


Abstract
The tRNA m1G9 methyltransferase (Trm10) is a member of the SpoU-TrmD (SPOUT) superfamily of methyltransferases, and Trm10 homologs are widely conserved throughout Eukarya and Archaea. Despite possessing the trefoil knot characteristic of SPOUT enzymes, Trm10 does not share the same quaternary structure or key sequences with other members of the SPOUT family, suggesting a novel mechanism of catalysis. To investigate the mechanism of m1G9 methylation by Trm10, we performed a biochemical and kinetic analysis of Trm10 and variants with alterations in highly conserved residues, using crystal structures solved in the absence of tRNA as a guide. Here we demonstrate that a previously proposed general base residue (D210 in Saccharomyces cerevisiae Trm10) is not likely to play this suggested role in the chemistry of methylation. Instead, pH-rate analysis suggests that D210 and other conserved carboxylate-containing residues at the active site collaborate to establish an active site environment that promotes a single ionization that is required for catalysis. Moreover, Trm10 does not depend on a catalytic metal ion, further distinguishing it from the other known SPOUT m1G methyltransferase, TrmD. These results provide evidence for a non-canonical tRNA methyltransferase mechanism that characterizes the Trm10 enzyme family.
INTRODUCTION
Despite an increased appreciation for the presence and function of modified nucleotides in biological RNAs, transfer RNA (tRNA) remain exceptional because of their extensive posttranscriptional modification, with modified nucleotides found throughout tRNA species in organisms from all domains of life (1–5). The importance of many tRNA modifications in the anticodon stem–loop (and their corresponding enzymes) is relatively well-documented, as loss of these modifications frequently affects translation efficiency or fidelity (6,7). In contrast, roles of modifications that occur outside of the anticodon region, in the body of the tRNA, are more difficult to define, as deletion of a single enzyme responsible for these modifications frequently does not typically cause an obvious growth phenotype. Nonetheless, stress-related fitness defects have been associated with loss of tRNA modifications and an increasing number of diseases have been attributed to mutations in human modification enzymes (8–15), both of which suggest important roles for tRNA modifications that remain to be fully demonstrated. To better understand the biological impact of these modifications, it is therefore important to understand the biochemical properties and reaction mechanisms of the associated enzymes.
Trm10 is responsible for one post-transcriptional modification found in the body of multiple tRNAs in Archaea and Eukarya. Trm10 methylates the N1 of guanosine at the ninth position of tRNAs (G9) using S-adenosyl methionine (SAM) as the methyl group donor (16,17) (Figure 1). In Saccharomyces cerevisiae, TRM10, like many other genes for tRNA modification, is non-essential (16); however, trm10Δ strains exhibit hypersensitivity to the common anti-tumor drug 5-fluorouracil (5FU) (18). Metazoa encode multiple paralogs of Trm10, including three homologs found in humans (TRMT10A, TRMT10B and TRMT10C). TRMT10C is the most well-characterized of these, which localizes to mitochondria and functions in tRNA 5′-end maturation as part of an unusual proteinaceous ribonuclease P (RNase P) complex (19,20). For the other two non-mitochondrial human homologs, less is known. Recently, however, disease syndromes characterized by neurodegenerative defects and glucose metabolic disorders were associated with two different familial mutations in human TRMT10A (hTRMT10A), indicating at least some non-redundant functions for TRMT10A and TRMT10B in human cells, further adding to the questions surrounding this enzyme family and its biochemical properties (10,11,21–23).
Figure 1.

Trm10 catalyzes m1G9 formation in tRNA. (A) Representative cloverleaf structure of tRNA showing position of the G9 residue that is the target for Trm10 methylation. (B) Trm10 uses S-adenosylmethionine (SAM) as the donor of the methyl group to form m1G9. A protein residue (D210 in ScTrm10) has been proposed to serve as a general base (indicated by GB:) to abstract the proton from the N1 target atom for methylation.
