. Since expression levels and significance in p-values were low, these sample specific expressions were not defined as differential expression. Regarding to observed alternative splicing patterns, it is tempting to speculate that each alternatively spliced isoforms have different expression profiles and might have differential functions during stress response.
Repeat-masking of stress-responsive lncRNAs against known Poaceae repeat elements revealed that 37% to 64% of stress-responsive lncRNA sequences contain repetitive elements in three of the replicates. The difference in the repeat content of stress-responsive lncRNAs stem from repeat elements from small RNAs found in T. dicoccoides varieties. Once small RNAs excluded from repeat library, percent of stress-responsive lncRNAs containing repeat elements were decreased to 33–34% in both TR39477 and TTD-22 varieties. Interestingly, lncRNAs which were from small RNA sequences, made up 34% or 61% of all repeats (Fig. 6), were detected in T. dicoccoides samples, TR39477 and TTD-22 whereas no siRNAs were detected in Kiziltan. These small RNA repeats were from closely related species including Zea mays, Triticum aestivum and Oryza sativa. The most common small RNA sequences found in both TR39477 and TTD-22 samples was ZRSiRGRR00000035, following ZRSiRGRR00000042 and TRSiRGRR00000062. These observations indicate that some lncRNAs involved in stress response may act as siRNA precursors. Besides, their corresponding siRNAs were, therefore, regulated in a stress dependent manner. Excluding small RNAs from repeat content, stress-responsive lncRNAs which were from DNA transposons were marked in all three samples in almost half percent of repeats.
Figure 6.

Repeat content of stress-responsive lncRNAs. Stress responsive lncRNAs were associated with both DNA transposons and retrotransposons. Each variety was represented with a different color (blue: Kiziltan, pink: TR39477 and orange: TTD-22).
miRNA-related functions of lncRNAs
miRNAs can regulate gene expression at the post-transcriptional level by interacting with the complementary binding sites on target sequences, resulting in cleavage, decoy, or translation repression8, 13, 40. Several studies have suggested that lncRNAs might have functions associated with miRNAs being either their targets or precursors19. To explore such functional roles of lncRNAs, in silico miRNA prediction was performed from all of three varieties by utilizing a list of 1,404 high confidence and/or experimentally verified plant miRNAs subtracted from miRBase release 2141. In silico miRNA identification process led to the identification of 54, 58 and 46 lncRNAs in Kiziltan, TR39477 and TTD-22, respectively, as putative precursors of miRNAs belonging to 38 miRNA families. Interestingly, only one of the precursor lncRNAs in each assembly exhibited differential expression during drought treatment. In TR39477 and TTD-22, the stress responsive lncRNAs TR_c65168_g7_i1 and TTD_c34631_g1_i1 were detected as the precursors of miR1127 which do not have any determined target in these transcriptome assemblies. On the other hand, in Kiziltan, the stress-responsive lncRNA, Kiz_c66393_g4_i7, was identified as the putative precursor of the two miRNAs, with 1 or 2 nt changes in mature miRNA sequences of Triticum aestivum miRNAs; miR1117 and miR1127a. LncRNAs which have ability to generate miRNA sequences might perform an indirect regulatory function through lncRNAs - generated miRNAs. In order to determine this indirect regulatory path, target transcripts of lncRNAs-derived miRNAs were analyzed and targets were identified only for miR1117. miR1117 was associated with one coding; Kiz_c69869_g4_i1: a coding transcript expressed in both control and drought-treated samples without any change in expression; and two noncoding RNA targets; drought-specific Kiz_c106327_g1_i1 and control specific Kiz_c85253_g1_i1. This indicates that lncRNA-derived miRNAs can perform multiple targeting potential which includes both coding and noncoding transcripts, indicating the complex regulatory mechanisms through noncoding RNAs even though the underlying regulatory network is not completely understood. Moreover, differential expression of precursor transcripts might result in the differential expression of corresponding mature miRNAs, leading to an increased regulation of expression; however, analysis of mature miRNAs at small RNA level is necessary for further validation of differential miRNA expression.
