


Abstract
Purification of cell type-specific RNAs remains a significant challenge. One solution involves biosynthetic tagging of target RNAs. RNA tagging via incorporation of 4-thiouracil (TU) in cells expressing transgenic uracil phosphoribosyltransferase (UPRT), a method known as TU-tagging, has been used in multiple systems but can have limited specificity due to endogenous pathways of TU incorporation. Here, we describe an alternative method that requires the activity of two enzymes: cytosine deaminase (CD) and UPRT. We found that the sequential activity of these enzymes converts 5-ethynylcytosine (EC) to 5-ethynyluridine monophosphate that is subsequently incorporated into nascent RNAs. The ethynyl group allows efficient detection and purification of tagged RNAs. We show that ‘EC-tagging’ occurs in tissue culture cells and Drosophila engineered to express CD and UPRT. Additional control can be achieved through a split-CD approach in which functional CD is reconstituted from independently expressed fragments. We demonstrate the sensitivity and specificity of EC-tagging by obtaining cell type-specific gene expression data from intact Drosophila larvae, including transcriptome measurements from a small population of central brain neurons. EC-tagging provides several advantages over existing techniques and should be broadly useful for investigating the role of differential RNA expression in cell identity, physiology and pathology.
INTRODUCTION
Cell type-specific transcription is an essential determinant of cell fate and function. While techniques that quantify mRNAs (RNA-seq, microarrays) allow investigation of gene expression, the quality and type of information obtained may be limited by the method of RNA purification. Ideally, cell type-specific RNA should be obtained under in vivo conditions, with no physical alteration of tissues. Additionally, analysis of newly transcribed mRNA is often more informative than analysis of bulk mRNA: newly transcribed mRNA can be used to determine synthesis and decay rates (1,2) and reveal rare transcripts (2). Techniques for obtaining cell type-specific mRNA generally fall into two categories: physical isolation or tagging and capture of RNAs (3). Methods of physical isolation (fluorescence-activated cell sorting (4), laser-capture microdissection (5), INTACT (6)) disrupt the cell’s environment and may affect mRNA transcription or decay. Methods of RNA tagging and capture often use mRNA-binding proteins that allow purification of bulk poly(A) mRNAs (7) or translating mRNAs (8), but do not enrich for newly transcribed mRNAs and miss non-coding RNAs (3).
TU-tagging is a cell type-specific RNA tagging method that allows analysis of newly transcribed RNAs (9,10) and has the potential to purify noncoding RNAs (11). TU-tagging relies on cell type-specific expression of uracil phosphoribosyltransferase (UPRT) to convert a modified uracil, 4-thiouracil (TU), into 4-thiouridine (4sUd) monophosphate that is subsequently incorporated into nascent RNAs. TU-tagging has been used to study cell type-specific gene expression in Drosophila (10,12), zebrafish (13,14), mammalian tissue culture cells (15) and mice (16,17). TU-tagging has also been used to measure cell type-specific mRNA decay in Drosophila embryos (18). While this technique has proven useful in many systems, the specificity of TU-tagging is limited in some cases. UPRT activity is primarily found in bacteria, fungi and protozoans but metazoan cells may salvage uracil via alternative pathways (potentially through the sequential activity of uridine phosphorylase and uridine kinase) (19) and an endogenous UPRT was recently identified in Drosophila (20). Another limitation of TU-tagging is the relative inefficiency of RNA purification based on disulfide bond formation, although optimized methods have been described (21). In contrast to thiol-containing nucleosides, other orthogonal handles may be more robust for RNA enrichment (22,23). The need for novel approaches for cell type-specific biosynthetic RNA tagging necessitates expanding the chemical toolkit and manipulating alternative metabolic pathways, all while achieving stringent cell type-specificity.
The cytosine deaminase (CD) enzyme is unique to bacteria and yeast: animals lack cytosine deaminase activity (24). Cytosine deaminase converts the ribonucleobase cytosine into uracil and the combined activity of CD and UPRT results in conversion of cytosine into uridine monophosphate. The CD-UPRT pathway has been used in suicide gene approaches where mammalian cells expressing CD and UPRT convert 5-fluorocytosine (5FC) into the cytotoxic nucleotide 5-fluorouridine monophosphate (5FUdMP) (25). 5FUdMP toxicity is primarily caused by inhibition of thymidylate synthetase and impaired DNA synthesis, although 5-fluorouridine triphosphate is also incorporated into tRNA and may interfere with tRNA aminoacylation (26). While 5FUdMP is cytotoxic, the nucleoside 5-ethynyluridine (5EUd) is a RNA polymerase substrate that is generally well tolerated by cells (27) (toxicity is only observed after prolonged exposure (28)). Additionally, the ethynyl group of 5EUd allows efficient click chemistry-based labeling and purification of RNA (29). We reasoned that the modified nucleobase 5-ethynylcytosine (5EC) might be useful for RNA tagging: if 5EC is a CD substrate (allowing production of 5-ethynyluracil (5EU)) and 5EU is a UPRT substrate (allowing production of 5-ethynyluridine monophosphate (5EUdMP), then 5EC could allow cell type-specific RNA tagging via the CD-UPRT pathway.
Here, we describe RNA tagging via the combination of 5EC exposure and cell type-specific expression of CD and UPRT. We call this technique ‘EC-tagging’ and demonstrate the specificity and sensitivity of EC-tagging by obtaining cell type-specific transcriptome data from distinct cell populations in Drosophila.
MATERIALS AND METHODS
5-ethynylcytosine synthesis
5EC was prepared by coupling 5-iodocytosine (1) with trimethylsilylacetylene under Sonogashira conditions to afford the intermediate 2 in 87% yield (Scheme 1) (30). The trimethylsilyl protecting group was removed with concentrated aqueous NH3 to give EC. The intermediate (2) and final product were spectroscopically characterized and the data for EC matched that reported.
Scheme 1.

5-ethynylcytosine synthesis.
Preparation of 2
A mixture of 5-iodocytosine (1, 105 mg, 0.44 mmol), Pd(PPh3)2Cl2 (15 mg, 0.021 mmol), CuI (8 mg, 0.042 mmol), anhydrous triethylamine (268 mg, 2.65 mmol) and 0.8 ml of dry dimethylformamide was degassed by bubbling argon at 25°C for 1 h. Trimethylsilyl acetylene (152 mg, 1.55 mmol) was added and the mixture was stirred at 25°C for 45 min. The reaction mixture was diluted with MeOH and filtered (5 ml). The precipitate was washed with H2O (3 × 10 ml), acetone (2 × 5 ml) and dried to give 2 as an off-white solid: yield 80 mg (87%). 1H NMR (dimethylsulfoxide-d6) δ 11.11 − 10.67 (m, 1H), 7.73 (s, 2H), 6.61 − 6.30 (m, 1H), 0.20 (s, 9H).
Preparation of 5EC
A suspension of 70 mg (0.34 mmol) of 2 in concentrated aqueous NH3 (2 ml) and MeOH (0.5 ml) was stirred in a sealed reaction vessel for 2 days. The reaction mixture was concentrated under reduced pressure to obtain 3 as a brown solid: yield 40 mg (87%). 1H NMR (DMSO-d6) δ 11.10 – 10.46 (m, 1H), 7.74 (s, 1H), 7.71 – 7.41 (m, 1H), 6.81 – 6.52 (m, 1H), 4.30 (s, 1H).
5-ethynyluracil synthesis
Preparation of 5EU
5EU synthesis steps are summarized in Scheme 2. 5-Iodouracil (1, 2000 mg, 8.4 mmol, 1 eq), TMS-acetylene (2.4 ml, 16.8 mmol, 2 eq), Et3N (4.7 ml, 33.6 mmol, 4 eq), Pd(PPh3)4 (196mg, 0.17 mmol, 0.02 eq) and CuI (65 mg, 0.34 mmol, 0.04 eq) were dissolved in 25 ml of degassed EtOAc. The suspension was stirred at r.t. for 3 h under Ar. The suspension was then filtered and washed with EtOAc. The extract was collected and dissolved in 10 ml of 1 M NaOH and stirred at r.t. for 2 h. The solution was then diluted with 10 ml of H2O and concentrated in vacuo. The residue was then redissolved in 10 ml of H¬2O and AcOH was added until a pH of 5 was reached. The suspension was then set on ice for 30 min and filtered. The extract was washed with H2O, acetone and Et2O. The extract was then dried in vacuo to give 5EU (823 mg, 72%) as an off white solid. Spectra are in agreement with those reported in the literature previously (31). HRMS Calcd for C6H4N2O2 [M-H-] 135.0195, found 135.0195; 1H NMR (400 MHz, DMSO) δ 11.29 (s, 2H), 7.78 (s, 1H), 3.99 (s, 1H).
Scheme 2.

5-ethynyluracil synthesis.
Cell culture and expression constructs
HeLa cells (American Type Culture Collection) and SH-SY5Y human neuroblastoma cells (provided by M. Kitazawa) were cultured using standard methods and transfected using Lipofectamine (ThermoFisher). A Saccharomyces cerevisiae (S.c.) CD:UPRT fusion construct, pSELECT-zeo-FcyFur (InvivoGen), was used to polymerase chain reaction (PCR) amplify S.c.CD, S.c.UPRT and S.c.CD:UPRT for the following constructs: pcDNA3.3_HA-CD:UPRT, pcDNA3.3_GFP-S.c.CD, pcDNA3.3_GFP-S.c.CD:UPRT and pcDNA3.1(zeo)_mCherry-S.c.UPRT. Split CD constructs were made by chemical synthesis (IDT) of the N-terminal CD(A23L)1–77 and C-terminal CD(V108I, I140L, T95S, K177E)57–158 fragments. The N-terminal CD was fused to C-terminal leucine zipper sequence ALKKELQANKKELAQLKWELQALKKELAQ and the C-terminal CD was fused to N-terminal leucine zipper sequence: EQLEKKLQALEKKLAQLEWKNQALEKKLAQ (32). The leucine zipper-fused split CD fragments were sub-cloned into pcDNA3.3.