Previous biochemical and structural characterization suggested several unusual mechanistic features associated with Trm10 methylation. First, although the originally-discovered S. cerevisiae Trm10 (ScTrm10) acts as an obligate G9 methyltransferase, an archaeal Trm10 homolog from Sufolobus acidocaldarius acts only on tRNA species with adenines at A9, and others, such as the enzyme from Thermococcus kodakarensis and human TRMT10C, methylate both G9 and A9 (16,20,24). The molecular basis for these variable target nucleotide specificities is unknown but suggests distinct catalytic features compared to other base-modifying RNA methyltransferases, which so far all exhibit specificity for a single target base (3). Second, although several crystal structures corroborated the original bioinformatics prediction that Trm10 is a member of the SpoU-TrmD (SPOUT) superfamily, there are notable differences that preclude inferring the mechanism of Trm10 by direct analogy to other more well-studied SPOUT family members (25–27). These differences include a unique quaternary structure; while Trm10 structures implicate a monomer as the catalytically relevant form of the enzyme, all other SPOUT family members (with the exception of Sfm1—a protein arginine methyltransferase (28)) function as dimers with the active site located at the dimeric interface. Sequence differences are also an issue, since sequence alignments fail to identify analogous residues to some highly conserved amino acids implicated in the mechanisms of other SPOUT methyltransferases (27,29–31). Finally, although a highly conserved aspartate (D210 in ScTrm10) has been proposed by several groups to serve as a general base during m1G9 methylation (20,25), this role has not been biochemically validated for any family member. For all of these reasons, the overall mechanism of Trm10 catalysis remains poorly understood. Here we use biochemical and kinetic methods to investigate the mechanism of human and S. cerevisiae Trm10, and provide evidence for a novel mode of catalysis that differs from several previous predictions.
MATERIALS AND METHODS
Mutagenesis and purification of Trm10
Trm10 variants (yeast and human) were generated using Phusion mutagenesis (Thermo Scientific) using previously described plasmids for heterologous expression and purification of wild type ScTrm10 and hTRMT10A from Escherichia coli (16,17). Mutations were confirmed by sequencing and wild-type and mutant proteins were purified using metal ion affinity chromatography, as previously described (17) and dialyzed into a storage buffer containing 50% glycerol, 20 mM Tris pH 7.5, 55 mM NaCl, 1 mM MgCl2, 1 μM ethylenediamine tetraacetic acid (EDTA) and 1 mM dithiothreitol. The resulting proteins were judged to be 75–90% pure by SDS-PAGE and stored at –20°C (Supplementary Figure S1).
In vitro methylation assay
Yeast tRNAGly and yeast G9-tRNAPhe (17) were prepared from in vitro transcripts and specifically labeled with 32P at the phosphate immediately 5′ to G9 using previously described methods (17). For specific activity assays, reactions containing G9-labeled tRNAGly, 50 mM Tris pH 8.0, 1.5 mM MgCl2 and 0.5 mM SAM were initiated with 10-fold serial dilutions of enzyme, as indicated. After incubation for 1–2 h at 30°C, reactions were stopped by addition of phenol: chloroform: isoamyl alcohol (25:24:1) and purified by phenol extraction and ethanol precipitation. The purified tRNA was then digested to single nucleotides using nuclease P1 (Sigma-Aldrich) leaving the 5′-32P label on methylated (p*m1G) or unmethylated (p*G) G9, which were resolved by cellulose thin-layer chromatography (TLC) in isobutyric acid: H2O:NH4OH (66:33:1) solvent. The plates were exposed to a phosphor screen and scanned using Typhoon™ imaging system (GE Healthcare) and quantified using using ImageQuant™ TL software (GE Healthcare).
SAM-dependence assays were performed using the above assay with 1 μM enzyme and 5 nM–500 μM SAM. The percent of methylated G9 (%P) was quantified and plotted as a function of SAM concentration and the data were fit to Equation (1) to yield the concentration of SAM required for half maximum product formation (K1/2) using Kaleidagraph software (Synergy).
