


Abstract
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause cystic fibrosis (CF), but are not good predictors of lung phenotype. Genome-wide association studies (GWAS) previously identified additional genomic sites associated with CF lung disease severity. One of these, at chromosome 11p13, is an intergenic region between Ets homologous factor (EHF) and Apaf-1 interacting protein (APIP). Our goal was to determine the functional significance of this region, which being intergenic is probably regulatory. To identify cis-acting elements, we used DNase-seq and H3K4me1 and H3K27Ac ChIP-seq to map open and active chromatin respectively, in lung epithelial cells. Two elements showed strong enhancer activity for the promoters of EHF and the 5′ adjacent gene E47 like ETS transcription factor 5 (ELF5) in reporter gene assays. No enhancers of the APIP promoter were found. Circular chromosome conformation capture (4C-seq) identified direct physical interactions of elements within 11p13. This confirmed the enhancer-promoter associations, identified additional interacting elements and defined topologically associating domain (TAD) boundaries, enriched for CCCTC-binding factor (CTCF). No strong interactions were observed with the APIP promoter, which lies outside the main TAD encompassing the GWAS signal. These results focus attention on the role of EHF in modifying CF lung disease severity.
INTRODUCTION
The power of genome-wide association studies (GWAS) to link regions of the genome with physiological or disease states is immense (reviewed in (1,2)). However, the challenges of moving from single nucleotide polymorphisms (SNPs) exhibiting high associations with a trait to causative variants are even greater (reviewed in (3)). In the simplest case, the variant lies in the coding region of a gene and compromises protein function by altering amino acids. More often, the SNPs map to non-coding regions of the genome and are hence assumed to impact regulatory elements for nearby genes (4). In rare cases, the most significant SNP alters the binding site of a critical transcription factor in a known cis-regulatory element (5). More commonly, the highest P-value SNPs merely mark an intergenic region and the associated haplotype then provides a starting point for exhaustive molecular studies aimed at deciphering the regulatory mechanisms and key gene targets. An example of this is the cystic fibrosis modifier locus on chromosome 11p13.
Cystic fibrosis (CF), the most common autosomal recessive disease among Caucasians is caused by mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR) (6–8). Lung and digestive system pathology are the major features of the disease, which is associated with more than 2000 different mutations within CFTR. One mutation, F508del (the deletion of a phenylalanine residue at amino acid 508) occurs on about 70% of CF chromosomes (9). However, even among CF patients homozygous for this mutation, there is no correlation with lung disease severity, the major predictor of lifespan (10). Despite this lack of concordance with CFTR mutation, CF lung function exhibits a strong genetic component, implicating the existence of other loci that modify the phenotype (11,12).
Extensive GWAS for lung disease severity in CF patients identified multiple regions of the genome that strongly associated with this trait (13,14). These sites include both known loci (the MUC4/MUC20 mucin genes at chr3q29 and the SLC9A3 solute carrier gene at chr5p15.3, among others) and an intergenic region at chr11p13, located between the Ets-homologous factor (EHF) and Apaf-1 interacting protein (APIP) genes. Here we investigate the chr11p13 region to reveal how SNPs underlying the GWAS findings contribute to the regulatory landscape. Of note is the very high linkage disequilibrium across the region (15), which adds substantial complexity to the search for causative variants (16).
The most significant SNP in the combined GWAS is rs10742326, which lies 3′ to the promoters of the 2 flanking genes and is almost equidistant from each of them (Figure 1) (14). On the 5′ side is EHF, an epithelial-specific Ets family transcription factor and on the 3′ side is APIP, which is involved in apoptosis and the methionine salvage pathway (17,18). EHF was recently shown to have an important role in lung epithelial function by regulating inflammation and pathways of response to injury (19,20). A second Ets family transcription factor, E47 like ETS transcription factor 5 (ELF5), which is expressed in glandular epithelium is located 5′ and adjacent to EHF (21). At the other end of the genetic interval, the X component of the Pyruvate dehydrogenase complex (PDHX) maps 3′ to APIP and may share a bidirectional promoter (Figure 1). Using a combination of methods to examine open chromatin, histone modifications, physical interactions across the region, and enhancer function, we establish the 3D-architecture of 11p13 and define regulatory mechanisms that are likely key to the role of this region in determining CF lung disease severity.
Figure 1.

The chromatin landscape of the 11p13 CF modifier region. UCSC genome browser graphic shows a 500 kb window surrounding the highest P-value SNPs at 11p13 between EHF and APIP, with the most significant GWAS I+II SNP (rs10742326) shown in grey. ChIP-seq for the active histone marks H3K27ac and H3K4me1 in Calu3 (20) and primary HBE (28) cells are shown at the top. Below are DNase-seq data showing open chromatin regions as DNase hypersensitive sites (DHS) in fibroblast (GM03348, http://www.ncbi.nlm.nih.gov/geo/GSM1008563 (33)), Caco2 (28), 16HBE14o-, Calu3 (20), HBE (28) and HTE (29) cells. The location of genes in the region is shown underneath, with cloned fragments assayed in reporter genes and key genomic features relevant to experiments shown below.
MATERIALS AND METHODS
Cell culture
Calu3 (22), 16HBE14o- (23), Caco2 (24) and BEAS-2B (25) cells were grown in Dulbecco's Modified Eagle's Medium (low glucose) with 10% fetal bovine serum (FBS). K562 cells (26) were grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS. Primary human bronchial epithelial cells (HBE) were donated by Dr Scott Randell (UNC) and grown in Bronchial Epithelial Cell Growth Medium (Lonza).
Luciferase-based reporter assays
Sequences encompassing gene promoters (∼1.5–2.0 kb upstream of TSS) and putative enhancers (∼400–800 bp centered on the DHS) were amplified using Phusion polymerase (New England Biolabs) with the primers shown in Supplementary Table S1A. Fragments were inserted into the promoter or enhancer sites of the pGL3 Basic luciferase vector (Promega) and transfected with Lipofectin or Lipofectamine 2000 (Life Technologies) into 16HBE14o-, Caco2 and BEAS2B cells. A modified pRL Renilla luciferase vector (Promega) was used as a transfection control. Cells were assayed for Renilla and firefly luciferase activity after 48 h using the Dual-Luciferase Reporter Assay System (Promega) (27).