Drosophila genetics
pUAS-HA-CD:UPRT-attB constructs (containing either a N-terminal HA-tagged S. cerevisiae S.c. CD:UPRT fusion gene or a Drosophila codon-optimized CD:UPRT fusion gene) were used to generate second and third chromosome UAS-CD:UPRT lines for both the S.c.CD:UPRT and optimized CD:UPRT. The following Gal4 lines were obtained from the Bloomington Drosophila Stock Center: Act5C-Gal4 (#25374), GMR12B08-Gal4 (#48489), ppk-Gal4 (#32078 and #32079), MB247-Gal4 (#50742), Canton-S-iso2B (#9514), da-Gal4; da-Gal4 (#55849), GMR32C12-Gal4 (#49708) and 10XUAS-IVS-mCD8-GFP (#32185). TH-Gal4 was provided by F. Wolf.
RNA-sequencing human cell lines
Library preparation (using oligo-dT priming), RNA-sequencing and data analysis were performed using the Beijing Genome Institute's RNA-Seq. service. Gene ontology (GO) analysis of differentially expressed genes was performed using DAVID (33) and only categories with an enrichment ≥ 2.0 and Bonferonni-corrected P-values of less than 0.001 were considered significant.
5EC toxicity assays
Cell lines
A total of 5000 control HeLa cells or CD:UPRT(+) HeLa cells were added per well in a 96-well plate. Cells were pre-incubated for 24 h prior to addition of the indicated concentration of 5EC or 5FC (InvivoGen). At the indicated timepoints, 10 μl of Cell Counting Kit-8 (CCK-8) solution (Sigma) was added to each well and 450 nm absorbance was measured 1 h. Percent cell viability was calculated as A450 nm treated cells/A450 nm untreated control ×100.
Flies
Act5C-Gal4 / CyO; UAS-CD:UPRT larvae and Canton-S controls were hatched onto standard fly media with or without 500 μM 5EC. Larval mortality was counted every 24 h.
5EC/5EUd treatment, RNA biotinylation and EU-RNA detection
5EC and 5EUd (ThermoFisher) were suspended in DMSO and added to cell culture media or yeast-free Drosophila media at the indicated concentrations. Drosophila media must be yeast-free to avoid yeast converting 5EC to 5EUd and transfer of 5EUd to Drosophila that ingest the yeast. Total RNA extraction was performed using the standard Trizol method. RNA to be biotinylated was first treated with RNAse-free DNAse (Qiagen) followed by RNeasy Mini column (Qiagen) clean-up. RNA biotinylation was performed using PEG4-carboxamide-6-azidohexanyl-biotin and Click-iT reagents according the manufacturer’s protocol (ThermoFisher) or biotin-dPEG7-azide (Sigma-Aldrich) and custom reagents as previously described (23). For Click-iT-based biotinylation, 10–30 μg of input RNA was mixed with 25 μl Click-iT EU buffer (buffer B), 4 μl CuSO4 and PEG4-carboxamide-6-azidohexanyl-biotin at a concentration of 1 mM (final volume adjusted to 47.25 μl with RNAse free water) then mixed by pipetting before adding 1.25 μl Click-iT EU reaction buffer additive 1 (buffer E). The reaction was immediately mixed by pipetting and incubated 3 min at room temperature before adding 1.5 μl Click-iT EU reaction buffer additive 2 (buffer F) followed by a final round of mixing by pipetting. For the custom biotinylation reaction, 10–30 μg of input RNA (up to 34 μl) was mixed with 5 μl of 20 mg/ml tris-(3-hydroxypropyltriazolylmethyl) amine (THPTA), 1 μl of 100 mM CuSO4, 5 μl of 200 mM sodium ascorbate, 5 μl of 10 mM biotin-dPEG7-azide and RNAse-free water to adjust the final volume to 50 μl. In both cases, the biotinylation reaction was incubated in a thermomixer at 700 rpm, 25°C for 30–45 min. The biotinylation reaction was stopped with addition of 450 μl HEPES buffer (10 mM HEPES pH7.5, 1 mM ethylenediaminetetraacetic acid (EDTA)) and 500 μl of chloroform followed by vigorous mixing, transfer to Phase Lock Gel Heavy tubes (ThermoFisher) and centrifugation at 16 000 × g for 10 min at 4°C. The aqueous phase was subjected to a second round of chloroform extraction using Phase Lock Gel Heavy tubes and centrifugation at 16 000 × g for 10 min at 4°C. RNA was precipitated from the final aqueous phase by adding 50 μl 5M NaCl and 450 μl isopropanol, mixing well, incubating at room temperature for a minimum of 10 min, then centrifuging at 12 000 × g for a minimum of 15 min at 4°C. Pelleted RNA was washed with 1.0 ml 75% ethanol twice then re-suspended in 20–30 μl RNAse-free water.
RNA blot detection of biotinylated RNA with streptavidin-HRP was performed as previously described (10). Dot blots and slot blots were loaded with either 1 or 5 μg of total RNA (equal loading for samples being compared on a single blot) following the biotinylation reaction and clean-up. RNA-transfer blots were loaded with 10 μg of total RNA treated with an equal volume of NorthernMax-Gly Sample Loading Dye (Ambion) following standard northern blot protocol.
EU-RNA capture on streptavidin beads
Biotinylated EU-RNA was captured using Dynabeads MyOne Strepatvidin T1 (Invitrogen) with 50 μl of beads for 20 μg of biotinylated input RNA (roughly equivalent to the biotin-reacted RNA obtained from 20–30 third instar larvae). The following buffers were used, based on a previous protocol (34): Solution A (0.1 M NaOH, and 0.05 M NaCl); Solution B (0.1 M NaCl); Tris.HCL-NaCl-EDTA (TNE) 2.0 buffer (10 mM Tris.HCl pH 7.5, 1 mM EDTA, and 2 M NaCl); Blocking & Washing (B&W) Buffer (5 mM Tris.HCl pH 7.5, 0.5 mM EDTA and 1 M NaCl); TNE 0.2 Buffer (10 mM Tris.HCl pH 7.5, 1 mM EDTA and 200 mM NaCl); and Wash Buffer 65 (100 mM Tris.HCl pH 7.5, 10 mM EDTA, 1 M NaCl and 0.1% Tween 20). Prior to adding RNA, beads were washed twice with B&W buffer at room temperature; twice with Solution A at room temperature; twice with Solution B at room temperature; twice with TNE 2.0 buffer at room temperature; once with Wash 65 Buffer at 65°C with vigorous mixing; and twice with TNE 0.2 at room temperature. Beads were subsequently incubated in a blocking solution (10 mM Tris.HCl, pH 7.5; 1 mM EDTA; 0.2 M NaCl; 2 mg/ml nuclease-free bovine serum albumin; and 1 μg/ml poly(deoxyinosinic-deoxycytidylic) acid) for 24 h at 4°C. Following the blocking steps, beads were washed three times with B&W buffer. RNA samples were denatured at 70°C for 5 min followed by an incubation period of 3 min on ice. Denatured RNA was incubated with the blocked beads in a mixture of B&W buffer, 2 μl of RNAseOUT Recombinant Ribonuclease Inhibitor (Invitrogen) and nuclease-free water for a final volume of 2.5 ml (for 10 μg of RNA) or 5 ml (for 20 μg of RNA). The RNA and beads mixture were incubated in the dark at room temperature for 30–45 min with gentle rotation to prevent the beads from settling. Beads were then subjected to a number of high stringency washes (1.0 ml each) to remove non-biotinylated RNA: four washes with TNE 2.0 at room temperature; four washes with B&W buffer at room temperature; four washes with Wash 65 Buffer at 65°C with vigorous mixing; and four washes with TNE 0.2 buffer at 65°C with vigorous mixing. After the last wash, the beads were directly used for RT-qPCR or cRNA synthesis.
TU-tagging
Larvae were fed 1.0 mM 4-thiouracil (Sigma-Aldrich) or 1.0 mM 4sUd (Sigma-Aldrich) as previously described (10). Whole larvae were used for total RNA extraction using Trizol reagent. Total RNA was reacted with MTSEA-biotin (Biotium) followed by streptavidin-bead purification and elution according to published protocols (21). A total of 20 μg of input RNA was used for all purifications (matching input amounts used for EC-tagging experiments) and 100 ng of eluted TU-RNA was used for cRNA probe generation, as described in the ‘Microarrays’ section.