In order to provide more insight into miRNA-lncRNA association, functions of lncRNAs were analyzed in the sense of acting as miRNA targets using psRNATarget webtool at the default settings. It was shown that 1,276 lncRNAs were targeted by 33 miRNAs in Kiziltan where 1,124 lncRNAs targeted by 24 miRNAs in TR39477 and 560 lncRNAs by 26 miRNAs in TTD-22. In Kiziltan, 9 of the lncRNAs targeted by miRNAs, further suggesting 13 stress-responsive miRNA-lncRNA target pairs, detected as differentially expressed in drought condition (Supplementary Table 2). In TR39477, 15 stress-responsive lncRNAs were detected as putative miRNA targets, building 27 unique miRNA-lncRNA target pairs (Supplementary File 2). Yet, only 4 of the target lncRNA transcripts that established 7 miRNA-lncRNA target pairs in TTD-22 showed differential expression between drought-stressed and control samples (Supplementary File 2). Intriguingly, miRNAs targetting stress-responsive lncRNAs were mostly dominated by miR1436 and miR1439, where miR1118, miR1122, miR1130, miR1137 and miR1139 possessed putative lncRNA targets in TR39477 samples only, and miR1133 and miR1136 targeted lncRNAs in Kiziltan and TR39477, moderate to high tolerant samples (Supplemental File 2). Additionally, it was shown that, in accordance with drought tolerance profiles of the samples, miR1436 and miR1439 mediated 8, 16 and 5 miRNA-lncRNA target pairs in Kiziltan, TR39477 and TTD-22 samples, respectively. These results suggested that gene regulation of miRNAs on stress-responsive lncRNAs are well corralated with the stress-tolerance profiles of the three genotypes such that stress-responsive miRNA-lncRNA target pairs were prevalent at most in TR39477, the most tolerant genotype and vice versa in TTD-22, the most sensitive genotype to drought. Moreover, the diversity of target lncRNAs in the most tolerant variety, TR39477, might be an indicator of additional regulatory mechanisms mediated by these lncRNAs. Thus, functional characterization of these target lncRNAs may shed light onto the drought tolerance mechanisms in T. turgidum species.
Functional characterization of lncRNAs through lncRNA-miRNA-mRNA networks
lncRNAs may interrupt miRNA-based regulation of gene expression by target mimicry where miRNAs bind to lncRNAs instead of their actual mRNA targets42. Thus, lncRNAs may indirectly enhance functioning of particular coding transcripts by preventing negative regulation of their translation by miRNAs. To explore stress specific association of miRNAs, lncRNAs and mRNAs, stress-responsive lncRNAs and their miRNA-mRNA network was particularly analyzed. lncRNAs-miRNA-mRNA networks were observed in all three varieties with different levels of complexities (Fig. 7, Supplementary File 2). The most complex interaction networks were established by miR1436 and miR1439. Intriguingly, these two miRNAs were detected as sharing similar target transcripts in all samples, indicating a dual-regulation of gene expression. Besides it was shown that miR437 and miR1135; miR1120, miR1122, miR1128 and miR1130; and miR1120 and miR1128 were also contributing to the interaction circuitry of miR1436 and miR1439 in Kiziltan, TR39477 and TTD-22 samples, respectively, suggesting additional players in these complex networks. Among these, interactions through miR1120 and miR1128 were conserved at the interspecies level. The putative functions of coding transcripts were elucidated through GO mapping annotations. Intriguingly, antioxidant, electron carrier and molecular transducer molecular functions and growth biological process were highly enriched in Kiziltan and TR39477 varieties but no evidence was found in TTD-22 varieties (Fig. 8). These results suggested that increased regulation in these functions might be involved in drought stress response in T. turgidum varieties. Among the interaction networks, expression profiles of miRNAs (mir1436–1 and miR1436–4) and their corresponding lncRNA (c70772_g2_i1 and c90557_g1_i1) and mRNA (c69036_g1_i1 and c9653_g1_i2) targets were shown in Fig. 9. The expression of mRNA and lncRNAs molecules were concordant with the RNA-Seq data where both mRNAs and lncRNAs are upregulated under drought stress. Additionally, Q-RT-PCR results proved the drought specific expression of lncRNA c90557_g1_i1 where no expression for this lncRNAs was detected under control condition in variety Kiziltan.
Figure 7.

miRNA regulated networks between lncRNAs and coding transcripts. miRNA-lncRNA and mRNA networks were represented for each variety, Kiziltan (A), TR39477 (B) and TTD-22 (C). miRNA nodes were presented as rectangle and colored by miRNA family names. lncRNAs and coding transcripts were presented as triangles and circles, respectively. Transcripts that were upregulated by drought were colored as red and downregulated transcripts were colored green.
Figure 8.

Distribution of Gene Ontology mapping results of coding targets of putative miRNAs. Targets from each variety was represented with a different color; blue: Kiziltan, red:TR39477, green: TTD-22. GO terms histogram was prepared through WEGO online tool.
Figure 9.

Relative normalized expression analysis results for lncRNA-miRNA-mRNA networks involved differentially expressed transcripts. The quantification of transcript expression was performed with both root and whole-seedling tissue. The error bars were constructed based on standard deviation across three replicates of each sample.
Discussion
The increased effects of drought stress, caused by climate change, compel the improvement of major crop species such as wheat. However, complex genome of hexaploid wheat, combined from three different sub-genomes, A, B and D, becloud the understanding of gene regulations and molecular pathways underlying stress adaptation mechanisms, which is essential for establishing better crop performance. Alternatively, tetraploid wheat species, possessing a less complex genomic organization, stand as good candidates to pave the way for a deeper understanding of such mechanisms in wheat. Several varieties of wild tetraploid wheat have already been shown to exhibit differential drought tolerance, which might enhance our understanding of the drought-tolerance mechanisms in bread wheat9. With the aim of providing further insights to drought response mechanisms and associated stress tolerance profiles of tetraploid wheat, transcriptomic changes in the roots of three different T. turgidum samples under slow drought imposition were analyzed at both coding and non-coding levels. Overall, this study showed the differential regulation of both coding and non-coding transcripts in response to drought stress, which might further be used for a better crop performance under drought conditions.