DNase-seq
DNase-seq data for Calu3 (20), Caco2 (28), HTE (29), and HBE (28) cells were generated previously. DNase-seq for 16HBE14o- cells was performed as previously described (28,30). All data are deposited at GEO (http://www.ncbi.nlm.nih.gov/geo; GSE52181, GSE63400, GSE74709, GSE94726).
Chromatin Immunoprecipitation (ChIP)
ChIP followed by quantitative polymerase chain reaction (ChIP-qPCR) was performed as described previously (31). Antibodies specific for CTCF (Millipore 07–729) or normal rabbit IgG (Millipore 12-370) were used. qPCR was performed using SYBR Green reagents (Life Technologies) and primers listed in Supplementary Table S1B.
ChIP and deep-sequencing (ChIP-seq) data was generated according to standard protocol as described previously (20). Antibodies used were from Abcam (ab) or Millipore (mi) and specific for H3K4me1 (ab8895), H3K27ac (ab4729), H3K4me3 (mi07–473), H3K27me3 (mi07–449), H3K9me2 (ab1220), H3K9me3 (ab8898) and H3K36me3 (ab9050). H3K4me1 and H3K27ac data in Calu3 and HBE cells used here were generated previously (Calu3 (20), HBE (55)) All data are deposited at GEO (http://www.ncbi.nlm.nih.gov/geo; GSE63400, GSE74709, GSE94726).
Reverse Transcription-qPCR (RT-qPCR)
RNA was extracted from cells using Trizol (Ambion) according to manufacturer's instructions. Reverse transcription used TaqMan Reverse Transcription Reagents (Life Technologies) by the standard protocol. qPCR assays were performed using SYBR Green reagents (Life Technologies) and primers listed in Supplementary Table S1C.
Circular chromosome conformation capture and deep-sequencing (4C-seq)
4C-seq was performed as previously reported (28) in Calu3, HBE, 16HBE14o- and K562 cells. The primers used to generate libraries for each viewpoint are listed in Supplementary Table S1D.
Two independent 4C libraries were generated for each viewpoint in each cell type. The sequencing data were processed using the 4Cseqpipe protocol (32). All 4C-seq images were generated using default parameters of the pipeline.
Statistical analysis and graphs
Error bars denote standard error of the mean (SEM). Statistical significance was performed using Student's unpaired t tests on Prism software (Graphpad).
RESULTS
Chromatin landscape of the 11p13 region
Open chromatin
The region identified by the GWAS for CF lung disease severity is shown in Figure 1 and encompasses ∼200 kb between EHF and APIP. Although these genes immediately border the intergenic segment with the highest P-value SNPs, they may not be the targets of cis-regulatory elements within it. Hence, we analyzed a 500 kb window surrounding the SNP with highest genome-wide significance (rs10742326) to extend across all four genes (ELF5, EHF, APIP and PDHX) at the modifier locus. To identify cell-type-selective control elements throughout the locus we first examined chromatin accessibility by DNase I digestion followed by deep sequencing (DNase-seq) in primary human tracheal (HTE (29)) and human bronchial (HBE (28)) epithelial cells. DNase-seq data from two lung cell lines, an immortalized bronchial epithelial line 16HBE14o- (23) and Calu3 lung adenocarcinoma cells (22), were also inspected (Figure 1). These results were compared to DNase-seq data from skin fibroblasts (GM03348, http://www.ncbi.nlm.nih.gov/geo/GSM1008563 (33)) and Caco2 colon carcinoma cells (28) to distinguish airway-selective from ubiquitous sites. Within the 500 kb window, there is substantial overlap in regions of open chromatin/DHS in HBE, HTE and Calu3 cells, consistent with their 11p13 gene expression profiles. All three cell types express abundant EHF in contrast to 16HBE14o- cells which exhibit few DHS across the region and express very low levels of EHF. After removing ubiquitous sites that are also seen in fibroblasts, all the EHF-expressing airway cells share 7 out of 28 sites (25%), Calu3 and HBE share 10 (35%), Calu3 and HTE share 7 (25%), while HBE and HTE share 60% of the peaks (17 DHS). Multiple DHS are seen in airway cells that are not evident in Caco2 cells, suggesting functions of 11p13 that are not conserved in all epithelial cell types. Of particular interest in Figure 1 are DHS marked with red arrows, that are restricted to HBE and HTE cells, and those marked with black arrows, which are evident in all three EHF expressing airway cell types. In addition to these common peaks of open chromatin, each cell type has some unique peaks. For clarity in functional analysis, the DHS were named according to the chromosome 11 peak numbers in the HTE DNase-seq data (Figure 1) (eg. Chr11.2516 = site 2516). Additional sites not evident in HTE cells were numbered according DNase-seq peak numbers in the relevant cell type (eg. HB11.1485 in HBE cells and Ca11.1327 in Calu3 cells).
Histone marks
Next, we analyzed ChIP-seq data for the active histone marks H3K4me1 and H3K27ac in Calu3 (20) and HBE (55) cells to determine epigenetic features of the CF modifier locus (Figure 1). The most notable feature was a ‘stretch-enhancer’ (34) over the EHF gene, marked by extensive H3K27ac. For most sites marked with H3K27Ac, coincident H3K4me1 was evident. H3K4me3, a mark of active and poised promoters, was enriched at multiple sites across the EHF locus and at the APIP/PDHX promoters (Supplementary Figure S1). ChIP-seq for repressive histone marks (H3K27me3, H3K9me2, H3K9me3 and H3K36me3) revealed relatively few sites of enrichment in the region in Calu3 and HBE cells, consistent with their active chromatin profile (Supplementary Figure S1). A peak of H3K9me3 enrichment in both cell types within the APIP gene coincides with an element that likely contributes to locus architecture and is discussed further below.
Since our goal was to determine the function of the 11p13 region in airway epithelium, we focused on airway cell DHS that were associated with active histone marks. We identified eight airway-selective DHS (11.2522, 11.2524–11.2530) and two ubiquitous DHS (11.2516 and 11.2521) (Figure 1). With the exception of 11.2524, all the DHS coincided with peaks of both H3K4me1 and H3K27ac enrichment in Calu3 or HBE cells, the majority in both cell types. None of the DHS overlapped with promoters (H3K4me3 peaks) or inactive chromatin (Supplementary Figure S1). The data suggest that active regulatory elements/enhancers may be associated with these sites.