RT-qPCR
Real-time PCR quantitation was performed on a Rotor-Gene Q (Qiagen) in 20 μl reactions using QuantiTect Primer Assays (Qiagen) and SYBR green detection. Ct values were normalized to an RpL32 internal reference and relative abundance calculated by the equation, fold-change = 2−Δ(ΔCt). RT-qPCR analysis was performed on biological replicate samples with cDNA synthesis performed using EU-RNA on beads for one sample and cDNA synthesis performed on eluted EU-RNA for the other sample. Dynabeads with bound EU-RNA or eluted EU-RNA were used to make cDNA using the SuperScript VILO cDNA Synthesis Kit (Invitrogen). For EU-RNA on beads, cDNA synthesis was performed with the recommended reaction volume scaled to 50 μl: After the final EU-RNA purification wash, the beads were re-suspended in 25 μl of TNE 0.2 buffer (10 mM Tris.HCl pH 7.5, 1 mM EDTA and 200 mM NaCl). A total of 10 μl 5× VILO reaction mix was then added to the bead solution, mixed by pipetting and incubated at 25°C for 10 min with continuous mixing on a thermomixer to prevent the beads from settling. Afterward, 10 μl RNAse-free water and 5 μl Superscript enzyme mix were added to the reaction then mixed by pipetting. The reaction was incubated at 42°C for 1 h with continuous mixing on a thermomixer. The reaction was then heated to 85°C for 5 min to terminate cDNA synthesis and to release cDNA from the beads. This cDNA was directly used in qPCR reactions. An alternative approach (used for one of the replicate RT-qPCR experiments) is to elute EU-RNA from the beads using an elution buffer (20 mM Tris.HCl, 1 mM EDTA, 0.5% SDS, 1 mM d-Biotin (Sigma-Aldrich), and 20 U Proteinase K (Life Technologies)), based on a previously described protocol (35). For elution, beads with bound EU-RNA were incubated in 300 μl of elution buffer for 30 min with vigorous mixing on a thermomixer at 65°C. Beads were then collected by magnet and the supernatant was aliquoted to a new tube. EU-RNA was extracted from the supernatant once with a mixture of 3M sodium acetate and acid phenol/chloroform; and twice with chloroform. EU-RNA was then precipitated with isopropanol and 2 μl of linear polyacrylamide (20 mg/ml). Pellets were washed twice with 75% ethanol and resuspended in nuclease-free water.
Microarrays
The Low Input Quick Amp Labeling Kit (Agilent) was used to make Cy3-labeled cRNA from EU-RNA bound to beads or 100 ng of eluted TU-RNA. cRNA synthesis from eluted TU-RNA followed the manufacturer’s protocol. The protocol was slightly modified for cRNA synthesis from EU-RNA on beads. Following the final EU-RNA purification step (described above), ∼6 μl of EU-RNA and beads remained. The EU-RNA plus beads mixture was combined with 3 μl of diluted One-Color Spike-In Control RNA (Agilent) and 5.4 μl of T7 primer mix. All subsequent cDNA synthesis steps were performed per the manufacturer’s protocol, but all reagent volumes were increased 3-fold: 6 μl 5× first-strand buffer, 3 μl dithiothreitol, 1.5 μl dNTP mix, 3.6 μl Affinity Script RNAse block mix. The cDNA synthesis reaction was incubated at 40°C for 2 h with continuous mixing on a thermomixer followed by incubation at 85°C for 5 min then immediate bead collection in the magnetic stand to remove the cDNA solution (∼22–25 μl cDNA). This cDNA was then used for cRNA synthesis according to the manufacturer’s protocol, with transcription mix reagent volumes increased 3-fold: 9.6 μl 5× transcription buffer, 1.8 μl DTT, 3 μl nucleotide triphosphate mix, 0.63 μl T7 RNA polymerase blend, 0.72 μl Cy3-CTP and RNAse-free water to bring final volume to 48 μl. cRNA purification for TU-RNA and EU-RNA samples was performed using RNeasy Mini Kit columns (Qiagen). Microarray analysis was performed using Agilent 4 × 44k Gene Expression Microarrays. All microarray data are based on pooled biological replicate RNA samples that were subsequently used for independent processing in duplicate microarrays (independent RNA biotinylation, EU-RNA or TU-RNA purification, and cRNA synthesis). A total of 1.65 μg of cRNA was hybridized to microarrays at 65°C for 17 h followed by washing according to standard Agilent protocols. Microarrays were scanned with a GenePix 4000B scanner. Post-processing of microarray data was performed using the computing environment R. Spots with fluorescence intensity <66% above background were excluded. Spot fluorescence minus background fluorescence signal was normalized by first excluding the bottom 10% lowest fluorescence spots and the top 10% highest fluorescence spots, calculating the mean fluorescence for the remaining spots, then applying a normalization factor to all spots so that the mean signal is equivalent across all microarrays directly compared to each other. Average signal intensity per spot was calculated from biological replicate microarrays and then used to calculate target EU-RNA/whole larvae EU-RNA (5EUd-fed) ratios and target TU-RNA/whole larvae TU-RNA (4sUd-fed) ratios. Ratios per spot were then used to calculate the average ratio and standard deviation per gene (most genes are represented by multiple spots on the microarray). For determination of enrichment in Gal4-driver targeted populations, spots with 12B08 EU-RNA, MB EU-RNA or MB TU-RNA normalized signal intensity <300 (cutoff determined by analysis of negative control genes that are not transcribed in larvae) were excluded from the per gene ratio calculations. For determining depletion, spots with whole larvae EU-RNA or whole larvae TU-RNA normalized signal intensity <300 were excluded from the per gene ratio calculations. All raw and normalized microarray data are available through the NCBI GEO series record GSE94346.
Transcriptome data analysis
GO analysis of Drosophila data (Figures 4 and 5) was performed using GO-Term Finder (36) and only included named genes (genes known only by an annotation symbol or ‘CG number’ were excluded as they tend to lack ontology information). Only categories with an enrichment ≥2.0 and Bonferonni-corrected P-values of <0.001 were considered significant. Redundant GO categories were identified based on nearly identical gene lists and similar category names. Alignment of all non-redundant significantly enriched GO categories from the MB247 and 12B08 datasets allowed identification of the overlapping and non-overlapping GO categories summarized in Figure 5D and listed in Supplementary Table S4. Positive control mushroom body genes shown in Figure 5A were selected from the Crocker et al. (37) list of neurotransmitter, neurotransmitter receptor, peptide and peptide receptor genes enriched in adult gamma mushroom body neurons with a z-score ≥ 0 (44 genes, as reported in Figure 4 of Crocker et al. (37)). From this set of 44, genes with microarray signal below background (no spots with normalized fluorescence intensity minus background >300) for the 12B08 EU-RNA dataset were excluded (as these may represent adult mushroom body-specific genes), yielding 25 genes for analysis. Negative control muscle genes shown in Figure 5A were selected from the Schnorrer et al. (38) list of embryonic RNAi targets (77 genes, as reported in Supplementary Table S3 of Schnorrer et al. (38)). From this set of 77, genes with microarray signal below background (no spots with normalized fluorescence intensity minus background >300) in the whole larvae EU-RNA dataset were excluded (as these may not be expressed or only weakly expressed in larvae) yielding 56 genes. An additional seven genes were removed from the muscle negative control set since they were also detected in adult mushroom body gamma neurons by RNA-seq (37), yielding 49 genes for analysis.
Figure 4.

Comparison of EC-tagging and dissection-based transcriptome profiling. (A) Expected specificity of RNA tagging in 12B08 > CD:UPRT larvae fed 5EC versus larvae fed 5EUd. (B) GO categories over-represented among 12B08 EU-RNA enriched and depleted genes. Observed/expected value = frequency of category genes in 12B08 EU-RNA/frequency in the Drosophila genome for the 1279 genes enriched in 12B08 EU-RNA (plotted as positive values) and the 405 genes depleted in 12B08 EU-RNA (plotted as negative values). Heatmap = Bonferroni-corrected P-values. (C) Relative expression levels for 609 genes enriched ≥2-fold in 12B08 EU-RNA with corresponding modENCODE Anatomy RNA-seq data. 12B08/whole larvae EU-RNA ratios are plotted on the y-axis and modENCODE CNS/carcass or imaginal disc/carcass ratios (the tissue with the highest RNA-seq expression level was selected) is plotted on the x-axis. The dashed line indicates 1.5-fold enrichment in the modENCODE data. (D) 12B08/whole larvae EU-RNA values (EC-tagging) and modENCODE Anatomy RNA-seq CNS/carcass (axon guidance category) or CNS/digestive system (small molecule metabolism category) ratios are shown as ‘Neural/Reference’ values (y-axis). EC-tagging data are the average and standard error of the mean for multiple measurements across replicate microarrays.
Figure 5.

EC-tagging in larval mushroom body neurons. (A) MB/whole larvae EU-RNA ratios and MB/whole larvae TU-RNA ratios for 25 mushroom body genes (MB) and 49 muscle genes (muscle). Wilcoxon rank sum test P-value is shown (n.s. = not significant). Values below zero represent genes that were not detected (n.d.) in MB TU-RNA. (B and C) Enrichment of mushroom body transcripts versus broadly expressed neuronal transcripts in MB EU-RNA, 12B08 EU-RNA. Data are the average and standard error of the mean for multiple measurements across replicate microarrays. Values for error bars that do not fit on the graph are: prt (±2.3), ey (±2.4), trio (±3.5). Small red bars below zero indicate the gene was not detected in 12B08 EU-RNA. (D) GO categories enriched in the 12B08 versus MB datasets. Venn diagram represents the 210 GO categories enriched in the 12B08 dataset (red, 151 unique), the 75 GO categories enriched in the MB dataset (blue, 16 unique) and the 59 overlapping categories (purple). Ppk is listed as a gene of interest in the MB-specific ‘ion transport’ category. (E) Confirmation of ppk expression in a small number of mushroom body neurons. Cell bodies (arrowhead) and partial dendrite projections (asterisk) are shown for GFP+ neurons in ppk>UAS-mCD8:GFP larval brains. Antibody staining for the mushroom body transcription factors Eyeless (Ey) and Dachshund (Dac) is shown in magenta. The dorsal region of a single brain hemisphere is shown (anterior down, medial left). The top figure includes a superimposed GFP+ cell from a slightly more ventral position that would otherwise be obscured (white outlined square).