Sequenced reads from both control and drought-treated samples were assembled together and analyzed in the sense of differentially expressed coding transcripts and lncRNAs. A stringent filtering of transcripts (Fig. 1) enabled the identification of 35, 36 and 39% of actively-expressed coding transcripts besides 26, 29 and 21% of actively-expressed lncRNAs over all actively-expressed transcripts in Kiziltan, TR39477 and TTD-22 varieties, respectively (Supplementary Table 3). Overall, 2 mRNA and 2 lncRNA transcripts were validated with qRT-PCR experiment and the expression trend of transcript showed a similar pattern with RNA-Seq data analysis even though the fold changes are different. Since the shock drought stress treatment was used for validation of presence of these transcripts, this is an expected result; particularly for lncRNAs, which the expression is highly dependent on condition. In the RNA-Seq analysis, interestingly, the number of transcripts was detected as decreased in all three varieties, regardless of their stress tolerance (Table 1). The stress-induced protein breakdown is a known phenomenon in plants where the accumulated amino acids support the osmotic balances in cells43. Thus, it might be possible that the number of transcripts which leads to translation of several proteins may decrease to further support this breakage and osmotic balance. Additionally, it was noted that although having the highest percent of coding transcripts, the transcriptome of TTD-22, the most sensitive genotype, contained the lowest percent of lncRNAs. As several studies have provided evidence of the functional importance of lncRNAs for drought stress response44, 45, the low abundance of lncRNAs in TTD-22 might be associated with its low drought tolerance; however, further characterization of stress responsive lncRNAs, particularly from drought tolerant variety TR39477, is essential to fully understand the role of lncRNAs in drought response.
To understand the function of differentially expressed genes under drought stress, the functional annotation was conducted via BlastX and Blast2GO. Analysis of homology patterns in T. turgidum proteins revealed a scattered homology of proteins across different Poaceae members. Although a high homology of T. urartu proteins is expected because of heritage of tetraploid wheat, high homology with A. tauchii stands as unexpected and further examination of these homolog proteins might provide insight into evolution of T. turgidum. The conservation of different coding transcripts was also observed between different accessions of T. turgidum. Additionally, more than 70% of transcripts were detected as common under control and drought-treated samples. Since plant cells tries to keep basal reaction rate for cellular maintenance, it is normal for high conservation of proteins under drought treatment46. Moreover, even though a small portion of transcripts was differentially expressed under drought stress, they might have serious effect on other proteins.
Drought specific transcripts from TR39477 and TTD-22 were further analyzed in regards to their function to further understand the differences in the drought tolerance. Comparison of these transcripts from two varieties revealed that only approximately 20% of these transcripts are conserved. These common transcripts, expressed in response to drought regardless of the degree of drought tolerance, might be associated with general proteins which are expressed in the stress conditions such as ABA-responsive transcription factors of ROS scavengers43. On the other hand, TR39477 revealed 36 transcripts which are specific to this cultivar and do not possess any degree of homology to TTD-22 transcripts. These transcripts were connected with several osmolytes and secondary metabolite such as ‘Glutathione’ and ‘Thiamine’ metabolisms regarding to KEGG maps. For instance, glutathione metabolism was associated with proline production, which is an important osmolytes accumulated in drought stress47. Thiamine is an important molecule which involves in to phenylpropanoid pathway and this pathway cause the generation of several secondary metabolites which enhance the performance of plants under drought stress43, 48. Thus, it is tempting to speculate that TR39477 utilize these transcripts to regulate osmolytes production together with secondary metabolites to survive under drought stress. Further characterization of these transcripts may provide more insights into the molecular mechanisms of these events.
Besides the stress responsive transcripts, lncRNAs exclusive to drought stress and their relationship with miRNAs and mRNAs provided further insight to the molecular mechanism of drought tolerance. The most complex networks were detected for miR1436 and miR1439, which indicates their important function in drought stress response. Among these, miR1439 was detected as targeting an aquaporins proteins which is conserved in wheat, rice and Brachypodium49. Under drought stress, lncRNAs might embed the inhibition of aquaporin translation, via target mimicry to miR1439 family members, and enhance the function of this protein for further transport of water from roots. Interestingly, miR1120 and miR1128 detected as conserved at interspecies level suggesting its important function. In another study, Yao and colleagues also detected ubiquitous expression pattern of miR1128 (misnamed as miR504 in the publication) even though no information about the targets of these miRNAs again not suggested50. Computational inspection and experimental validation of targets of these miRNAs might shed light onto their presence in these networks. Here, with Q-RT-PCR, we validated expression of miRNA1436-1 and miR1436-4 and their corresponding targets, supporting the existence of lncRNA-miRNA-mRNA networks (Fig. 9).