Enhancer elements within multiple DHS at 11p13 activate the EHF and ELF5 promoters
To determine the enhancer activity of 11p13 DHS carrying active histone marks, we tested their impact on the promoters of ELF5, EHF and APIP, which are all biologically relevant to CF lung phenotype. Based on H3K4me3 enrichment (Supplementary Figure S1) and the open chromatin map of 11p13 (Figure 1), APIP and PDHX may share a common, bidirectional promoter. However, because of the high ubiquitous expression levels of PDHX and its critical role in mitochondria, we considered this an unlikely target of airway epithelial-selective enhancer elements in this genomic interval. Hence, we only pursued studies on this shared promoter region in the APIP direction. First, the activities of the ELF5, EHF and APIP promoters were assayed after transient transfection into 16HBE14o- cells (Figure 2A). Fragments of about 1 kb 5′ and adjacent to each transcription start site were cloned into the promoter site of the pGL3B luciferase reporter vector. Transfections were done in triplicate with a Renilla vector control, and promoter construct activity was measured after 48 hrs and normalized to the pGL3B empty vector. The APIP promoter was strongest (∼125-fold over pGL3B alone), while EHF and ELF5 showed ∼60-fold and ∼30-fold values respectively. These data are consistent with endogenous expression levels for each gene in 16HBE14o- cells (Supplementary Figure S2). Here, APIP transcripts are the most abundant, EHF levels are lower and ELF5 transcripts are barely detectable. In this context, enhancers of the APIP promoter may not be quantifiable in 16HBE14o- cells. Expression of ELF5, EHF, APIP and PDHX relative to β2-microglobulin (β2M) were measured by RT-qPCR in all the cell lines used in the current analyses (HBE, Calu3, 16HBE14o-, BEAS-2B and K562) and is shown in Supplementary Figure S2. In contrast, RNA-seq data from undifferentiated primary HTE and HBE cells revealed that endogenous EHF expression levels were most abundant in the primary cells, with lower APIP and ELF5 levels (Gillen et al., unpublished).
Figure 2.

The 11.2516 and 11.2521 DHS encompass enhancer elements. (A) The ELF5, EHF and APIP promoters in the pGL3B vector were transfected into 16HBE14o- cells with a Renilla vector as transfection control. Luciferase expression is normalized to Renilla values. n = 3. Open chromatin peaks from 11p13 were inserted into the enhancer sites of the (B) pGL3B-ELF5, (C) pGL3B-EHF and (D) pGL3B-APIP promoter constructs and transfected as in (A). For DHS showing >3-fold luciferase expression relative to the promoter-only construct, n = 3. (E), (F) show transfections into Caco2 cells (n = 2 with each sample assayed in triplicate). (E) The ELF5, EHF, and APIP promoters in the pGL3B vector, (F) the 11.2516 and 11.2521 elements in the enhancer site of pGL3B-EHF. The 11.2516 and 11.2521 elements were cloned in tandem into the (G) pGL3B-ELF5 and (H) pGL3B-EHF promoter constructs and transfected into 16HBE14o- cells, n = 3. For all panels, ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns = not significant.
Next, we tested the enhancer activities of the subset of DHS 11.2516, .2521, .2522, .2524, .2525, .2526, .2527, .2528, .2529 and .2530 with each promoter. Depending on the extent of the DHS, 400–800 bp encompassing each element was cloned into the enhancer site of the pGL3B vector containing the ELF5 (Figure 2B), EHF (Figure 2C), or APIP (Figure 2D) promoters. As before, constructs were transiently transfected in triplicate into 16HBE14o- cells and normalized to the Renilla control. In the first screen we identified elements with >3-fold enhancer activity relative to the promoter-only constructs for further validation, and did not pursue the remainder. Enhancer-containing elements were assayed at least three times, and also in both the forward and reverse orientations (Supplementary Figure S3A, EHF; data not shown, ELF5). Of particular note were 11.2516 and 11.2521, which reproducibly functioned as strong enhancers of both the ELF5 and EHF promoters (Figure 2B and C). Orientation-independent increases of 20–40-fold were seen for the EHF promoter and 40–50-fold for ELF5. A third element 11.2526 modestly enhanced ELF5 gene promoter activity, but had no effect on the EHF promoter. Other elements, including 11.2525, the DHS closest to the highest P-value SNP rs10742326 (∼2 kb away), had no impact on the ELF5 or EHF promoters. Furthermore, none of the DHS elements had a strong effect on APIP promoter activity in 16HBE14o- cells (<3-fold, Figure 2D). The 11.2516 and 11.2521 ELF5 and EHF constructs were also tested in another airway epithelial cell line, BEAS-2B, where the promoters were equally active in luciferase reporter genes (Supplementary Figure S3B), and the Caco2 intestinal epithelial cell line (Figure 2E). The results confirmed the enhancer properties of both elements in BEAS-2B cells, though in this line 11.2516 was much more active than 11.2521 with both promoters (Supplementary Figure S3C, D). Neither element was active with the EHF promoter in Caco2 cells (Figure 2F), suggesting an airway-specific effect.
Of additional interest was whether the 11.2516 and 11.2521 enhancers could function cooperatively to promote gene expression. These elements were cloned together into the enhancer site of pGL3B containing either the ELF5 or the EHF promoter and relative luciferase activity was measured after transfection into 16HBE14o- cells as above (Figure 2G and H). The combination of 11.2516 and 11.2521 had an additive effect on the activity of both ELF5 and EHF promoters. This result suggests these elements may cooperate to regulate gene expression at 11p13. Since 11.2516 and 11.2521 are ∼80 kb apart these data implicate a complex higher order chromatin structure that is required for proper gene regulation.
The 11p13 locus is organized by complex long-range chromatin interactions between known and novel regulatory elements
To examine the long range chromatin interactions across 11p13 in different cell types we used both publicly available data and our own de novo analyses using circular chromosome conformation capture followed by deep sequencing (4C-seq) (28,32). We also examined the location of CCCTC-binding factor (CTCF) and cohesin occupancy sites within the region (Myers, Hardison, Snyder Labs; ENCODE (33)), since they are key interacting architectural proteins organizing the chromatin topologically associating domains (TADs) (35–38). Inspection of Hi-C data in IMR-90 cells (Ren/Yue Labs; http://www.3dgenome.org), suggests that the entire 11p13 modifier region is contained within a single TAD or sub-TAD, between the 3′ end of ELF5 (5′) and the APIP/PDHX promoter (3′) (Figure 3A, apex denoted by black arrow). Since long-range chromosome interactions occur much more frequently within TADs than between them (39), we next looked for 3D proximity of the enhancers at 11.2516 and 11.2521 with other regions across the locus using 4C-seq. Figure 3B shows interaction profiles locus-wide in Calu3 cells from viewpoints at the EHF and APIP promoters, the strong enhancers at 11.2516 and 11.2521, the inactive site at 11.2525 closest to rs10742326, and the weak enhancer at 11.2526. Two independent 4C libraries were generated for each viewpoint.