RESULTS
EC-tagging in cell lines
5EC was synthesized and characterized as described in the ‘Materials and Methods’ section. The pathway of 5EC conversion to 5EUdMP (5-ethynyluridine monophosphate) by CD and UPRT is summarized in Figure 1A. To test RNA tagging via this pathway, we expressed a CD-UPRT fusion gene (CD:UPRT) in human SH-SY5Y neuroblastoma cells. CD:UPRT expression conferred 5EC dose-dependent RNA tagging and no RNA tagging was detected in cells lacking CD:UPRT (Figure 1B and Supplementary Figure S1). We also tested CD and UPRT individually or in combination and found that optimal RNA tagging occurs in cells expressing CD and UPRT (Figure 1C). No RNA tagging occurred in cells expressing mCherry-UPRT while relatively weak RNA tagging occurred in cells expressing GFP-CD. These results revealed that SH-SY5Y cells can convert the nucleobase 5EU produced by CD to 5EUdMP (likely via UPRT-independent pathways (19)) and confirmed that conversion of 5EC to 5EU is a necessary first step in EC-tagging. These results also demonstrated that conversion of 5EU to 5EUdMP is augmented by transgenic UPRT expression.
Figure 1.

RNA tagging via combined CD expression, UPRT expression and 5EC delivery. (A) Pathway of 5EC conversion to 5EUdMP (5-ethynyluridinemonophosphate). 5EUdMP is phosphorylated by nucleoside kinases to form 5EUdTP (dashed arrow) prior to incorporation into nascent RNA. (B) 5EC dose-dependent RNA tagging. CD:UPRT+ SH-SY5Y cells and control SH-SY5Y cells were exposed to the indicated concentration of 5EC for 6 h prior to RNA extraction, biotinylation and slot-blot probing with streptavidin-HRP (SA-HRP). In this and subsequent figures, the ‘RNA’ panel shows total RNA based on methylene blue staining and the top ‘SA-HRP probe’ panel shows the streptavidin-HRP signal specific for RNA containing biotinylated 5-ethynyluridine nucleotides. (C) Optimal EC-tagging in cells expressing CD and UPRT. SH-SY5Y cells were transfected with empty vector, a vector expressing CD, a vector expressing UPRT, or a combination of CD and UPRT expressing vectors. Cells were exposed to 500 μM 5EC for 6 h. (D) 5EC-tagging versus 5EU-tagging. CD:UPRT+ HeLa cells and control HeLa cells were exposed to 5EC or 5EU for the indicated time. (E) RNA tagging via ‘split CD’. UPRT(+) SH-SY5Y cells were transfected with empty vector, vectors expressing N-terminal and C-terminal CD fragments lacking leucine zipper domains (CD-n, CD-c), vectors expressing N-terminal and C-terminal CD fragments with complementary leucine zipper domains (CD-n-zip, zip-CD-c), or a vector expressing full-length CD.
We next compared RNA tagging by 5EC (the CD-UPRT pathway) and 5EU (the UPRT pathway) in control and CD:UPRT(+) HeLa cells (Figure 1D). Robust CD:UPRT-dependent RNA tagging was observed for both nucleobases. 5EU RNA tagging was more rapid, possibly due to the single enzymatic step required for 5EU conversion to 5EUdMP as opposed to the two steps required for 5EC conversion to 5EUdMP. No RNA tagging occurred in CD:UPRT(−) cells exposed to 5EC while relatively weak RNA tagging occurred in CD:UPRT(−) cells exposed to 5EU, similar to the RNA tagging observed in CD(+) SH-SY5Y cells treated with 5EC. We also tested if CD:UPRT(+) cells exposed to 5EC excrete 5EU, since uracil excretion has been described in cultured fibroblasts (39). CD:UPRT(+) HeLa cells were exposed to 5EC for 2 h and cell-free media was transferred to CD:UPRT(−) HeLa cells. Exposure to conditioned media caused relatively weak RNA tagging in CD:UPRT(−) cells compared to robust RNA tagging in CD:UPRT(+) cells (Supplementary Figure S2), suggesting that 5EU (or possibly 5-ethynyluridine) is excreted from CD:UPRT(+) cells. From these comparisons of 5EC RNA tagging and 5EU RNA tagging, we conclude that RNA tagging via 5EC and CD-UPRT is much more sensitive and stringent. EC-tagging achieves cell type-specificity even when 5EU may be shared between CD:UPRT(+) and CD:UPRT(−) cells, although the relative contribution of RNA tagging via 5EC and excreted 5EU should be considered in such mixed cell culture experiments.
Previously described RNA tagging and purification methods rely on cell type-specific expression of a transgene (such as UPRT (10) or an epitope-tagged ribosomal protein (8)) and the resolution of tagging is therefore largely determined by the specificity of the enhancer used to drive transgene expression. EC-tagging has the potential to provide greater resolution via combinatorial control of CD and UPRT expression. Our experiments described above suggest that CD may be the primary determinant of specificity due to the ability of UPRT-negative cells to incorporate 5EU into RNA. We therefore sought a method of placing CD under combinatorial control. Split protein systems have been used to restrict various activities to specific cell types, including targeted reconstitution of the Gal4 transcription factor in Drosophila (40). Work in yeast has shown that functional CD can be reconstituted from N-terminal and C-terminal fragments fused to complementary leucine zipper domains (41). We used similar leucine zipper-CD fusions and expressed the complementary fragments in UPRT(+) SH-SY5Y cells (Figure 1E). Co-expression of the leucine zipper-CD fragments enabled EC-tagging, while co-expression of CD fragments lacking the leucine zippers did not enable EC-tagging. These results establish an experimental platform that may be used to fine-tune EC-tagging specificity via combinatorial control of split CD expression.
To be useful for RNA analysis, EC-tagging should have minimal effects on cell physiology and gene expression. To assay toxicity in tissue culture cells, we measured the viability of control cells and CD:UPRT(+) cells exposed to 5EC over 72 h. CD:UPRT(+) cells cultured in 100 μM 5EC had no loss of viability and CD:UPRT(+) cells cultured in 500 μM 5EC were unaffected over 48 h followed by a decline in viability to ∼60% of controls by 72 h (Figure 2A). The toxic compound 5FC had a markedly different effect on viability: 100 μM 5FC caused approximately 50% loss of viability by 48 h (Figure 2A). To test if EC-tagging alters gene expression, we compared RNA from CD:UPRT(+) cells exposed to 500 μM 5EC for 6 h to RNA from untreated control cells. The mRNA profiles of control and CD:UPRT(+) cells were nearly identical (Supplementary Figure S3). Only 148 genes had reproducible differences in transcript abundance (Supplementary Table S1). We performed GO analysis to determine if these minor changes were indicative of altered cell physiology but did not find any significant GO category enrichment. We interpret these results as evidence that EC-tagging does not significantly alter cell physiology or gene expression.
Figure 2.

Analysis of EC-tagging effects on viability and gene expression. (A) Viability of CD:UPRT(+) cells exposed to 5EC or 5FC. Percent viability is plotted relative to untreated CD:UPRT(−) control cells. (B) Viability of ubiquitous CD:UPRT larvae and CD:UPRT(−) larvae with or without continuous 500 μM 5EC feeding, relative to day 0. ALH = after larval hatching. (C) Gene expression in ubiquitous CD:UPRT larvae fed 1.0 mM 5EC for 24 h (CD:UPRT + 5EC) compared to CD:UPRT-negative larvae fed 1.0 mM 5EC for 24 h (control + 5EC). Expression levels for 7434 genes are plotted as the log2 of the gene-specific signal/mean signal for all genes.
EC-tagging in Drosophila
To apply EC-tagging in an animal model, we made UAS-CD:UPRT transgenic Drosophila to allow targeted expression of CD:UPRT when combined with cell type-specific Gal4 lines (42). We first tested the effects of EC-tagging on Drosophila development using Act5C-Gal4 to ubiquitously express CD:UPRT. Ubiquitous CD:UPRT larvae and negative control larvae were fed 500 μM 5EC from the time of hatching. The viability of ubiquitous CD:UPRT larvae gradually began to decline after 24 h and reached 70% of controls by 5 days after larval hatching (Figure 2B). Ubiquitous CD:UPRT larvae continuously fed 5EC transitioned between larval instars with normal timing (based on size and morphological characteristics) but did not develop beyond the late third instar (L3) stage. Ubiquitous CD:UPRT larvae reared in the absence of 5EC developed normally, indicating that expression of the enzyme does not affect development. To test if EC-tagging alters gene expression in Drosophila, we compared RNA from L3 larvae that ubiquitously express CD:UPRT (da-Gal4 >CD:UPRT) and control larvae (no CD:UPRT) that were fed 1.0 mM 5EC for 24 h. Gene expression between the EC-tagged and control larvae was nearly identical (Figure 2C and Supplementary Table S2). Only 159 genes had reproducible changes in gene expression of 2-fold or more (9 genes with increased expression, 150 genes with decreased expression). GO analysis did not reveal any functional relationships among these genes, suggesting that their altered expression is not indicative of a specific response to EC-tagging. We conclude that EC-tagging for periods as long as 24 h does not adversely affect gene expression in Drosophila larvae.