Gene regulation is not limited to protein-coding genes where most of the genes transcribed in complex organisms are in fact non-protein-coding genes with important regulatory functions. Increasing number of studies has showed that both sRNAs and lncRNAs are important players of gene regulation in various vital biological processes, including stress responses in plants. Drought is a major stress factor to crops, causing serious yield losses to wheat (Triticum ssp.), and an important food source worldwide. On top of being an important limiting factor to the yield already, the effect of drought has been expected to increase by climate changes. Improved crop varieties that are tolerant to drought could sustain increased yield and quality of crops. In order to obtain improved varieties with enhanced productivity and stress tolerance, introgression of favorable elements into domesticated crop varieties has been suggested as a viable approach for decades. However, understanding of the molecular mechanisms behind drought response is crucial in determining these elements. The current study provides a comprehensive transcriptome analysis of tetraploid wild wheat varieties with diverse stress tolerance profiles, revealing drought-responsive genes and lncRNAs, thereby enriching the genetic information available for T. turgidum varieties. Further in silico predictions of miRNAs and their target interactions exploited the putative functional roles of lncRNAs. Besides, identification and characterization of lncRNAs in the present study expands the current knowledge of lncRNAs and their regulatory roles in drought response in plants in general.
Methods
Total transcriptome sequencing, assembly and identification of differentially expressed transcripts
In a previous study of our group, a number of wild wheat varieties were subjected to slow drought imposition where two wild emmer wheat (T. turgidum ssp. dicoccoides) varieties, TR39477 and TTD-22, exhibited contrasting responses as the most tolerant and the most sensitive compared to the cultivated durum wheat (T. turgidum ssp. durum) variety Kiziltan with moderate response8. Total RNA isolation from a pool of three biological replicates of the root samples of control and drought-stressed modern durum wheat, Kiziltan, and wild emmer wheats, TR39477 and TTD-22, conducted with TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) following the manufacturer’s recommendations and RNA integrity was controlled using Agilent Bioanalyzer 2100 RNA 6000 Nano Kit (Agilent Technologies, Santa Clara, CA, USA8. Following, high-throughput sequencing with Illumina HiSeq. 2000 were performed with the libraries prepared by using TruSeq RNA Sample Prep Kit v2 8. Illumina HiSeq. 2000 paired end reads can be accessible at ENA database with the run ID: ERR1987529. Raw paired-end reads from RNA sequencing of these six samples (three genotypes × two conditions) were quality trimmed using Trimmomatic (v0.32) with default parameters (LEADING:5, TRAILING:5, MINLEN:36)26. De novo assembly for each genotype was generated by Trinity platform27 (release 2014-07-17) from combination of paired-end Illumina reads of control and drought-stressed samples. Assembled transcripts were aligned back to the raw reads using bowtie aligner and the abundance estimation of all transcripts was quantified as FPKM with utilization of RSEM under Trinity pipeline. Individual assembly files for each control and drought-stressed samples were separated based on their corresponding abundance estimates for further analysis. Differential expression analysis was conducted using EdgeR pipeline35 with the default threshold parameters of p-value = 0,001 and log2(fold_change) = 2.
Annotation of transcripts and identification of long non-coding RNAs
Following transcriptome assembly, annotation of transcripts and identification of lncRNAs were performed through following rigorous criteria: exclusion of contaminants, open reading frame (ORF) size prediction, ab initio predictions and homology screenings. As the first layer of analyses, transcripts were excluded from the assemblies if defined as contaminants after blast screenings against Triticum turgidum non-coding RNAs deposited at NCBI and ENA databases (1E-05, -pident 95, -length 30); rRNA, tRNA, snoRNA, snRNA sequences of Triticum families deposited in NCBI database (1E-05, -pident 95, -length 30); Triticum aestivum mitochondrion complete genome (NC_007579.1) and Triticum turgidum organellar RNAs deposited at NCBI and ENA databases (1E-15, -pident 95, -length 30). Remaining analyses evaluated the coding potential of transcripts and aid to determine either lncRNAs or coding transcripts.
Subsequent to contaminant analysis, the ORF size prediction for each assembly was conducted in order to differentiate between protein coding and non-coding transcripts. Since many transposons have similar ORFs to host genes which may corrupt the coding gene annotation, all assemblies were subjected to repeat-masking prior to ORF content predictions, against the repeat library of MIPS Repeat Element Database v9.3 p for Poaceae (ftp://ftp.mips.helmholz-muenchen.de/plants/REdat/)51 using RepeatMasker v4.0.5 software52. Ability of repeat-masked transcripts to construct a full-length protein was evaluated by employing two different software, Transdecoder (-m 80) and EMBOSS:getorf53. Transcripts with a continuous ORF > 240 nucleotide in length were accepted as transcripts with a functional ORF. Coding potentials of transcripts were predicted with several ab initio methods; CPC online tool (options: reverse strand mode was included)29, CNCI (version 2, options: -m pl)30 and AUGUSTUS online tool31 with the pre-established system trained for Triticum/wheat. Transcripts identified as ‘coding’ by at least one of these tools satisfy the ab initio prediction criterion.