Figure 3.

Three dimensional chromatin architecture of 11p13 in Calu3 cells. (A) Hi-C data (Ren/Yue labs, http://www.3dgenome.org) in IMR90 cells spanning a 2.5 Mb window encompassing the 11p13 modifier region. The TAD (red triangle) containing the SNPs identified in the GWAS is marked by an arrow. (B) 4C-seq data in Calu3 cells with viewpoints at the EHF promoter, 11.2516, 11.2521, 11.2525, 11.2526, and the APIP promoter. The genomic map across 500 kb presented at the top and key DHS identified in Figure 1 are shown. The 4C-seq data are in two parts: (i) the main trend of interaction profile set to a 5 kb window (upper panel, black line), with relative interaction frequencies normalized to the strongest point; and (ii) the domainogram (40), which shows relative interactions based on a color-coded scale with a window size ranging from 2 to 50 kb. Red denotes the strongest interactions, while dark blue, to turquoise, to gray illustrate decreasing interaction frequencies. Red lines highlight the viewpoints and arrows show important data features discussed in the results. For all 4C data shown in Figures 3–5 each viewpoint was assayed twice in separate experiments to demonstrate reproducibility of interactions and one of them is shown.
For each 4C-seq panel, the black line shows the main trend of interactions across the locus while below it is the domainogram (40), which uses color-coded intensity values to show relative interactions with window sizes varying from 2 to 50 kb. Here, red denotes the strongest interactions and dark blue, through turquoise, to gray represent gradually decreasing frequencies. Arrows denote important data features described in the results. In Calu3 cells, the EHF promoter viewpoint showed interactions with the 11.2516, 11.2521 and 11.2525 elements. Also, novel elements were found to interact with this viewpoint, including a region 5′ to 11.2516 (marked I; site HB11.1485 in Figure 1), regions within the EHF gene body (particularly Ca11.1058/11.1327 in intron 6), the 11.2526 element, and a region within the third intron of APIP (marked II) that is not associated with a DHS in any cell type examined here. Viewpoints at the 11.2516 and 11.2521 enhancers confirmed their cooperative interactions with the EHF promoter; 11.2516 showed a strong association while 11.2521 exhibited a more modest interaction. Both enhancers also appeared to have reciprocal associations, interactions with 11.2525, and the novel elements 5′ to EHF and within APIP intron 3. The 11.2525 element did not show any defined points of association, but rather broad, moderate levels of interaction across the locus. In contrast, the 11.2526 enhancer was weakly associated with 11.2521, elements 5′ to EHF, and APIP intron 3, but apparently not with the EHF promoter. This is consistent with the luciferase assays showing a much weaker enhancer at DHS 11.2526 than at 11.2516 and 11.2521. A viewpoint at the APIP promoter also showed modest interaction frequencies, but these were localized only to the 5′ and 3′ ends of the 11p13 region. These results suggest that the APIP promoter is not controlled by specific cis-regulatory elements within the 11p13 region in Calu3 cells, and concur with enhancer assays in 16HBE14o- cells that failed to reveal any activators of the APIP promoter.
Another important observation from the 4C-seq in Calu3 cells (Figure 3B) is that the 11p13 modifier region is contained within a single TAD or sub-TAD, extending from the novel element 5′ to EHF (I, DHS HB11.1485) to the APIP intronic element (II). Critically, the APIP promoter lies outside these TAD boundaries. This suggests that 11p13 SNPs associated with CF lung disease severity and cis-regulatory elements within the EHF/APIP intergenic region are more likely to impact EHF expression than APIP.
Cell-type specific conformations organize the 11p13 locus
Since cis-regulatory elements are often cell-type specific, we next examined the 3D chromatin architecture of 11p13 in HBE cells, which express abundant EHF, and 16HBE14o- and K562 erythroleukemia cells in which the EHF locus is poorly expressed. 4C-seq was performed with viewpoints at the promoters of EHF (Figure 4A) and APIP (Figure 4B). The EHF promoter showed similar interactions in HBE cells to those seen in Calu3 cells at 11.2516, 11.2521 and the novel HB11.1485 (I) and II elements. Additional interactions were also seen at 11.2526 and near 11.2528. In contrast, associations with the EHF promoter in K562 cells were diffuse. Though a weak association with 11.2516 was evident, there appeared to be few specific points of strong interaction across the intergenic region. These results suggest that the looped 3D chromatin structure at 11p13 is associated with airway epithelial cell types, though no direct correlation with EHF expression levels is apparent. Additional data using different viewpoints in HBE (Supplementary Figure S4) and 16HBE14o- (Supplementary Figure S5) cells confirm these observations. There appear to be many more specific interactions across 11p13 in the HBE cells than are seen in the 16HBE14o- cell line, though in both cell types the enhancers at 11.2516 and 11.2521 interact with the EHF promoter and boundary sites I and II. In K562 cells, these same viewpoints showed few specific points of interaction across the locus, indicating a less active chromatin structure (Supplementary Figure S6).
Figure 4.

Chromatin architecture at 11p13 is cell type specific. 4C-seq data are presented as in Figure 3 and show Calu3, HBE, 16HBE14o- and K562 cells. Interactions are from (A) the EHF promoter viewpoint and (B) the APIP promoter viewpoint. Arrows denote features discussed in the results.
In contrast to the dynamic associations of active regulatory elements across 11p13, cis-interactions across the locus with the APIP promoter viewpoint are quite similar in diverse cell types, irrespective of EHF expression (Figure 4B, Supplementary Figures S4–S6). All cell types exhibit similar patterns of diffuse associations localized to both ends of the 11p13 interval with no strong interaction in the central intergenic region.