We tested EC-based RNA tagging in larvae by expressing CD:UPRT broadly in the nervous system and imaginal discs using GMR12B08-Gal4 (43,44). GMR12B08 > CD:UPRT larvae and UAS-CD:UPRT larvae (without any Gal4 activation of CD:UPRT expression) were fed 5EC for 24 h. As a positive control, UAS-CD:UPRT larvae were fed 5-ethynyluridine (5EUd) for 24 h. 5EUd is incorporated into RNA in all cells, independent of CD or UPRT expression. RNA blots revealed strong RNA tagging in 5EUd-fed larvae, weaker RNA tagging in GMR12B08 > CD:UPRT larvae (as expected, since fewer cells are capable of incorporating the RNA tag) and no RNA tagging in negative control larvae (Figure 3A). Next we tested dose-dependent EC-tagging in a small population of neurons, using TH-Gal4 to express CD:UPRT in ∼75 dopaminergic neurons of the central nervous system (CNS) (45). 5EC dose-dependent RNA tagging occurred in TH-Gal4 > UAS-CD:UPRT larvae and no tagging was detected in negative control larvae, even after feeding high doses of 5EC (Figure 3B). Similarly, we observed time-dependent RNA tagging in larvae, with EU-RNA detected after 30 min in 5EUd fed larvae and after 3 h in 5EC fed larvae that express CD:UPRT in a small population of mushroom body neurons (MB247-Gal4 (46) > UAS-CD:UPRT) (Supplementary Figure S4). In addition to EC-tagging in larval stages, we found that 5EC feeding resulted in robust RNA tagging in adult flies with MB247-Gal4 expressing CD:UPRT primarily in mushroom body neurons (Figure 3C) and in embryos with string-Gal4 (GMR32C12) expressing CD:UPRT primarily in the developing nervous system (Supplementary Figure S5).
Figure 3.
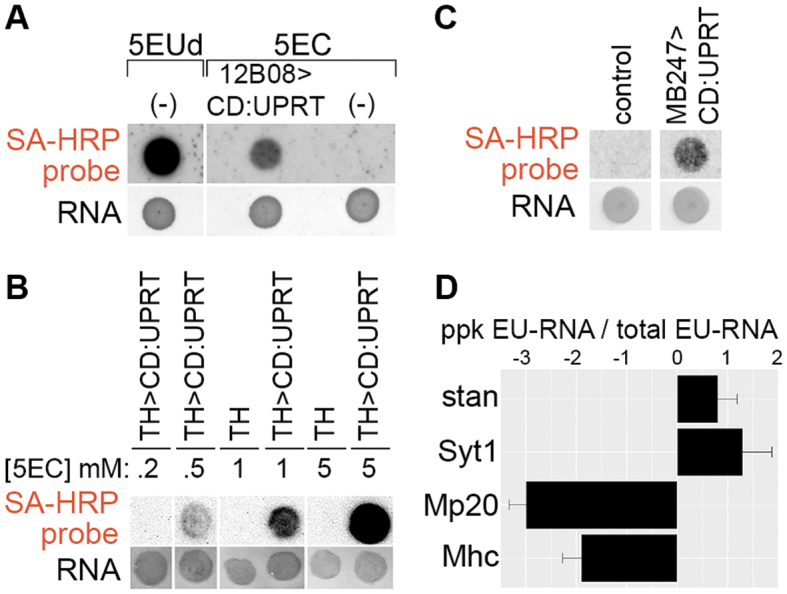
EC-tagging in Drosophila. (A) EC-tagging in larvae. CD:UPRT(−) larvae were fed either 500 μM 5EUd (positive control for RNA tagging) or 500 μM 5EC (negative control for RNA tagging) for 24 h. Larvae expressing CD:UPRT under control of GMR12B08-Gal4 were fed 500 μM 5EC for 24 h. In this and subsequent figures, total RNA was analyzed by dot blot and the ‘RNA’ and ‘SA-HRP’ panels are as described for Figure 1. (B) 5EC dose-dependent EC-tagging in larval dopaminergic neurons. Larvae expressing CD:UPRT under control of TH-Gal4 or TH-Gal4 larvae alone (no CD:UPRT) were fed the indicated concentration of 5EC for 6 h. (C) EC-tagging in adult brains. Adult flies expressing CD:UPRT under control of MB247-Gal4 driver or control flies (UAS-CD:UPRT alone) were fed 1.0 mM 5EC for 16 h prior to RNA extraction from whole flies. (D) Cell type-specific mRNA enrichment from larval peripheral nervous system neurons. Larvae expressing CD:UPRT under control of ppk-Gal4 were fed 500 μM 5EC (ppk EU-RNA) and matched larvae were fed 500 μM 5EUd (total EU-RNA). mRNA abundance for the indicated genes was measured by RT-qPCR. Data are the average and standard error of the mean from two biological replicates (separate 5EC feeding, RNA purification and RT-qPCR analysis).
To further test the specificity and sensitivity of EC-tagging, we used ppk-Gal4 to express CD:UPRT in a very small subset of larval peripheral nervous system cells (47). Ppk > CD:UPRT L3 larvae were fed 5EC for 24 h prior to carcass dissection and RNA extraction. Carcass dissection provided a defined tissue population composed of known cell types: the rare CD:UPRT(+) neurons (ppk-Gal4 is expressed in only three multidendritic neurons per hemisegment) and a large excess of CD:UPRT(−) cells including muscle (30 muscle fibers per hemisegment), oenocytes and epidermis. To obtain reference tagged RNA from all cells, we fed larvae 5EUd for 24 h prior to performing the same carcass RNA extraction. RNA purified from ppk > CD:UPRT larvae and 5EUd-fed larvae was compared using reverse transcription—quantitative PCR (RT-qPCR) for two neural-specific transcripts, Synaptotagmin 1 (Syt1) and starry night (stan), and two muscle-specific transcripts, Myosin heavy chain (Mhc) and Muscle protein 20 (Mp20). These genes were selected because their cell type-specific expression is well known and their mRNA abundance in L3 carcasses has been measured by the modENCODE Anatomy RNA-seq Project (48). The modENCODE RNA-seq data (linear values, scaled to maximum expression level) show that in L3 carcasses the neural-specific transcripts are present at low levels (expression values of 11 (Syt1) and 24 (stan)) and the muscle-specific transcripts are present at very high levels (expression values of 218 (Mhc) and 582 (Mp20). In our EC-tagging experiments Syt1 and stan were enriched in the ppk > CD:UPRT EU-RNA while Mhc and Mp20 were depleted from the ppk > CD:UPRT EU-RNA (Figure 3D). The ability to enrich for mRNAs transcribed in a small number of target neurons and deplete much more abundant mRNAs transcribed in non-target cells suggested that EC-tagging is sensitive and cell type-specific, prompting us to further test EC-tagging using transcriptome-wide measurements.
Cell type-specific transcriptome analysis
To evaluate the use of EC-tagging in transcriptome analysis, we compared EU-RNA purified from GMR12B08 > CD:UPRT larvae to EU-RNA purified from larvae fed 5EUd. We refer to RNA purified from GMR12B08 > CD:UPRT larvae as 12B08 EU-RNA and RNA purified from the 5EUd fed sample as whole larvae EU-RNA (Figure 4A). Independently-processed replicates were prepared for each condition and used to compare 12B08 EU-RNA and whole larvae EU-RNA by microarray analysis. Microarray signals between replicates correlated well, while correlations between 12B08 EU-RNA and 5EUd samples were much lower, as expected (Supplementary Figure S6). We first used 12B08 EU-RNA and whole larvae EU-RNA microarray data to test for any correlation between the efficiency of EU-RNA purification and the number of uridines per mRNA. Transcripts with more uridines are expected to incorporate more EU residues and this could favor their purification relative to transcripts with fewer uridines. We did not find any correlation between uridine number and mRNA yields (Supplementary Figure S7), suggesting that all mRNAs are equally likely to incorporate the minimum number of EU tags required for efficient biotinylation and purification.
Comparison of 12B08 EU-RNA and whole larvae EU-RNA identified 1279 mRNAs enriched 2-fold or more in 12B08 EU-RNA and 405 mRNAs depleted two-fold or more in 12B08 EU-RNA (Supplementary Table S3). We performed GO analysis and found significant over-representation of categories associated with GMR12B08-positive tissues (nervous system and imaginal discs) among the enriched genes and significant over-representation of categories associated with GMR12B08-negative tissues (epidermis and digestive system) among the depleted genes (Figure 4B). GO categories depleted in 12B08 EU-RNA also include categories associated with mitochondrial activity. We previously observed decreased abundance of mitochondria-associated mRNAs in the embryonic nervous system (18) and this may reflect the unique mitochondrial homeostasis needs of neurons (49). Next we compared 12B08 EC-tagging results to data obtained by sequencing RNA from dissected larval tissues, as reported in the modENCODE Anatomy RNA-seq database (48). Of the 1279 mRNAs enriched two-fold or more by 12B08 EC-tagging, we identified 609 genes with corresponding RNA-seq counts above the ‘very low expression’ threshold (Flybase annotation) in at least one of the following tissues: CNS, imaginal discs and carcass (composed of muscle, epidermis, oenocytes and peripheral neurons). We used these RNA-seq data to calculate CNS/carcass and imaginal disc/carcass ratios and found that 524 genes (86%) were enriched 1.5-fold or more in the CNS or imaginal discs according to the modENCODE data (Figure 4C). Examples of EC-tagging versus modENCODE RNA-seq data are shown for enriched CNS genes in the ‘axon guidance’ category and depleted digestive system genes in the ‘small molecule metabolism’ category in Figure 4D. These comparisons of EC-tagging and modENCODE data suggest that EC-tagging is similarly effective at identifying cell type-specific mRNAs without the need for any tissue dissection.