In order to identify homolog coding transcripts with other species, assemblies were aligned to a dataset of coding sequences using Blast tool kit (version 31)28. All transcripts were initially blasted against several datasets; Uniprot/Swissprot database (http://web.expasy.org/docs/swiss-prot_guideline.html) (parameters: -evalue 1E-05, -pident 80, -length 30); Triticum aestivum UniGenes (https://www.ncbi.nlm.nih.gov/unigene, build#63) (parameters: -evalue 1E-30, -pident 98, -length 90, -max-target-seqs. 1); Triticum turgidum ESTs and coding sequences deposited at NCBI (https://www.ncbi.nlm.nih.gov/) and ENA (http://www.ebi.ac.uk/ena) databases (parameters: -evalue 1E-05, -pident 95, -length 30) together with fully annotated proteins from Brachypodium distachyon (annotation: v1.2, http://mips.helmholtz-muenchen.de/plant/brachypodium)54, Oryza sativa (assembly: IRGSP-1.0, http://rapdb.dna.affrc.go.jp(download/irgsp1.html)55, Sorghum bicolor (annotation: v1.4, http://mips.helmholtz-muenchen.de/plant/sorghum)56, high confidence proteins from Hordeum vulgare (http://mips.helmholtz-muenchen.de/plant/barley/)57, and Triticum UniProt sequences (144,397 entries, http://uniprot.org/) (parameters: -evalue 1E-05, -pident 95, -length 30). Additionally, Transdecoder (-m 30) predicted peptide sequences of each transcript were screened against Swissprot entries (parameters: -blastp, -evalue 1E-05, -pident 80, -length 30). Conserved protein domains preserved in these peptides were also controlled with Hmmer (v.3.1b1) against Pfam domains (-evalue 1E-05)58. Transcripts with homology evidence from any of these screenings satisfy homology-based prediction criterion.
As described in Fig. 1, after exclusion of contaminants, transcripts qualifying remaining criteria were defined as coding transcripts where transcripts with no evidence of coding in ORF size predictions, ab initio predictions and homology screenings were defined as lncRNAs. Final functional annotation of coding transcripts were carried out using Blast2GO software32 with the initial blast screen run locally against all Viridiplantae (taxid: 33090) proteins from the NCBI nr protein database (parameters: blastx, -evalue 1E-5, -outfmt 5, -max_target_seq. 1).
Genome mapping and splicing
Wild emmer wheat genome was obtained from WEWseq consortium (Zavitan_v2_pseudomolecule: http://wewseq.wixsite.com/consortium 37). Transcripts were mapped to Zavitan genome using GMAP (version 2016–07–11, with all parameters set to default except –min-identity = 90 –cross-species -f 2). Obtained GFF files were converted into GTF format using gffread. GTF files were submitted to ASTALAVISTA database at default settings to identify alternative splicing events.
Quantitative Real Time PCR (QRT-PCR) analysis for miRNA, mRNA and lncRNA transcripts
In order to show the accordance of differential expression analysis based on RNA sequencing with wet lab, quantitative Real Time PCR (qRT-PCR) experiment was performed. Prior to experiment, a subset of lncRNAs and mRNAs were selected. The differentially expressed transcript across Kiziltan, TR39477 and TTD-22 were compared via blast analysis and transcript sequences showing similarity with 80% identity and query coverage between whole samples was defined as ‘common’. Between common transcripts, 2 mRNA (Kiziltan_ mRNA_c17408_g1_i1, Kiziltan_ mRNA_c55246_g1_i1) and 2 lncRNA sequences (Kiziltan_ lncRNA_c118446_g1_i1 and lncRNA_c47700_g1_i1) were chosen randomly and qRT-PCR primers were designed (Supplementary Table 4). Kiziltan seed were surface sterilized in 4% sodium hypochlorite and grown in tall plastic jars for 15 days at 23 °C with adequate amount of water. At the end of two week, plant seedlings reached the four-leaf stage were dehydration shocked for 4 hours by removing them from plastic jars and leaving on paper towels under the same lighting conditions, while control plants were immediately fast frozen in liquid nitrogen. Both root and whole seedling tissues were collected and stored at −80 °C. Total RNA isolation from whole collected tissues from control and drought treated samples was performed with TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) following the manufacturer’s recommendations. First strand cDNA synthesis was performed on 1 µg of total RNA using RevertAid H Minus Reverse Transcriptase (QuantiTect Reverse Transcription Kit, Qiagen) according to manufacturer’s protocols. For quantification of mRNA and lncRNAs transcripts from control/ drought stressed root/whole seedling samples, the reaction mix containing 3 µl of 5X diluted of cDNA, 1 µl of forward primers, 1 µl of reverse primer and 5 µl of iTaqTM Universal SYBR® Green Supermix (Bio-Rad) incubated in Bio-Rad CFX 96 Thermal Cycler with following conditions: 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 1 min and then 95 °C for 15 s. The constitutive gene of Triticum aestivum Actin (TaActin)59 was used as internal standard to normalize the transcripts using a gene-specific primer (Supplementary Table 4). The 2-ΔCt method was used to calculate the difference in expression of chosen genes60.