The 11p13 locus forms a TAD that is coordinated by CTCF
Regulatory elements are known to interact much more frequently with gene promoters in the same TAD than with those in adjacent TADs (39). Inspection of the Hi-C data in Figure 3 provides a guide to the likely organization of the chr11p13 region. Also our 4C-seq data shows strong interactions between an element in intron 3 of APIP and several sites between ELF5 and EHF. Together these data suggest the likely location of the TAD boundaries encompassing EHF and the high P-value SNPs from the GWAS. To confirm these predictions, 4C-seq was performed in Calu3 cells using viewpoints at the HB11.1485 site (see Figure 1) 5′ to EHF (site I) and site II within APIP intron 3. Strong interactions were seen between these 2 sites in Calu3 cells and both were also closely associated with a site in intron 6 of EHF and a site near 11.2525 at APIP +84 kb (Figure 5A), that lies very close to the highest P-value rs10742326 SNP. However, inspection of the limits of 5′ interaction with site II in both HBE (Supplementary Figure S4) and 16HBE14o- (Supplementary Figure S5) suggest these may extend to regions upstream of site I, particularly with DHS 11.1512 at ∼ –55 kb 5′ to EHF (see Figure 1).
Figure 5.

The topologically associating domain encompassing the 11p13 CF modifier region. (A) 4C-seq data are presented as described in Figure 3. Viewpoints are from the HB11.1485 (I) and APIP intron 3 (II) elements. (B) ChIP-qPCR for CTCF enrichment across the 11p13 locus in Calu3 cells. Amplicons are at 11.2512 (EHF -55kb), HB11.1485 (I), DHS 11.2516, EHF intron 6, 11.2521, APIP +84kb, APIP intron 2 (II), and APIP intron 1. Data are shown as percent recovery over input for CTCF (black bars) and IgG control (gray bars). Arrows denote CTCF motif orientation. n = 3. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05.
To further define the TAD structure at 11p13, we next investigated enrichment of CTCF across the 11p13 locus by using ChIP with a CTCF-specific antibody followed by quantitative PCR. CTCF is known to function at insulator elements and often defines the boundaries of TADs (41). A site 48.9 kb downstream of the CFTR translational stop site previously identified to have strong CTCF occupancy was used as a positive control (38), and a region on chromosome 11 showing no chromatin marks or transcription factor binding according to ENCODE was used as a negative control (20) (see Supplementary Table S1B for primers). In agreement with our hypothesis that elements I and II function as boundary elements, we found significant enrichment of CTCF at these sites in Calu3 (Figure 5B), 16HBE14o- and K562 cells (both in Supplementary Figure S7), as well as at the +48.9 kb CFTR site. In all three cell types, CTCF enrichment at site II was substantially higher than at site I [primers for site II were located ∼1 kb from the 4C-seq viewpoint at an ENCODE-documented CTCF site in APIP intron 2]. The APIP +84 kb element (near 11.2525), which displayed 4C interactions with the EHF promoter, element I, and element II, was also found to have significant CTCF enrichment in Calu3 and K562 cells, though not in 16HBE14o- cells. Other elements, such as 11.2516 and 11.2521, which we found to function as enhancer elements, did not show substantial CTCF enrichment in Calu3 cells, though 11.2516 had minor yet significant CTCF occupancy (Figure 5B). None of the enhancer sites bound CTCF in the other cell types assayed (Supplementary Figure S7). Since the 4C-seq boundaries of interaction with several viewpoints appeared to extend beyond sites I and II, we also evaluated CTCF occupancy at the neighboring sites predicted from ENCODE data (33,42–44). DHS 11.2512 (EHF-55kb) and a site in APIP intron 1 both showed highly significant levels of CTCF binding. These results concur with our 4C data and predict that the 11.2512/HB11.1485 and APIP intron 1/intron 2 sites are the invariant TAD or sub-TAD boundaries encompassing EHF and its regulatory elements at the 11p13 locus. Consistent with this hypothesis is the orientation of the CTCF motifs at 11.2512/HB11.1485 and APIP intron 1/intron 2 which are convergent (Figure 5B) (45–47). A redundancy in CTCF sites at TAD boundaries is often observed (28,48,49). However, the broad interactions observed upstream of 11.2512 indicate that site I may also have a structural role in cell type-specific regulation of ELF5.
Based on our data, we established a model for how the 11p13 region may function in vivo (Figure 6). In cell types, such as 16HBE14o-, Calu3, and HBE where the locus is active and EHF expressed, 11p13 is organized by CTCF into a TAD limited by the EHF upstream and APIP intronic elements. A third site, near 11.2525 (APIP +84 kb) aids in holding the 3D structure together, while enhancers 11.2516, 11.2521 and 11.2526 within the EHF/APIP intergenic region recruit the transcription factors that promote EHF expression. Though we show these enhancers also interact with the ELF5 gene promoter in reporter gene assays, this locus appears to be located within an adjacent TAD, so is less likely to be a target in vivo. The APIP/PDHX promoter lies outside of the main regulatory region, either requiring fewer or entirely different enhancer elements to drive gene expression. In contrast, the 3D organization of the 11p13 locus in cell types that do not express EHF, such as K562 cells, is much more compact. Though the TAD is invariant and reliant on CTCF binding, regulatory elements may remain buried in the chromatin.
Figure 6.
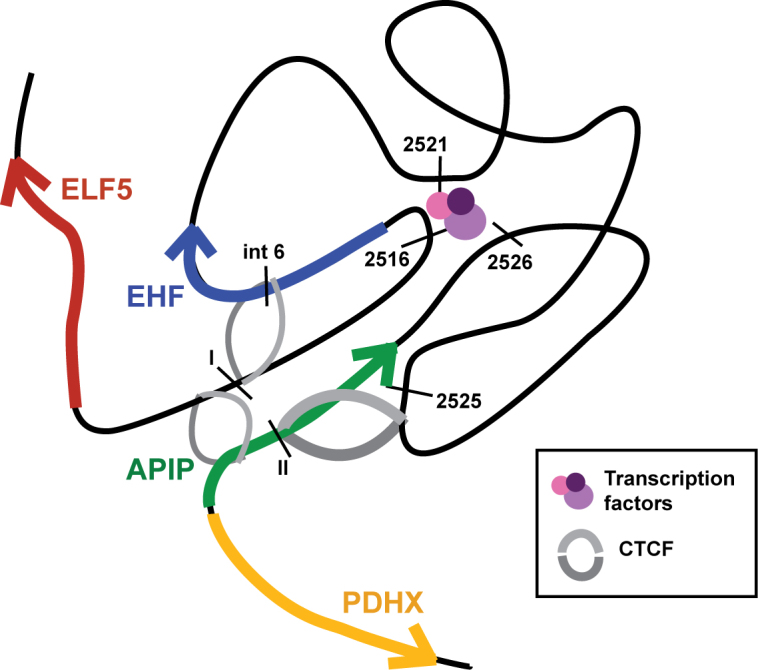
A model of the 11p13 region in lung epithelial cells. A putative model of the 11p13 locus in an active state, as found in Calu3 and primary HBE cells. A TAD is anchored by CTCF while chromatin is open and accessible to transcription factors that promote gene expression. Genes are represented by arrows, CTCF by grey semi-circles, transcription factors by colored circles, and cis-regulatory elements identified in the results section by black bars.