To test our prediction that EC-tagging provides greater sensitivity and specificity than TU-tagging, we compared the ability of these methods to purify mRNA from mushroom body neurons of the larval brain. Mushroom body neurons comprise an important learning and memory center (50) and while gene expression in the adult mushroom body has previously been investigated (37,51), the transcriptional program of larval mushroom body neurons is less well-defined. We used MB247-Gal4 to express CD:UPRT in mushroom body neurons and fed L3 larvae 1.0 mM 5EC or 1.0 mM 4-thiouracil (TU) prior to purification of tagged RNAs. CD:UPRT-transgenic larvae work for EC-tagging and TU-tagging since the CD:UPRT fusion enzyme converts 4-thiouracil as efficiently as the UPRT enzyme alone (Supplementary Figure S8). We refer to potential mushroom body-specific RNA samples as MB EU-RNA and MB TU-RNA. MB EU-RNA was compared to whole larvae EU-RNA purified from EUd fed larvae and MB TU-RNA was compared to whole larvae TU-RNA purified from 4sUd fed larvae. Replicate microarrays were analyzed for all samples and there was little variation between replicates but considerable variation between EC-tagging and TU-tagging samples (Supplementary Figure S9). EC-tagging identified 1011 mRNAs enriched two-fold or more in MB EU-RNA and TU-tagging identified 639 mRNAs enriched 2-fold or more in MB TU-RNA (Supplementary Table S4). There was very little overlap in the set of enriched genes identified by EC-tagging and TU-tagging: only 51 genes were enriched in MB EU-RNA and MB TU-RNA (Supplementary Table S4) and we did not identify any functional relationship or cell type-specificity shared by these genes (according to GO analysis and Flybase annotations). To test for mushroom body mRNA enrichment, we analyzed 25 signaling pathway genes expressed in the adult mushroom body (37) (positive control genes, selection criteria described in ‘Materials and Methods’ section) and 49 predicted muscle-specific genes (38) (negative control genes, selection criteria described in ‘Materials and Methods’ section). As shown in Figure 5A, 23 positive control genes were enriched >1.8-fold by EC-tagging and only one negative control gene was enriched by EC-tagging (genes annotated in Supplementary Table S4). In contrast, TU-tagging did not yield any enrichment of positive control mRNAs (signals were similar to those obtained for negative control genes) and 8 of the 25 mushroom body mRNAs were below the limit of detection in MB TU-RNA (Figure 5A).
Since MB247 expresses Gal4 in a small group of brain neurons and GMR12B08 expresses Gal4 broadly in the nervous system, we predicted that MB EU-RNA would have greater enrichment of mushroom body mRNAs and 12B08 EU-RNA would have greater enrichment of widely-expressed neuronal mRNAs. This was indeed the case: mRNAs expressed primarily in the mushroom body (based on published larval expression data (52–57)) were strongly enriched in MB EU-RNA but absent or non-enriched in 12B08 EU-RNA (Figure 5B). In contrast, mRNAs expressed broadly in the nervous system were strongly enriched in 12B08 EU-RNA and weakly enriched in MB EU-RNA (Figure 5C). Neither mushroom body genes nor broadly expressed nervous system genes were enriched in MB TU-RNA (Supplementary Table S4). We next compared GO categories enriched in 12B08 EU-RNA, MB EU-RNA and MB TU-RNA. Surprisingly, the only non-redundant GO category enriched by MB TU-tagging was ‘chitin-based cuticle development’, suggesting that non-specific incorporation of 4-thiouracil may be particularly strong in the epidermis (data not shown). In contrast, MB EU-RNA had significant enrichment of categories associated with neural function (Figure 5D and Supplementary Table S5). Aligning MB EU-RNA and 12B08 EU-RNA GO data revealed distinct gene expression categories. Of 210 non-redundant GO categories enriched in 12B08 EU-RNA, 151 were unique to this dataset and 59 were shared between the 12B08 and MB datasets. 12B08-specific GO categories include ‘motor neuron axon guidance’, ‘mitotic cell cycle’ and ‘imaginal disc pattern formation’. The absence of these categories from the MB dataset is expected since MB247-Gal4 is not expressed in motor neurons, mitotic progenitors or imaginal discs (58). Of 75 non-redundant GO categories enriched in MB EU-RNA, 16 were unique to the MB dataset. MB-specific GO categories are associated with mushroom body properties such as neuropeptide signaling, G protein-coupled receptor signaling and behavior. Neuropeptide and G protein-mediated signaling pathways regulate activity in the adult mushroom body (59) and our data suggest these signaling systems also function in larval learning and memory. In contrast, Wnt and Notch signaling categories were only enriched in the 12B08 dataset. This likely reflects the fact that Wnt and Notch signaling is widespread in the nervous system (60,61) and imaginal discs (62) and not restricted to or elevated in the mushroom body like the neuropeptide and G protein-mediated pathways.
‘Ion transport’ is another GO category unique to the MB dataset. Within this category, we were surprised to find the pickpocket gene identified as a MB-enriched transcript. Pickpocket is a voltage-insensitive ion channel involved in larval locomotion (47) and mechanical nociception (63). Pickpocket is best known for its expression and function in the peripheral nervous system and so we sought to confirm ppk expression in mushroom body neurons. Ppk-Gal4 matches endogenous ppk expression and is expressed in a small number of brain cells (47), but the identity of these ppk-Gal4+ brain cells was not previously determined. We used ppk-Gal4 to express UAS-mCD8-GFP (membrane-anchored GFP) and identified between two and four GFP+ mushroom body neurons per brain hemisphere. Mushroom body identity was determined by co-localization with Eyeless and Dachshund (transcription factors expressed in the mushroom body (56)) (Figure 5E), and neuron projections into the mushroom body calyx, peduncle and lobes (Supplementary Figure S10). Our discovery of a small population of ppk-expressing mushroom body neurons confirms the sensitivity and specificity of EC-tagging.
DISCUSSION
Here, we show that combinatorial control of CD expression, UPRT expression and 5EC delivery allows purification of cell type-specific RNAs without the need to physically isolate cells of interest. While dissection-based transcriptome profiling yields valuable information, such approaches are often labor intensive and may alter gene expression as a result of tissue manipulations. Here we show that EC-tagging yields tissue-specific gene expression data similar to those obtained by the dissection-based modENCODE Anatomy RNA-seq project. We also show that EC-tagging provides a significant improvement over its methodological precursor TU-tagging. EC-tagging successfully enriched for rare mushroom body mRNAs from a mixture of all larval mRNAs, while TU-tagging failed to identify mushroom body mRNAs in parallel experiments. The lack of specificity in our mushroom body TU-tagging experiments was likely due to widespread RNA-tagging via endogenous Drosophila UPRT (20). Background RNA labeling is a known limitation of TU-tagging and may be partially avoided by dissection of relevant tissues, as previously described (10,12,64–65). MB247-Gal4-positive mushroom body neurons comprise ∼700 cells out of at least 1 million total cells in L3 stage larvae. We therefore estimate that in these RNA tagging experiments, the target cells constitute as little as 0.07% of the population. Given this low percentage, it is not surprising that TU-tagging applied to whole larvae failed to enrich mushroom body mRNAs. In contrast, EC-tagging identified known or predicted larval mushroom body transcripts and led to the discovery of a novel mushroom body-expressed gene, ppk.
While target mRNA enrichment by EC-tagging was robust, depletion of transcripts from non-target tissues was variable. As shown for MB EC-tagging, several muscle-specific genes had MB/whole larvae EU-RNA ratios close to 1.0. These mRNAs are highly abundant in larvae (based on modENCODE RNA-seq data) and we expect such transcripts to be difficult to completely remove during EU-RNA purification. EC-tagging is more effective at depleting less abundant off-target transcripts, as demonstrated by the depletion of ‘cuticle development’ and ‘small molecule metabolism’ mRNAs in the 12B08 EC-tagging experiments. Another source of off-target transcripts may be RNA tagged via 5EU excreted from CD:UPRT+ cells, as observed in SH-SY5Y cells. However, depletion of muscle transcripts in our ppk-Gal4 >CD:UPRT experiments (where 5EU could potentially be excreted from CD:UPRT(+) neurons and taken up by the surrounding muscle) and the lack of off-target transcript enrichment in our transcriptome profiling experiments argues against significant 5EU excretion during EC-tagging in Drosophila. We conclude that while enrichment of cell type-specific transcripts by EC-tagging is sensitive and robust, depletion of off-target transcripts may be variable and is likely due to non-specific capture of untagged RNAs during the purification step.
There are multiple parameters to consider when designing an EC-tagging experiment, including the duration of 5EC exposure. Long exposure increases the abundance of tagged RNAs in target cells and allows cell type-specific mRNA discovery starting from a small amount of input material: in our 24 h EC-tagging experiments, EU-RNA for microarray analysis was obtained from 20 μg of biotinylated input RNA (starting from 20–30 L3 larvae). While we did not detect any mortality or major changes in gene expression after 24 h of ubiquitous EC-tagging, future users of EC-tagging may want to investigate potential side effects in the context of their experimental system (particularly if feeding 5EC for more than 24 h). Long periods of 5EC exposure are not a requirement for effective EC-tagging: we detected tagged RNA from CD:UPRT(+) mushroom body neurons after 3 h of 5EC feeding and tagged RNA from all cells after 30 min of 5EUd feeding. The rapid incorporation of 5EUd suggests that short exposure times will work for EC-tagging but will require increased amounts of input RNA (compared to what we used following 24 h exposure) for detection and purification of EU-RNA.