For validation of mRNA, miRNA and lncRNAs, miR1436-1 and miR1436-4 were chosen and validated together with their mRNA and lncRNA targets. For qRT-PCR analysis of mRNA and lncRNAs, the same method was utilized described in above. In order to obtain cDNA belonging to mature miRNAs, miRNA-specific stem–loop reverse transcription reactions were performed using RevertAid H Minus Reverse Transcriptase with iScriptTM Select cDNA Synthesis Kit (BioRad) following the manufacturer’s recommendations with slight modifications. Prior to cDNA synthesis, a mix of 500 ng of DNAase treated RNA and 1 µl of miRNA-specific stem loop PCR was incubated at 65 °C. After incubation, 2 µl of GSP enhancer solution, 4 µl of 5x iScript select reaction mix and 1 µl of iScript reverse transcriptase were added and reaction mix (10 µl) was incubated at 42 °C for 1 hour followed by 5 minutes of 85 °C incubation to heat-inactivate the reverse transcriptase. For validation of miRNA expression, the reaction mix containing 3 µl of 5X diluted of cDNA, 1 µl of forward primers, 1 µl of universal reverse primer and 5 µl of iTaqTM Universal SYBR® Green Supermix (Bio-Rad) incubated in Bio-Rad CFX 96 Thermal Cycler with following conditions: 95 °C for 5 min, followed by 35–45 cycles of 95 °C for 5 s, 60 °C for 10 s, and 72 °C for 1 s. For melting curve analysis, samples were denaturated at 95 °C, then cool to 65 °C at 20 °C per second. The fluorescence signals were collected at 530 nm wavelengthcontinuously from 65 °C to 95 °C at 0.2 °C per second. The constitutive gene of Triticum aestivum rRNA26 homolog61 was used as internal standard to normalize the miRNA expression (Supplementary Table 4). For internal control, several control genes including TaU6 were attempted and the rRNA26 was chosen because of its expressional stability under different conditions and tissues. The 2-ΔCt method was used to calculate the difference in expression of chosen miRNAs.
Construction of lncRNA-miRNA-mRNA networks
High confidence and/or experimentally identified mature miRNA sequences from 72 Viridiplantae species were collected from miRBase (v21, June 2014)62, suggesting a dataset of 1,404 non-redundant mature miRNA sequences. A homology-based in silico miRNA prediction analysis was performed using this miRNA dataset as query, as previously described40, 63, 64. Following, a list of lncRNA transcripts and a list of coding transcripts are retrieved as target datasets for each transcriptome. These datasets were screened for relative gene targets of miRNAs, predicted from the assemblies, using psRNAtarget web-tool, with user-defined query and target options at default parameters (http://plantgrn.noble.org/psRNATarget/)65. lncRNAs functioning as coding-target mimics were evaluated based on the complementary pairs between miRNA-to-coding transcript targets and miRNAs-to-lncRNA targets. Cytoscape 3.3.066 was used for the visualization of lncRNA-miRNA-mRNA interaction networks.
Electronic supplementary material
Acknowledgements
We acknowledged TUBITAK-BIDEB for providing PhD scholarship for Halise Busra Cagirici. This project partially funded Montana Plant Science Endowment.
Author Contributions
H.B.C. and B.A. performed the analysis and drafted the manuscript. H.B. conceived the idea of the study, designed the experiments, supervised all analyses and drafted the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-11170-8
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.FAO. FAO Statistics. The Food and Agriculture Organization of the United Nations (2014).