DISCUSSION
Using a combination of data generated de novo and by mining the ENCODE database (33) we reveal key features of the 11p13 region that encompasses a modifier of cystic fibrosis lung disease severity (13,14). Since the most significant SNPs lie in an intergenic interval and thus may influence regulatory elements, we specifically focused our studies on primary airway cells and airway cell lines. The genomic tools we used identified both ubiquitous features of 11p13 and airway-selective sites. Of note, two regions of open chromatin functioned as enhancer elements for the nearby EHF and ELF5 promoters in lung epithelial cell types. One of these sites (11.2516) is enriched for RNA polymerase (Pol2), as shown by ChIP-seq with an antibody specific for the largest subunit of Pol2 (Pol2–4H8; Myers, Hardison Labs, ENCODE (33)). This is consistent with the presence of enhancer RNAs (eRNAs) transcribed at these elements (50,51). Circular chromosome conformation capture experiments confirmed that these enhancers associated with each other in airway cells and also interacted with multiple cis-elements at the EHF locus including a site in the sixth intron that is similarly enriched for Pol2. In addition, 4C-seq revealed the topologically associating domains across the 11p13 region and showed that the EHF locus is separated from the APIP promoter by a boundary element located within the APIP gene body. The highest P-value SNPs in the GWAS lie within this TAD implicating aspects of EHF regulation and/or function to underlie the phenotypic association with lung disease severity.
In previous studies we identified EHF as a critical regulator of responses to injury in the lung (20). EHF may activate or repress gene expression, and it is enriched in intronic and intergenic regions coinciding with histone marks associated with enhancers (20). EHF depletion delays wound closure and increases transepithelial resistance of lung epithelial cells. Hence, alterations in its expression from the 11p13 locus due to regulatory variants could modify wounding responses in the CF lung. Furthermore, EHF is regulated by NF-κB signaling, which is a known mediator of chronic inflammation in CF (19,52). The CFTR p.Phe508del mutation has been shown to constitutively activate NF-κB through a misfolded protein stress response in the ER (53). Consistent with these observations are recent findings from our group and others showing that rare EHF alleles or EHF depletion impacts genes involved in protein folding and trafficking (54,55), thereby linking EHF regulation to p.PheF508del CFTR. Combined with recent findings that the 11p13 GWAS signal is driven by the F508del genotype (15), an axis combining mutant CFTR, enhanced NF-κB signaling, and altered EHF expression provides a likely explanation for disease modification in CF lung epithelia.
Our data support a primary role for EHF in modifying CF lung disease severity; however, it does not exclude the participation of other genes at 11p13 or nearby. To date, a search for expression quantitative trait loci (eQTL) using data from the Genotype-Tissue Expression (GTEx) project (56) (www.gtexportal.org) has not revealed any loci corresponding to GWAS I+II SNPs that significantly alter gene expression at the region. However, in several tissue types (including the lung), eQTLs were identified just 5′ to the GWAS SNPs that correlate with expression of APIP. Located towards the 5′ side of the EHF/APIP intergenic region and over the APIP gene body, the significant eQTLs do not overlap with the GWAS signal and therefore are unlikely to fall within regulatory elements or the main TAD we identified. It is still possible that SNPs altering levels of other non-coding RNAs (lncRNAs) could underlie the disease-association of the 11p13 region.
A search for causal variant(s) among the SNPs identified in the CF modifier GWAS has not proven fruitful. Overlapping the location of SNPs from the GWAS with chromatin features at 11p13 was not informative. The highest p-value SNP, rs10742326, coincides with a peak of H3K27ac in the Calu3 lung adenocarcinoma line but not in primary airway (bronchial or tracheal) cells. The nearest region of open chromatin (DHS) in any cell type is >2 kb away and the closet peaks of H3K4me1 are >3 kb proximal or distal to the site. Further, rs1074236 does not exactly coincide with any negative histone marks or with binding sites for the architectural proteins CTCF and cohesin. Altogether, these data suggest that the SNP is unlikely to create or destroy a key transcription factor binding site or impact enhancer activity. Thus, we predict that the top SNP(s) are indicative of the causative haplotype rather than conferring direct functionality. The high linkage disequilibrium (LD) across the 11p13 region and location of SNPs in LD with one another across regions of open chromatin supports this hypothesis.
Our data focus attention on the likely role of EHF in CF lung disease. Moreover, the identification of enhancers for this gene at 11p13 is consistent with a cell-specific expression profile for EHF. Though the two strong EHF enhancers we identified coincided with ubiquitous DHS, luciferase assays showed enhancer activity was restricted to lung epithelial cells. In contrast, no substantial enhancers of the APIP promoter were found in this genomic interval, as might be expected for a gene with constitutively high expression levels. In addition, a viewpoint at the APIP promoter revealed very few discrete sites of interaction across 11p13 in 4C-seq. Of course it is possible that other regulatory mechanisms for APIP are housed at 11p13 in some cell types, but after extensive data mining in the region, we have no supportive evidence for this.
The 4C-seq data generated with the EHF promoter viewpoint showed many highly specific interactions in several epithelial cell types, suggesting the existence of cis-elements that regulate EHF expression. These interacting elements include the 11.2516 and 11.2521 enhancers, but also other sites with currently undefined functions. This is particularly evident in lung epithelial cells where multiple sites of strong interaction are located within the EHF gene and correspond to an extended enhancer marked by broad H3K27Ac enrichment (34). In agreement, imputed chromatin state models from the Roadmap Epigenomics Project indicate active enhancer elements throughout a ∼50 kb region spanning EHF introns specifically in epithelial cell types (57).