The type of RNA populations to be compared is another experimental variable in EC-tagging. Here we compared purified EU-RNA from CD:UPRT(+) cells to purified EU-RNA from all cells (via 5EUd feeding). This design allowed comparison of equivalent biosynthetically tagged transcripts and ensured that each sample underwent identical processing steps. An alternative is to compare purified EU-RNA to input (pre-purification) RNA, but this approach may introduce biases. One potential problem is that the EU tag is only incorporated into RNAs made during 5EC exposure while input RNA contains transcripts made prior to 5EC exposure. Therefore, weakly transcribed long-lived mRNAs will be more abundant in input RNA, even if transcription and decay rates are equal in target and non-target cells. Conversely, rapidly degraded mRNAs will be more abundant in the purified EU-RNA pool, particularly when using short labeling times and enriching for nascent mRNAs. Another potential problem when comparing purified EU-RNA and input RNA is that the samples are processed differently: the biotinylation and purification steps may alter EU-RNA relative to input RNA. These potential problems are avoided when comparing purified EU-RNA from CD:UPRT(+) cells to purified EU-RNA from all cells. Another design consideration is the potential to express CD and UPRT, or the split-CD halves plus UPRT, from different enhancers. Such intersectional approaches could refine cell type targeting in mixed cell cultures and in vivo. In Drosophila and other organisms with endogenous uracil incorporation pathways, we predict that combinatorial control of split-CD expression combined with enhanced 5EU incorporation via targeted UPRT expression will give the greatest intersectional control. As a final experimental design option, we found it useful to compare EC-tagging results from related cell populations with well defined distinctions. 12B08 EC-tagging and MB EC-tagging enriched for neural transcripts but comparing data from each Gal4 line allowed us to distinguish broadly expressed neural genes from mushroom body-specific neural genes. These data should prove useful for identifying novel mushroom body properties, as demonstrated by our discovery of pickpocket-expressing mushroom body neurons. The discovery of these neurons reveals previously unknown cellular heterogeneity in the larval mushroom body and suggests that the pickpocket-expressing neurons respond to modalities that are distinct from those previously described in the mushroom body (66).
The list of biological functions performed by RNA continues to expand. In parallel, genomic research is moving away from isolated cultures of cells and into more complex environments like tissues and whole animals. In order to fully understand RNA expression and function in complex environments, new and highly stringent methods are needed to purify cell type-specific RNAs. Our results herein overcome nearly all the remaining hurdles for cell type-specific metabolic tagging of RNA. First, we show that a dual-enzyme system is highly robust and results in very specific RNA tagging. We also demonstrate that our method is amenable to split-enzyme designs, which will be valuable for fine-tuning cell targeting. Most importantly, we show that our approach allows in vivo analyses, even within complex tissue environments such as the nervous system. It is worth noting that use of EC-tagging is not limited to studies of differential gene expression. For example, noncoding RNAs are expected to incorporate the 5-ethynyluridine tag and temporal control of 5EC exposure may be used to measure RNA synthesis and decay. Based on the sensitivity and specificity of this method, we anticipate that EC-tagging will be useful for wide-ranging studies of gene expression and RNA biology.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the UC Merced Neurobiology Group (Wolf, Saha, Kitazawa and Cleary labs) for feedback during the development of this technique. We are also grateful to Fred Wolf for helpful comments on the manuscript and to Gil dos Santos at Flybase for assistance in acquiring scaled modENCODE data. Sai Prabhakar and Elijah Maxfield assisted with fly husbandry and molecular biology for this project.
Footnotes
Present address: Dana A. Burow, Department of Reproductive Medicine, School of Medicine, University of California, San Diego, La Jolla, CA 92093, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
California Blueprint for Research to Advance Innovations in Neuroscience (Cal-BRAIN) [350153 to M.D.C.]; National Institute of Child Health and Human Development [R01HD076927 to M.D.C.]; National Institute of General Medical Sciences [GM063028 to M.M.G.]; University of California start-up funds (to R.C.S.); National Institutes of Health [DP2GM119164 RCS, RO1MH109588 to R.C.S.]. Funding for open access charge: External grant; Internal university support.
Conflict of interest statement. M.D.C. and N.H. have filed a United States Patent Application covering EC-tagging technology.
REFERENCES
- 1. Dolken L., Ruzsics Z., Radle B., Friedel C.C., Zimmer R., Mages J., Hoffmann R., Dickinson P., Forster T., Ghazal P. et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 2008; 9:1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kenzelmann M., Maertens S., Hergenhahn M., Kueffer S., Hotz-Wagenblatt A., Li L., Wang S., Ittrich C., Lemberger T., Arribas R. et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:6164–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Handley A., Schauer T., Ladurner A.G., Margulies C.E.. Designing cell-type-specific genome-wide experiments. Mol. Cell. 2015; 58:621–631. [DOI] [PubMed] [Google Scholar]
- 4. Lobo M.K., Karsten S.L., Gray M., Geschwind D.H., Yang X.W.. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat. Neurosci. 2006; 9:443–452. [DOI] [PubMed] [Google Scholar]
- 5. Vincent V.A., DeVoss J.J., Ryan H.S., Murphy G.M. Jr. Analysis of neuronal gene expression with laser capture microdissection. J. Neurosci. Res. 2002; 69:578–586. [DOI] [PubMed] [Google Scholar]
- 6. Henry G.L., Davis F.P., Picard S., Eddy S.R.. Cell type-specific genomics of Drosophila neurons. Nucleic Acids Res. 2012; 40:9691–9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang Z., Edenberg H.J., Davis R.L.. Isolation of mRNA from specific tissues of Drosophila by mRNA tagging. Nucleic Acids Res. 2005; 33:e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heiman M., Schaefer A., Gong S., Peterson J.D., Day M., Ramsey K.E., Suárez-Fariñas M., Schwarz C., Stephan D.A. et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008; 135:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cleary M.D., Meiering C.D., Jan E., Guymon R., Boothroyd J.C.. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat. Biotechnol. 2005; 23:232–237. [DOI] [PubMed] [Google Scholar]
- 10. Miller M.R., Robinson K.J., Cleary M.D., Doe C.Q.. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 2009; 6:439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tallafuss A., Washbourne P., Postlethwait J.. Temporally and spatially restricted gene expression profiling. Curr. Genomics. 2014; 15:278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weng R., Cohen S.M.. Drosophila miR-124 regulates neuroblast proliferation through its target anachronism. Development. 2012; 139:1427–1434. [DOI] [PubMed] [Google Scholar]
- 13. Tallafuss A., Kelly M., Gay L., Gibson D., Batzel P., Karfilis K.V., Eisen J., Stankunas K., Postlethwait J.H., Washbourne P.. Transcriptomes of post-mitotic neurons identify the usage of alternative pathways during adult and embryonic neuronal differentiation. BMC Genomics. 2015; 16:1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Erickson T., Nicolson T.. Identification of sensory hair-cell transcripts by thiouracil-tagging in zebrafish. BMC Genomics. 2015; 16:842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rinn J.L., Wang J.K., Allen N., Brugmann S.A., Mikels A.J., Liu H., Ridky T.W., Stadler H.S., Nusse R., Helms J.A. et al. A dermal HOX transcriptional program regulates site-specific epidermal fate. Genes Dev. 2008; 22:303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gay L., Miller M.R., Ventura P.B., Devasthali V., Vue Z., Thompson H.L., Temple S., Zong H., Cleary M.D., Stankunas K. et al. Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes Dev. 2013; 27:98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chatzi C., Zhang Y., Shen R., Westbrook G.L., Goodman R.H.. Transcriptional profiling of newly generated dentate granule cells using TU tagging reveals pattern shifts in gene expression during circuit integration. eNeuro. 2016; 3, doi:10.1523/ENEURO.0024-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burow D.A., Umeh-Garcia M.C., True M.B., Bakhaj C.D., Ardell D.H., Cleary M.D.. Dynamic regulation of mRNA decay during neural development. Neural. Dev. 2015; 10:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carter D., Donald R.G., Roos D., Ullman B.. Expression, purification, and characterization of uracil phosphoribosyltransferase from Toxoplasma gondii. Mol. Biochem. Parasitol. 1997; 87:137–144. [DOI] [PubMed] [Google Scholar]