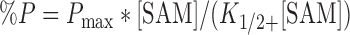 |
(1) |
Single-turnover kinetics and pH-rate analysis
Single turnover reactions contained 32P-G9 labeled tRNA under the same reaction conditions used for the specific activity assays above, but with ≥2 μM of each enzyme (to achieve >100-fold excess over [tRNA]). Aliquots were taken at desired time points and quenched with phenol: chloroform: isoamyl alcohol (25:24:1), purified, digested and analyzed by TLC as described above. The percent m1G9 (%P) formed at each time point was quantified and plotted as a function of time (t), and kobs was determined by fitting to a single exponential equation (Equation 2) using Kaleidagraph software (Synergy).
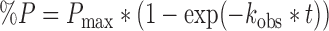 |
(2) |
For the pH-rate profile, single turnover rate constants were determined as above under varied pH conditions using an overlapping buffer system with one or more buffering agent in each reaction to control for buffer-dependent effects. The buffers used were: 25 mM sodium acetate + 50 mM Bis–Tris + 50 mM Tris (pH 5.0, 6.0, 7.0, 8.0, 9.0), 25 mM sodium acetate (pH 5.2), 50 mM Bis–Tris (pH 6.3, pH 6.5, pH 6.6), 50 mM Bis–Tris + 50 mM Tris (pH 7.2, pH 7.7, pH 8.3, pH 8.5, pH 8.8) and 50 mM Tris (pH 7.5). The kobs values obtained from three independent experiments were plotted as a function of pH and the pKa was calculated by fitting to Equation (3) using Kaleidagraph (Synergy), with kmax representing the pH-independent maximal rate.
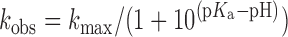 |
(3) |
To ensure that all kobs values were independent of the concentration of Trm10, rates were measured using at least 2 different excess enzyme concentrations for each wild type enzyme (hTRMT10A and ScTrm10) and for ScTrm10 D210A. An enzyme concentration of at least 1 μM was established to be saturating for all three enzymes, and the rates for all other variants were determined accordingly based on the similar KD,app for tRNA exhibited by these enzymes, using at least 2 μM enzyme in the assays.
Yeast complementation assay
Two previously described, otherwise isogenic strains of Saccharomyces cerevisiae, TRM10 and trm10Δ (16), were transformed with 2μ (high copy) LEU2 plasmids expressing various Trm10 genes (ScTRM10, hTRMT10A or D210N mutant of hTRMT10A) under the control of a galactose inducible promoter (17). Two independently isolated Leu+ transformants were each tested for 5-fluorouracil (5FU) sensitivity by replica plating onto SGal–leu plates containing varied concentrations of 5FU (0.1, 1, 25 μg /ml). Plates were grown at 30 or 37°C for 2–3 days before being photographed.
Fluorescence anisotropy
tRNA binding assays were performed by fluorescence anisotropy (FA) using the indicated enzymes and 5′-6-carboxyfluorescein labeled tRNAGly prepared as described previously (11). Anisotropy observed from two independent experiments was plotted as a function of enzyme concentration and fit to Equation (4) (FAmin – minimum anisotropy observed limit, FAmax – maximum anisotropy observed limit, n – Hill coefficient, [E] – enzyme concentration) using Kaleidagraph (Synergy) to obtain KD.
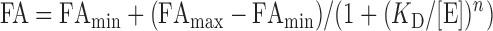 |
(4) |
Isothermal titration calorimetry (ITC)
ITC was used to evaluate the KD for SAM exhibited by wild type and variant (D210N, G206R) hTRMT10A enzymes. The experiment was carried out using a Microcal VP-ITC calorimeter. Purified enzymes were dialyzed overnight in a buffer containing 20 mM Tris–HCl pH 7.5 and 150 mM NaCl prior to use in the ITC experiment. The same dialysis buffer was used as the SAM dilution buffer. The experiment was carried out at 25°C by titrating SAM at a concentration of 2 mM into enzyme at a concentration of 100 μM. The isotherms obtained were analyzed and fit to equations corresponding to one binding site per enzyme on Origin 7.0 (Microcal) analysis software.