- 2.Marcussen T, et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science. 2014;345 doi: 10.1126/science.1250092. [DOI] [PubMed] [Google Scholar]
- 3.Budak H, Hussain B, Khan Z, Ozturk NZ, Ullah N. From Genetics to Functional Genomics: Improvement in Drought Signaling and Tolerance in Wheat. Front. Plant Sci. 2015;6 doi: 10.3389/fpls.2015.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akpinar, B. A., Lucas, S. J. & Budak, H. Genomics approaches for crop improvement against abiotic stress. Sci. World J. 2013, (2013). [DOI] [PMC free article] [PubMed]
- 5.Fang Y, Xiong L. General mechanisms of drought response and their application in drought resistance improvement in plants. Cell. Mol. Life Sci. 2015;72:673–689. doi: 10.1007/s00018-014-1767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alptekin, B. & Budak, H. Wheat miRNA ancestors: evident by transcriptome analysis of A, B, and D genome donors. Funct. Integr. Genomics (2016). [DOI] [PubMed]
- 7.Merchuk-Ovnat L, et al. Ancestral QTL Alleles from Wild Emmer Wheat Improve Drought Resistance and Productivity in Modern Wheat Cultivars. Front. Plant Sci. 2016;7 doi: 10.3389/fpls.2016.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akpinar, B. A., Kantar, M. & Budak, H. Root precursors of microRNAs in wild emmer and modern wheats show major differences in response to drought stress. Funct. Integr. Genomics (2015). [DOI] [PubMed]
- 9.Ergen NZ, Budak H. Sequencing over 13 000 expressed sequence tags from six subtractive cDNA libraries of wild and modern wheats following slow drought stress. Plant, Cell Environ. 2009;32:220–236. doi: 10.1111/j.1365-3040.2008.01915.x. [DOI] [PubMed] [Google Scholar]
- 10.The International Wheat Genome Sequencing Consortium, (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science345, 1251788 (2014). [DOI] [PubMed]
- 11.Budak H, Kantar M. Harnessing NGS and Big Data Optimally: Comparison of miRNA Prediction from Assembled versus Non-assembled Sequencing Data—The Case of the Grass Aegilops tauschii Complex Genome. Omi. A J. Integr. Biol. 2015;19:407–415. doi: 10.1089/omi.2015.0038. [DOI] [PubMed] [Google Scholar]
- 12.Budak H, Kantar M, Bulut R, Akpinar BA. Stress Responsive miRNAs and isomiRs in Cereals. Plant Sci. 2015;235:1–13. doi: 10.1016/j.plantsci.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Budak, H., Khan, Z. & Kantar, M. History and current status of wheat miRNAs using next-generation sequencing and their roles in development and stress. Brief. Funct. Genomics elu021- (2014). [DOI] [PubMed]
- 14.Griffiths-Jones S. Annotating noncoding RNA genes. Annu. Rev. Genomics Hum. Genet. 2007;8:279–98. doi: 10.1146/annurev.genom.8.080706.092419. [DOI] [PubMed] [Google Scholar]
- 15.Guleria P, Mahajan M, Bhardwaj J, Yadav SK. Plant Small RNAs: Biogenesis, Mode of Action and Their Roles in Abiotic Stresses. Genomics, Proteomics Bioinforma. 2011;9:183–199. doi: 10.1016/S1672-0229(11)60022-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budak H, Bulut R, Kantar M, Alptekin B. MicroRNA nomenclature and the need for a revised naming prescription. Briefings in Functional Genomics. 2016;15:65–71. doi: 10.1093/bfgp/elv026. [DOI] [PubMed] [Google Scholar]
- 17.Budak, H. & Akpinar, B. A. Plant miRNAs: biogenesis, organization and origins. Funct. Integr. Genomics (2015). [DOI] [PubMed]
- 18.Akpinar BA, Budak H. Dissecting miRNAs in Wheat D Genome Progenitor, Aegilops tauschii. Front. Plant Sci. 2016;7:1–17. doi: 10.3389/fpls.2016.00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chekanova JA. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015;27:207–216. doi: 10.1016/j.pbi.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, et al. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell. 2012;24:4333–45. doi: 10.1105/tpc.112.102855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2015;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 22.Swiezewski S, Liu F, Magusin A, Dean C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature. 2009;462:799–802. doi: 10.1038/nature08618. [DOI] [PubMed] [Google Scholar]
- 23.Lai F, Shiekhattar R. Where long noncoding RNAs meet DNA methylation. Cell Res. 2014;24:263–4. doi: 10.1038/cr.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin H, Shin HS, Chen R, Harrison MJ. Loss of At4 function impacts phosphate distribution between the roots and the shoots during phosphate starvation. Plant J. 2006;45:712–726. doi: 10.1111/j.1365-313X.2005.02629.x. [DOI] [PubMed] [Google Scholar]
- 25.Lu, X. et al. Genome-wide analysis of long noncoding rnas and their responses to drought stress in cotton (gossypium hirsutum l.). PLoS One11, (2016). [DOI] [PMC free article] [PubMed]
- 26.Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. 8 (2013). [DOI] [PMC free article] [PubMed]
- 28.Camacho C, et al. BLAST + : architecture and applications. BMC Bioinformatics. 2009;10 doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kong, L. et al. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35 (2007). [DOI] [PMC free article] [PubMed]
- 30.Sun, L. et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41 (2013). [DOI] [PMC free article] [PubMed]
- 31.Hoff, K. J. & Stanke, M. WebAUGUSTUS–a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 41 (2013). [DOI] [PMC free article] [PubMed]
- 32.Conesa A, Götz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics. 2008;2008 doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogata H, et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen, L. J. et al. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 36 (2008). [DOI] [PMC free article] [PubMed]