- 20. Ghosh A.C., Shimell M., Leof E.R., Haley M.J., O’Connor M.B.. UPRT, a suicide-gene therapy candidate in higher eukaryotes, is required for Drosophila larval growth and normal adult lifespan. Sci. Rep. 2015; 5:13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duffy E.E., Rutenberg-Schoenberg M., Stark C.D., Kitchen R.R., Gerstein M.B., Simon M.D.. Tracking distinct RNA populations using efficient and reversible covalent chemistry. Mol. Cell. 2015; 59:858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Curanovic D., Cohen M., Singh I., Slagle C.E., Leslie C.S., Jaffrey S.R.. Global profiling of stimulus-induced polyadenylation in cells using a poly(A) trap. Nat. Chem. Bio. 2013; 9:671–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nainar S., Beasley S., Fazio M., Kubota M., Dai N., Correa I.R. Jr, Spitale R.C.. Metabolic incorporation of azide functionality into cellular RNA. Chembiochem. 2016; 17:2149–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mullen C.A., Kilstrup M., Blaese R.M.. Transfer of the bacterial gene for cytosine deaminase to mammalian cells confers lethal sensitivity to 5-fluorocytosine: a negative selection system. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miyagi T., Koshida K., Hori O., Konaka H., Katoh H., Kitagawa Y., Mizokami A., Egawa M., Ogawa S., Hamada H. et al. Gene therapy for prostate cancer using the cytosine deaminase/uracil phosphoribosyltransferase suicide system. J. Gene. Med. 2003; 5:30–37. [DOI] [PubMed] [Google Scholar]
- 26. Waldorf A. R., Polak A.. Mechanisms of action of 5-fluorocytosine. Antimicrob. Agents Chemother. 1983; 23:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jao C.Y., Salic A.. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:15779–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tani H., Mizutani R., Salam K.A., Tano K., Ijiri K., Wakamatsu A., Isogai T., Suzuki Y., Akimitsu N.. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res. 2012; 22:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hao N., Bhakti V.L., Peet D.J., Whitelaw M.L.. Reciprocal regulation of the basic helix-loop-helix/Per-Arnt-Sim partner proteins, Arnt and Arnt2, during neuronal differentiation. Nucleic Acids Res. 2013; 41:5626–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kielkowski P., Pohl R., Hocek M.. Synthesis of acetylene linked double-nucleobase nucleos(t)ide building blocks and polymerase construction of DNA containing cytosines in the major groove. J. Org. Chem. 2011; 76:3457–3462. [DOI] [PubMed] [Google Scholar]
- 31. Liang Y., Pitteloud J. P., Wnuk S. F.. Hydrogermylation of 5-ethynyluracil nucleosides: formation of 5-(2-germylvinyl)uracil and 5-(2-germylacetyl)uracil nucleosides. J. Org. Chem. 2013; 78:5761–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ghosh I., Hamilton A.D., Regan L.. Antiparallel leucine zipper-directed protein reassembly: application to the green fluorescent protein. J. Am. Chem. Soc. 2000; 122:5658–5659. [Google Scholar]
- 33. da Huang W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 34. Yildirim O. Isolation of nascent transcripts with click chemistry. Curr. Protoc. Mol. Biol. 2015; 111:4.24.1–4.24.13. [DOI] [PubMed] [Google Scholar]
- 35. Spitale R.C., Flynn R.A., Zhang Q.C., Crisalli P., Lee B., Jung J.W., Kuchelmeister H.Y., Batista P.J., Torre E.A., Kool E.T. et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015; 519:486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boyle E.I., Weng S., Gollub J., Jin H., Botstein D., Cherry J.M., Sherlock G.. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004; 20:3710–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crocker A., Guan X.J., Murphy C.T., Murthy M.. Cell-type-specific transcriptome analysis in the drosophila mushroom body reveals memory-related changes in gene expression. Cell Rep. 2016; 15:1580–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schnorrer F., Schönbauer C., Langer C.C., Dietzl G., Novatchkova M., Schernhuber K., Fellner M., Azaryan A., Radolf M., Stark A. et al. Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature. 2010; 464:287–291. [DOI] [PubMed] [Google Scholar]
- 39. Chan T.S., Meuth M., Green H.. Pyrimidine excretion by cultured fibroblasts: effect of mutational deficiency in pyrimidine salvage enzymes. J. Cell Physiol. 1974; 83:263–266. [DOI] [PubMed] [Google Scholar]
- 40. Luan H., Peabody N.C., Vinson C.R., White B.H.. Refined spatial manipulation of neuronal function by combinatorial restriction of transgene expression. Neuron. 2006; 52:425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ear P.H., Michnick S.W.. A general life-death selection strategy for dissecting protein functions. Nat. Methods. 2009; 6:813–816. [DOI] [PubMed] [Google Scholar]
- 42. Brand A.H., Perrimon N.. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993; 118:401–415. [DOI] [PubMed] [Google Scholar]
- 43. Li H.H., Kroll J.R., Lennox S.M., Ogundeyi O., Jeter J., Depasquale G, Truman J.W.. A GAL4 driver resource for developmental and behavioral studies on the larval CNS of Drosophila. Cell Rep. 2013; 8:897–908. [DOI] [PubMed] [Google Scholar]
- 44. Manning L., Heckscher E.S., Purice M.D., Roberts J., Bennett A.L., Kroll J.R., Pollard J.L., Strader M.E., Lupton J.R., Dyukareva A.V. et al. A resource for manipulating gene expression and analyzing cis-regulatory modules in the Drosophila CNS. Cell Rep. 2012; 2:1002–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Friggi-Grelin F., Coulom H., Meller M., Gomez D., Hirsh J., Birman S.. Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J. Neurobiol. 2003; 54:618–627. [DOI] [PubMed] [Google Scholar]
- 46. Schulz R.A., Chromey C., Lu M.F., Zhao B., Olson E.N.. Expression of the D-MEF2 transcription in the Drosophila brain suggests a role in neuronal cell differentiation. Oncogene. 1996; 12:1827–1831. [PubMed] [Google Scholar]
- 47. Ainsley J.A., Pettus J.M., Bosenko D., Gerstein C.E., Zinkevich N., Anderson M.G., Adams C.M., Welsh M.J., Johnson W.A.. Enhanced locomotion caused by loss of the Drosophila DEG/ENaC protein Pickpocket1. Curr. Biol. 2003; 13:1557–1563. [DOI] [PubMed] [Google Scholar]
- 48. Brown J.B., Boley N., Eisman R., May G.E., Stoiber M.H., Duff M.O., Booth B.W., Wen J., Park S., Suzuki A.M. et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014; 512:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kann O., Kovács R.. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007; 292:C641–C657. [DOI] [PubMed] [Google Scholar]
- 50. Davis R.L. Mushroom bodies and Drosophila learning. Neuron. 1993; 11:1–14. [DOI] [PubMed] [Google Scholar]
- 51. Kobayashi M., Michaut L., Ino A., Honjo K., Nakajima T., Maruyama Y., Mochizuki H., Ando M., Ghangrekar I., Takahashi K. et al. Differential microarray analysis of Drosophila mushroom body transcripts using chemical ablation. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:14417–14422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Silva B., Goles N.I., Varas R., Campusano J.M.. Serotonin receptors expressed in Drosophila mushroom bodies differentially modulate larval locomotion. PLoS One. 2014; 9:e89641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Selcho M., Pauls D., Han K. A., Stocker R. F., Thum A. S.. The role of dopamine in Drosophila larval classical olfactory conditioning. PLoS One. 2009; 4:e5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. El-Kholy S., Stephano F., Li Y., Bhandari A., Fink C., Roeder T.. Expression analysis of octopamine and tyramine receptors in Drosophila. Cell Tissue Res. 2015; 361:669–684. [DOI] [PubMed] [Google Scholar]
- 55. Brooks E.S., Greer C.L., Romero-Calderón R., Serway C.N., Grygoruk A., Haimovitz J.M., Nguyen B.T., Najibi R., Tabone C.J., de Belle J.S. et al. A putative vesicular transporter expressed in Drosophila mushroom bodies that mediates sexual behavior may define a neurotransmitter system. Neuron. 2011; 72:316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Noveen A., Daniel A., Hartenstein V.. Early development of the Drosophila mushroom body: the roles of eyeless and dachshund. Development. 2000; 127:3475–3488. [DOI] [PubMed] [Google Scholar]
- 57. Awasaki T., Saito M., Sone M., Suzuki E., Sakai R., Ito K., Hama C.. The Drosophila trio plays an essential role in patterning of axons by regulating their directional extension. Neuron. 2000; 26:119–131. [DOI] [PubMed] [Google Scholar]
- 58. Pauls D., Selcho M., Gendre N., Stocker R.F., Thum A.S.. Drosophila larvae establish appetitive olfactory memories via mushroom body neurons of embryonic origin. J. Neurosci. 2010; 30:10655–10666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Keene A.C., Waddell S.. Drosophila olfactory memory: single genes to complex neural circuits. Nat. Rev. Neurosci. 2007; 8:341–354. [DOI] [PubMed] [Google Scholar]
- 60. Udolph G. Notch signaling and the generation of cell diversity in Drosophila neuroblast lineages. Adv. Exp. Med. Biol. 2012; 727:47–60. [DOI] [PubMed] [Google Scholar]
- 61. Yoshikawa S., McKinnon R.D., Kokel M., Thomas J.B.. Wnt-mediated axon guidance via the Drosophila Derailed receptor. Nature. 2003; 422:583–588. [DOI] [PubMed] [Google Scholar]
- 62. Hayward P., Kalmar T., Arias A.M.. Wnt/Notch signalling and information processing during development. Development. 2008; 135:411–424. [DOI] [PubMed] [Google Scholar]
- 63. Zhong L., Hwang R.Y., Tracey W.D.. Pickpocket is a DEG/ENaC protein required for mechanical nociception in Drosophila larvae. Curr. Biol. 2010; 20:429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Inagaki H.K., Ben-Tabou de-Leon S., Wong A.M., Jagadish S., Ishimoto H., Barnea G., Kitamoto T., Axel R., Anderson D.J.. Visualizing neuromodulation in vivo: TANGO-mapping of dopamine signaling reveals appetite control of sugar sensing. Cell. 2012; 148:583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lai S.L., Miller M.R., Robinson K.J., Doe C.Q.. The Snail family member Worniu is continuously required in neuroblasts to prevent Elav-induced premature differentiation. Dev. Cell. 23:849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yagi R., Mabuchi Y., Mizunami M., Tanaka N.K.. Convergence of multimodal sensory pathways to the mushroom body calyx in Drosophila melanogaster. Sci. Rep. 2016; 6:29481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.