RESULTS
The proposed aspartate general base D210 is not critical for methylation activity
A mechanism involving acid-base catalysis (Figure 1B) has been proposed, with an aspartate (D210 in S. cerevisiae) acting as a general base to extract a proton from the target N-1 atom of guanosine for subsequent methylation, based on absolute conservation of this residue in all Trm10 sequences of both eukaryal and archaeal origin that have been examined to date (see representative alignment in Figure 2) and its location near the predicted active site in multiple crystal structures (20,25). To explicitly test this role for D210, we altered it to each of several other amino acids (A, N, K) in the context of ScTrm10, and measured the effects on m1G9 methylation activity with a previously characterized yeast tRNAGly substrate (Figure 3A) (16,17). Surprisingly, none of these alterations abolished the methylation activity on substrate tRNAGly, and only a modest decrease in activity was observed compared to wild-type, as judged by similar specific activities exhibited by the variant enzymes, with the greatest effect (∼10-fold) observed for D210K ScTrm10 (Figure 3A).
Figure 2.

Multiple sequence alignment of Trm10 homologs. Alignment of selected Trm10 homologs from Eukarya (SpTrm10, ScTrm10 plus the three human paralogs) and Archaea (Trm10 from S. acidocaldarius, SaTrm10) was performed using Clustal Omega (41–43). Conserved residues are highlighted by shading (using BoxShade), with the positions of the three absolutely conserved carboxylate-containing residues (D100, E111, D210) indicated by arrows and the location of the SPOUT SAM binding motif (referred to as Motif II in the original analysis of SPOUT sequences) indicated by the solid bar under the sequence. Numbers to the left of each sequence indicate the relevant residue numbering for each homolog; some N-terminal and C-terminal residues are omitted from the alignment for clarity.
Figure 3.

D210 is not essential for m1G9 activity of Trm10. (A) Activity assay of ScTrm10 with G9-labeled tRNAGly. Reactions contain 10-fold serial dilutions of purified ScTrm10; WT (wild-type) (3 μM-0.3 nM); D210N (3–0.003 μM); D210K (3–0.03 μM); D210A (3–0.003 μM); or no enzyme (–). Positions of the labeled m1G product (p*m1G) and unreacted substrate (p*G) are indicated. Specific activity of each enzyme was calculated based on the amount of each enzyme required to produce 20% product in one hour at 30°C and is labeled under the relevant panel for each enzyme. (B) Complementation of the yeast trm10Δ phenotype. An S. cerevisiae trm10Δ strain was transformed with plasmids expressing either wild-type ScTrm10 or human TRMT10A, or the human TRMT10A D210N variant, as indicated. Control strains (TRM10 and trm10Δ) transformed with empty vector are shown in the top two rows of each panel. Two independently isolated colonies for each strain were analyzed by replica plating onto S-Gal-leu media containing the indicated concentrations of 5-fluorouracil (0–25 ug/ml) and grown at either 30 or 37°C for 2–3 days before photographing.
To fully quantify these effects on catalysis, single turnover rates (kobs) of methylation were measured at saturating enzyme concentrations for the D210A and D210K variants, chosen to represent the least and most defective variants at this position, respectively. The effects (4-fold and 20-fold, respectively) on the observed rates with these variants were consistent with the overall defects judged by specific activity (Table 1). These effects were not restricted to ScTrm10, since alteration of the analogous residue to D210 in hTRMT10A to asparagine caused a similarly modest (5-fold) decrease in observed rate of the enzyme with the same tRNAGly substrate (Table 1). These relatively small defects more or less prove that D210 cannot play an essential role in driving methylation as a general base. To further demonstrate the functionality of the variant enzymes, we performed an in vivo complementation assay to test the ability of the hTRMT10A D210N variant to suppress the 5FU hypersensitive phenotype of trm10 deletion in S. cerevisiae. Growth of the trm10Δ strain is inhibited compared to a TRM10 wild-type strain in media containing 1 μg/ml 5FU (Figure 3B) (18). This growth defect is due to the loss of TRM10, since expression of a wild-type copy of either ScTrm10 or hTRMT10A complements this phenotype at either 30 or 37°C, while expression of the empty vector does not restore wild-type growth to the trm10Δ strain. Cells expressing the hTRMT10A D210N variant were similarly resistant to the effects of 5FU as either wild-type enzyme, consistent with the in vitro biochemical results (Figure 3B). These results provide in vitro and in vivo evidence against the essential nature of the D210 residue, making it unlikely that D210 functions as an obligate general base during catalysis.