Other novel chromatin interactions that we identified, particularly with viewpoints at APIP intron 3 and the EHF promoter, involved CTCF-binding elements across the region. As we and others observed previously, occupancy of CTCF at TAD boundaries is invariant between different cell types (28,39,41,58). However, other CTCF sites within the TAD show different levels of CTCF occupancy and associated interaction profiles in diverse cell types, consistent with a known role of CTCF in cell-specific intra-TAD loop formation (41,59,60). An example is the site at APIP +84 kb, which is highly enriched for CTCF in Calu3 cells but has very low occupancy in 16HBE14o- cells, where the intra-TAD interactions are less prominent. Moreover, airway epithelial cells that express abundant EHF have a different interaction profile than K562 cells where the gene is inactive. It is also possible that ELF5 and EHF exhibit coordinate regulation, which influences the chromatin conformation across 11p13. This would correlate well with both promoters being activated by the 11.2516 and 11.2516 elements in reporter gene assays and with additional interactions upstream of EHF identified by 4C.
Current data do not exclude the possibility that epithelial cells are not the functional cell type underlying the association of 11p13 with CF lung disease severity, and in those cell types EHF/ELF5 might not be the key loci. However, our results suggest that cis-regulatory elements at 11p13 are the key feature of the GWAS modifier region and posit EHF as their likely target.
ACCESSION NUMBERS
The GEO accession number for the data reported is GSE94726.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs M. Knowles, W. O’Neal and H. Dang for helpful discussions and sharing data on the 11p13 region prior to publication; also Drs C. Cotton and S. Randell for primary tracheal and bronchial epithelial cells; Drs A. Safi, L. Song and G. Crawford for generating DNAse-seq libraries.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01HL117843 to A.H., F31HL126458 to L.R.S., F30HL124925 to S.F.]; HBE cells were provided with support from NIH [P30 DK065988]; Cystic Fibrosis Foundation [BOUCHE15R0]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Manolio T.A. Genomewide association studies and assessment of the risk of disease. N. Engl. J. Med. 2010; 363:166–176. [DOI] [PubMed] [Google Scholar]
- 2. Visscher P.M., Brown M.A., McCarthy M.I., Yang J.. Five years of GWAS discovery. Am. J. Hum. Genet. 2012; 90:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Edwards S.L., Beesley J., French J.D., Dunning A.M.. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 2013; 93:779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J. et al. . Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gaulton K.J., Nammo T., Pasquali L., Simon J.M., Giresi P.G., Fogarty M.P., Panhuis T.M., Mieczkowski P., Secchi A., Bosco D. et al. . A map of open chromatin in human pancreatic islets. Nat. Genet. 2010; 42:255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kerem B., Rommens J.M., Buchanan J.A., Markiewicz D., Cox T.K., Chakravarti A., Buchwald M., Tsui L.C.. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989; 245:1073–1080. [DOI] [PubMed] [Google Scholar]
- 7. Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L.. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989; 245:1066–1073. [DOI] [PubMed] [Google Scholar]
- 8. Rommens J.M., Iannuzzi M.C., Kerem B., Drumm M.L., Melmer G., Dean M., Rozmahel R., Cole J.L., Kennedy D., Hidaka N.. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989; 245:1059–1065. [DOI] [PubMed] [Google Scholar]
- 9. Kerem E., Corey M., Kerem B.S., Rommens J., Markiewicz D., Levison H., Tsui L.C., Durie P.. The relation between genotype and phenotype in cystic fibrosis–analysis of the most common mutation (delta F508). N. Engl. J. Med. 1990; 323:1517–1522. [DOI] [PubMed] [Google Scholar]
- 10. The Cystic Fibrosis Genotype-Phenotype Consortium Correlation between genotype and phenotype in patients with cystic fibrosis. N. Engl. J. Med. 1993; 329:1308–1313. [DOI] [PubMed] [Google Scholar]
- 11. Kerem E., Reisman J., Corey M., Canny G.J., Levison H.. Prediction of mortality in patients with cystic fibrosis. N. Engl. J. Med. 1992; 326:1187–1191. [DOI] [PubMed] [Google Scholar]
- 12. Vanscoy L.L., Blackman S.M., Collaco J.M., Bowers A., Lai T., Naughton K., Algire M., McWilliams R., Beck S., Hoover-Fong J. et al. . Heritability of lung disease severity in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007; 175:1036–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wright F.A., Strug L.J., Doshi V.K., Commander C.W., Blackman S.M., Sun L., Berthiaume Y., Cutler D., Cojocaru A., Collaco J.M. et al. . Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet. 2011; 43:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Corvol H., Blackman S.M., Boëlle P.-Y., Gallins P.J., Pace R.G., Stonebraker J.R., Accurso F.J., Clement A., Collaco J.M., Dang H. et al. . Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 2015; 6:8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dang H., Gallins P.J., Pace R.G., Guo X.-L., Stonebraker J.R., Corvol H., Cutting G.R., Drumm M.L., Strug L.J., Knowles M.R. et al. . Novel variation at chr11p13 associated with cystic fibrosis lung disease severity. Hum. genome Var. 2016; 3:16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reich D.E., Cargill M., Bolk S., Ireland J., Sabeti P.C., Richter D.J., Lavery T., Kouyoumjian R., Farhadian S.F., Ward R. et al. . Linkage disequilibrium in the human genome. Nature. 2001; 411:199–204. [DOI] [PubMed] [Google Scholar]
- 17. Cho D.-H., Hong Y.-M., Lee H.-J., Woo H.-N., Pyo J.-O., Mak T.W., Jung Y.-K.. Induced inhibition of ischemic/hypoxic injury by APIP, a novel Apaf-1-interacting protein. J. Biol. Chem. 2004; 279:39942–39950. [DOI] [PubMed] [Google Scholar]
- 18. Kang W., Hong S.H., Lee H.M., Kim N.Y., Lim Y.C., Le L.T.M., Lim B., Kim H.C., Kim T.Y., Ashida H. et al. . Structural and biochemical basis for the inhibition of cell death by APIP, a methionine salvage enzyme. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E54–E61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu J., Duan R., Cao H., Field D., Newnham C.M., Koehler D.R., Zamel N., Pritchard M.A., Hertzog P., Post M. et al. . Regulation of epithelium-specific Ets-like factors ESE-1 and ESE-3 in airway epithelial cells: potential roles in airway inflammation. Cell Res. 2008; 18:649–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fossum S.L., Mutolo M.J., Yang R., Dang H., O’Neal W.K., Knowles M.R., Leir S.-H., Harris A.. Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res. 2014; 42:13588–13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oettgen P., Kas K., Dube A., Gu X., Grall F., Thamrongsak U., Akbarali Y., Finger E., Boltax J., Endress G. et al. . Characterization of ESE-2, a novel ESE-1-related Ets transcription factor that is restricted to glandular epithelium and differentiated keratinocytes. J. Biol. Chem. 1999; 274:29439–29452. [DOI] [PubMed] [Google Scholar]
- 22. Shen B.Q., Finkbeiner W.E., Wine J.J., Mrsny R.J., Widdicombe J.H.. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl- secretion. Am. J. Physiol. 1994; 266:L493–L501. [DOI] [PubMed] [Google Scholar]
- 23. Cozens A.L., Yezzi M.J., Kunzelmann K., Ohrui T., Chin L., Eng K., Finkbeiner W.E., Widdicombe J.H., Gruenert D.C.. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994; 10:38–47. [DOI] [PubMed] [Google Scholar]
- 24. Fogh J., Wright W.C., Loveless J.D.. Absence of HeLa cell contamination in 169 cell lines derived from human tumors. J. Natl. Cancer Inst. 1977; 58:209–214. [DOI] [PubMed] [Google Scholar]
- 25. Reddel R.R. Immortalized human bronchial epitherial mesothelial cell lines. 1989.