- 35.Robinson MD, McCarthy DJ, Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao Y, et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016;44:D203–D208. doi: 10.1093/nar/gkv1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Avni R, et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science. 2017;357:93–97. doi: 10.1126/science.aan0032. [DOI] [PubMed] [Google Scholar]
- 38.Tang W, et al. Comprehensive Transcriptome Profiling Reveals Long Noncoding RNA Expression and Alternative Splicing Regulation during Fruit Development and Ripening in Kiwifruit (Actinidia chinensis) Front. Plant Sci. 2016;7:1–15. doi: 10.3389/fpls.2016.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao H, et al. Genome-wide identification of long noncoding RNA genes and their potential association with fecundity and virulence in rice brown planthopper, Nilaparvata lugens. BMC Genomics. 2015;16 doi: 10.1186/s12864-015-1953-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurtoglu KY, Kantar M, Budak H. New wheat microRNA using whole-genome sequence. Funct. Integr. Genomics. 2014;14:363–379. doi: 10.1007/s10142-013-0357-9. [DOI] [PubMed] [Google Scholar]
- 41.Kozomara, A. & Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42 (2014). [DOI] [PMC free article] [PubMed]
- 42.Franco-Zorrilla JM, et al. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007;39:1033–1037. doi: 10.1038/ng2079. [DOI] [PubMed] [Google Scholar]
- 43.Krasensky J, Jonak C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012;63:1593–1608. doi: 10.1093/jxb/err460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muthusamy M, Uma S, Backiyarani S, Saraswathi MS. Genome-wide screening for novel, drought stress-responsive long non-coding RNAs in drought-stressed leaf transcriptome of drought-tolerant and -susceptible banana (Musa spp) cultivars using Illumina high-throughput sequencing. Plant Biotechnol. Rep. 2015;9:279–286. doi: 10.1007/s11816-015-0363-6. [DOI] [Google Scholar]
- 45.Qi X, Xie S, Liu Y, Yi F, Yu J. Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol. Biol. 2013;83:459–473. doi: 10.1007/s11103-013-0104-6. [DOI] [PubMed] [Google Scholar]
- 46.Kantar, M., Lucas, S. J. & Budak, H. Drought Stress. Molecular Genetics and Genomics Approaches. Advances in BotanicalResearch57, (Elsevier Ltd, 2011).
- 47.Liang X, Zhang L, Natarajan SK, Becker DF. Proline mechanisms of stress survival. Antioxid. Redox Signal. 2013;19:998–1011. doi: 10.1089/ars.2012.5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boubakri H, et al. Thiamine modulates metabolism of the phenylpropanoid pathway leading to enhanced resistance to Plasmopara viticola in grapevine. BMC Plant Biol. 2013;13 doi: 10.1186/1471-2229-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su C, et al. Identification and characterization of a subset of microRNAs in wheat (Triticum aestivum L.) Genomics. 2014;103:298–307. doi: 10.1016/j.ygeno.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Yao Y, et al. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.) Genome Biol. 2007;8 doi: 10.1186/gb-2007-8-6-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nussbaumer, T. et al. MIPS PlantsDB: A database framework for comparative plant genome research. Nucleic Acids Res. 41 (2013). [DOI] [PMC free article] [PubMed]
- 52.Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. 2009;4:1–14. doi: 10.2174/157489309787158161. [DOI] [PubMed] [Google Scholar]
- 53.Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 54.International T. & Initiative, B. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka T, et al. The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res. 2008;36:D1028–33. doi: 10.1093/nar/gkm978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paterson AH, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 57.Mayer, K. F. X. et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 1–83 (2012). [DOI] [PubMed]
- 58.Zhang Z, Wood WI. A profile hidden Markov model for signal peptides generated by HMMER. Bioinformatics. 2003;19:307–308. doi: 10.1093/bioinformatics/19.2.307. [DOI] [PubMed] [Google Scholar]
- 59.Yue J, et al. Wheat homologs of yeast ATG6 function in autophagy and are implicated in powdery mildew immunity. BMC Plant Biol. 2015;15:1–15. doi: 10.1186/s12870-015-0472-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 61.Tenea GN, Peres Bota A, Cordeiro Raposo F, Maquet A. Reference genes for gene expression studies in wheat flag leaves grown under different farming conditions. BMC Res. Notes. 2011;4 doi: 10.1186/1756-0500-4-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kozomara, A. & Griffiths-Jones, S. MiRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, (2011). [DOI] [PMC free article] [PubMed]
- 63.Lucas, S. J. & Budak, H. Sorting the wheat from the Chaff: Identifying miRNAs in genomic survey sequences of Triticum aestivum chromosome 1AL. PLoS One7, (2012). [DOI] [PMC free article] [PubMed]
- 64.Alptekin B, Akpinar BA, Budak H. A Comprehensive Prescription for Plant miRNA Identification. Front. Plant Sci. 2017;7 doi: 10.3389/fpls.2016.02058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dai, X. & Zhao, P. X. PsRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 39 (2011). [DOI] [PMC free article] [PubMed]
- 66.Shannon P, et al. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.