Table 1. Kinetic parameters for m1G9 methylation of tRNAGly by Trm10 enzymes.
| Enzyme | k obs (min−1) | Fold defecta | pKa | K 1/2 (SAM) (μM) | K D (tRNA) (nM) |
|---|---|---|---|---|---|
| ScTrm10 | |||||
| Wild-type | 1.00 ± 0.13 | — | 8.1 ± 0.1 | UDc | 113.0 ± 3.2 |
| D210A | 0.24 ± 0.03 | 4 | 7.1 ± 0.1 | 4.3 ± 0.4 | ND |
| D210K | 0.05 ± 0.006 | 20 | 7.2 ± 0.1 | 91.8 ± 8.7 | 108.0 ± 6.9 |
| D100N | 0.44 ± 0.01 | 2 | 6.9 ± 0.1 | 0.03 ± 0.01 | 91.8 ± 14.7 |
| E111A | 0.18 ± 0.08 | 6 | 8.0 ± 0.1 | 1.4 ± 0.1 | 86.4 ± 9.9 |
| D100N+D210A (DM) | <0.002 | >500 | NDb | ND | 33.9 ± 3.6 |
| D100N+D210A+E111A (TM) | ND | ND | ND | ND | 43.7 ± 8.0 |
| G206A G207A | ND | ND | ND | 77.6 ± 9.9 | ND |
| hTRMT10A | |||||
| Wild-type | 0.34 ± 0.05 | — | 7.9 ± 0.2 | UDKD (ITC) = 17.7 ± 4.0 | 216.9 ± 40.6 |
| D210N | 0.065 ± 0.03 | 5 | 7.3 ± 0.1 | 7.84 ± 1.25KD (ITC) = 60.6 ± 10.3 | 127.7 ± 13.6 |
aFold defect calculated by kobs(wild-type)/kobs(variant).
bND = not determined.
cUD = undefined.
Analysis of the putative SAM binding site of Trm10
The crystal structures of ScTrm10 and Schizosaccharomyces pombe Trm10 (SpTrm10) were solved in the presence of a SAM analog, S-adenosyl homocysteine (SAH) and therefore aided in identifying residues that interact with SAM, which include the proposed general base residue D210 (25,26). D210 is embedded in a sequence motif that is characteristic of the SAM binding sequence of several other SPOUT family enzymes (Figure 2) (27). The crystal structure shows that D210 participates in an ionic interaction between the α-NH3+ group in the homocysteine moiety of SAH and its carboxylate side chain (25). To test the role of D210 as well as other nearby residues in the interaction with SAM, we made alterations to two highly conserved glycine residues in this sequence (G206, which interacts with the SAH ribose 2′-OH in the cofactor-bound structure (25), and G207) to alanine and tested the dependence of each variant enzyme on the concentration of SAM provided in the assay with tRNAGly. The constant K1/2 was defined as the concentration of SAM required to achieve half maximal activity for each enzyme. The parameter K1/2 could not be defined for either wild type enzyme (ScTrm10 and hTRMT10A) since significant levels of activity were observed even in the absence of added SAM for these two enzymes (Table 1, Supplementary Figure S2A and B). This result is likely due to co-purification of SAM with some fraction of these enzymes and the use of single-turnover enzyme excess conditions, as observed previously (16).