- 26. Lozzio C.B., Lozzio B.B.. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood. 1975; 45:321–334. [PubMed] [Google Scholar]
- 27. Phylactides M., Rowntree R., Nuthall H., Ussery D., Wheeler A., Harris A.. Evaluation of potential regulatory elements identified as DNase I hypersensitive sites in the CFTR gene. Eur. J. Biochem. 2002; 269:553–559. [DOI] [PubMed] [Google Scholar]
- 28. Yang R., Kerschner J.L., Gosalia N., Neems D., Gorsic L.K., Safi A., Crawford G.E., Kosak S.T., Leir S.-H., Harris A.. Differential contribution of cis-regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res. 2016; 44:3082–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bischof J.M., Ott C.J., Leir S.-H., Gosalia N., Song L., London D., Furey T.S., Cotton C.U., Crawford G.E., Harris A.. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax. 2012; 67:385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E.. High-Resolution Mapping and Characterization of Open Chromatin across the Genome. Cell. 2008; 132:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ott C.J., Blackledge N.P., Kerschner J.L., Leir S.-H., Crawford G.E., Cotton C.U., Harris A.. Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:19934–19939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van de Werken H.J.G., Landan G., Holwerda S.J.B., Hoichman M., Klous P., Chachik R., Splinter E., Valdes-Quezada C., Oz Y., Bouwman B.A.M. et al. . Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods. 2012; 9:969–972. [DOI] [PubMed] [Google Scholar]
- 33. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parker S.C.J., Stitzel M.L., Taylor D.L., Orozco J.M., Erdos M.R., Akiyama J.A., van Bueren K.L., Chines P.S., Narisu N., Black B.L. et al. . Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rubio E.D., Reiss D.J., Welcsh P.L., Disteche C.M., Filippova G.N., Baliga N.S., Aebersold R., Ranish J.A., Krumm A.. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8309–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H.C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T. et al. . Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008; 132:422–433. [DOI] [PubMed] [Google Scholar]
- 37. Wendt K.S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T. et al. . Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008; 451:796–801. [DOI] [PubMed] [Google Scholar]
- 38. Gosalia N., Neems D., Kerschner J.L., Kosak S.T., Harris A.. Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res. 2014; 42:9612–9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dekker J., Mirny L.. The 3D genome as moderator of chromosomal communication. Cell. 2016; 164:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. de Wit E., Braunschweig U., Greil F., Bussemaker H.J., van Steensel B.. Global chromatin domain organization of the Drosophila genome. PLoS Genet. 2008; 4:e1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.-K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R. et al. . Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y. et al. . Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012; 22:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J., Zhuang J., Iyer S., Lin X.-Y., Greven M.C., Kim B.-H., Moore J., Pierce B.G., Dong X., Virgil D. et al. . Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013; 41:D171–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Narendra V., Bulajić M., Dekker J., Mazzoni E.O., Reinberg D.. CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev. 2016; 30:2657–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. . A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo Y., Xu Q., Canzio D., Shou J., Li J., Gorkin D.U., Jung I., Wu H., Zhai Y., Tang Y. et al. . CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015; 162:900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guo Y., Xu Q., Canzio D., Shou J., Li J., Gorkin D.U., Jung I., Wu H., Zhai Y., Tang Y. et al. . CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015; 162:900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Van Bortle K., Nichols M.H., Li L., Ong C.-T., Takenaka N., Qin Z.S., Corces V.G.. Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol. 2014; 15:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. De Santa F., Barozzi I., Mietton F., Ghisletti S., Polletti S., Tusi B.K., Muller H., Ragoussis J., Wei C.-L., Natoli G.. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010; 8:e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim T.-K., Hemberg M., Gray J.M., Costa A.M., Bear D.M., Wu J., Harmin D.A., Laptewicz M., Barbara-Haley K., Kuersten S. et al. . Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010; 465:182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bodas M., Vij N.. The NF-kappaB signaling in cystic fibrosis lung disease: pathophysiology and therapeutic potential. Discov. Med. 2010; 9:346–356. [PMC free article] [PubMed] [Google Scholar]
- 53. Knorre A., Wagner M., Schaefer H.-E., Colledge W.H., Pahl H.L.. DeltaF508-CFTR causes constitutive NF-kappaB activation through an ER-overload response in cystic fibrosis lungs. Biol. Chem. 2002; 383:271–282. [DOI] [PubMed] [Google Scholar]
- 54. Stanke F., van Barneveld A., Hedtfeld S., Wölfl S., Becker T., Tümmler B.. The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells. Eur. J. Hum. Genet. 2014; 22:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fossum S.L., Mutolo M.J., Tugores A., Ghosh S., Randell S., Jones L.C., Leir S.-H., Harris A.. Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J. Biol. Chem. 2017; doi:10.1074/jbc.M117.775304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N. et al. . The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J. et al. . Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Smith E.M., Lajoie B.R., Jain G., Dekker J.. Invariant TAD boundaries constrain cell-type-specific looping interactions between promoters and distal elements around the CFTR locus. Am. J. Hum. Genet. 2016; 98:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Splinter E., Heath H., Kooren J., Palstra R.-J., Klous P., Grosveld F., Galjart N., de Laat W.. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2006; 20:2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hou C., Dale R., Dean A.. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:3651–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.