For all of the variants, however, methylation activity depended on the addition of SAM and comparison of the K1/2 suggests differences in these residues' interaction with the methyl donor. A strong dependence on [SAM] was observed with the G206A/G207A double mutant, as evident by its K1/2 of 77.6 ± 9.9 μM, compared with the relatively low K1/2 value (0.03 ± 0.01 μM) exhibited by D100N Trm10 (Table 1, Figure 4 and Supplementary Figure S2A). This result is consistent with the role of the two conserved glycine residues in SAM binding, as suggested by the position of SAH in the crystal structures and the severe catalytic defect exhibited by one of the human disease-associated alterations at G206 (11,25,26). Variants at D210 exhibited the next most SAM-dependent activity, with alterations to A or N resulting in intermediate values of K1/2, while the SAM-dependence of the D210K variant was similar to that of the double G206/G207 variant (Table 1, Figure 4). To directly assess the effects of alteration at D210 on SAM binding, we used isothermal titration calorimetry (ITC) to measure the KD,SAM of wild type and D210N variants of hTRMT10A (Table 1, Supplementary Figure S3). The human enzyme was used for ITC due to the high concentrations yielded from its purification (as necessary for ITC), but the similar behavior of hTRMT10A and ScTrm10 in our biochemical assays suggest that the results are likely relevant to ScTrm10 as well. The G206R variant of hTRMT10A that was previously shown to ablate methylation activity, likely due to an inability to bind SAM (11), was used as a control and exhibited no detectable binding by ITC, as expected (Supplementary Figure S3). The modest (∼3.5-fold) effect on KD,SAM of the D210N mutant enzyme measured by ITC is consistent with the SAM-dependence observed with the activity assay (Table 1). Thus we conclude that although D210 likely plays the previously suggested role in interaction with SAM, the presence of the wild-type carboxylate side chain is not essential for a productive interaction with SAM during methylation.
Figure 4.

Alteration of conserved residues in the SAM-binding motif affects SAM-dependence of methylation by ScTrm10. Methylation activity of each ScTrm10 enzyme with G9-labeled tRNAGly was quantified as described in methods and plots of percent conversion to m1G9 versus [SAM] were fit to Equation (1) to determine K1/2 (SAM) for each enzyme: G206A/G207A (open diamonds; K1/2 = 77.6 ± 9.9 μM), D210K (squares; K1/2 = 91.8 ± 8.7 μM), D210A (circles; K1/2 = 4.3 ± 0.4 μM).
Roles for two other conserved carboxylate residues near the presumed active site
Given the non-essential nature of D210, several alternatives to the originally-proposed D210 general base-mediated mechanism could be envisioned. Possibly another residue (instead of D210) functions as a general base. Alternatively, there could be redundancy in the active site, such that other residue(s) can function as a general base in the absence of D210. Finally, Trm10 may use another mechanism entirely that does not rely on a general base for catalysis. We looked for other conserved residues that could potentially act as an N1-proton abstracting general base and identified two candidates: a strictly conserved aspartate (D100) and a highly conserved glutamate (E111), which are both observed in reasonable proximity to the proposed active site in crystal structures (Figure 2) (25). Variants at each of these two positions were constructed to remove the carboxylate-containing side chain that could potentially accept a proton from N1. Interestingly, the D100A variant was insoluble when expressed in E. coli, suggesting a possible structural role for this residue and necessitating use of the readily purified D100N variant along with the E111A enzyme in these assays. Nonetheless, like the alterations at D210, variants at either of these positions again exhibited only modest defects in the single turnover rate of catalysis, effectively ruling out a unique role for any of these as a single general base (Table 1).
To test the idea of redundant function for the conserved acidic residues, we constructed double (D210A + D100N) and triple (D210A + D100N + E111A) variants of ScTrm10. The double mutant (DM) exhibited significant loss of activity compared to the individual mutants with some residual activity, while alteration of all three acidic residues (TM) completely abolished detectable methylation activity (Figure 5A). A theoretical kobs for the D210A + D100N double mutant, assuming independent effects of altering the two aspartates, was calculated using a double mutant cycle to be 0.11 min−1 based on the kobs measured for each single variant (Supplementary Figure S4). From the limited product formed in the assay with the DM variant (Figure 5A), an upper limit to kobs of 0.002 min−1 is estimated using the method of linear initial rates. Since this estimated kobs for ScTrm10 DM is at least 50-fold lower than that expected from an additive effect due to these alterations, D210 and D100 do not function completely independently in catalysis by Trm10. To address the possibility that the severe defects observed with the double and triple variants could be due to global structural effects, tRNA binding affinity was determined for each variant by fluorescence anisotropy (Figure 5B). These measurements revealed no significant difference from the wild-type enzyme, suggesting that although structural changes in the active site cannot be ruled out, significant misfolding is not the apparent cause of the low activity of the variants. The relatively weak activity associated with the double variants precluded measurement of K1/2 to assess whether the synergistic effect on catalysis is due in part to effects of these combined alterations on SAM-binding, although we note that neither D100 or E111 is predicted to interact directly with the cofactor in any available structure, possibly diminishing the likelihood of a SAM-binding contribution to the observed defects in catalysis. These results point toward an active site in which multiple residues are involved in catalysis, and argue against a single general base driven mechanism.
Figure 5.

Combinations of conserved D/E alterations in ScTrm10 severely affect Trm10 methylation activity. (A) Activity assay of ScTrm10 with G9-labeled tRNAGly. Reactions contain 10-fold serial dilutions of purified enzyme; WT (3–0.003 μM); DM (3–0.003 μM); TM (5.5–0.0055 μM); or no enzyme (–). Positions of the labeled m1G product (p*m1G) and unreacted substrate (p*G) are indicated. (B) Binding of indicated enzymes to tRNAGly measured with fluorescence anisotropy. Anisotropy was measured as described in methods with 5′-fluorescein modified tRNA and plotted as a function of increasing enzyme. KD,tRNA was determined by fit to the binding isotherm (Equation 4) using WT ScTrm10 (open circles; KD = 113 ± 3.2 nM), ScTrm10 DM (squares, KD = 33.9 ± 3.6 nM), ScTrm10 TM (diamonds, KD = 43.7 ± 8.0 nM). BSA was used as negative control (triangles).
pH-rate analysis suggests a single ionization dependent mechanism that is maintained in the absence of the conserved D/E residues
We performed pH-rate profile analyses to better understand the nature of the ionization(s) that were involved in m1G9 formation by Trm10. Measurements of the single turnover rate constant (kobs) of wild-type ScTrm10 and hTRMT10A were performed at various pH ranging from 5.0 to 9.0. Single turnover assay conditions with saturating concentrations of enzymes were used to isolate the maximal rate constant associated with the rate-limiting step for the reaction of the enzyme-substrate (ES) complex (either chemistry itself, or a conformational change that precedes a faster chemical step). Rate measurements at each pH value were independent of the concentration of Trm10 based on assays performed with at least two different concentrations of excess enzyme, thus ensuring that the kobs corresponds to the kmax for the rate-limiting step for reaction of the ES complex under these conditions. The concentration of SAM was 500 μM, well in excess of the measured SAM affinity (Table 1). The best fit to the pH-rate profile for wild-type ScTrm10 (Equation 3) was most consistent with the occurrence of a single basic ionization with a pKa of 8.1 ± 0.1 that is relevant for catalysis (Figure 6A). The same pH-rate analysis was performed with wild-type hTRMT10A, which showed a strikingly similar single ionization-dependent profile with a corresponding pKa of 7.9 ± 0.2 (Figure 6C).
Figure 6.

m1G9 formation by Trm10 is associated with a single basic ionization. Single turnover kobs (min−1) for tRNAGly methylation were measured at saturating enzyme concentrations at varied pH for (A) ScTrm10 WT and D210 variants; (B) ScTrm10 D100N and E111A variants and (C) hTRMT10A WT and D210N variant. WT enzymes are represented by open circles and solid fits and are repeated in all panels for better comparison with each variant. Note the difference in the Y-axis scale (all rates are reported as min−1) for each variant The fits correspond to the best fit to Equation (4) corresponding to a single ionization for each reaction and yield the following pKa values: ScTrm10 WT (pKa = 8.1 ± 0.1); ScTrm10 D210A (pKa = 7.1 ± 0.1); ScTrm10 D210K (pKa = 7.2 ± 0.1); ScTrm10 D100N (pKa = 6.9 ± 0.1); ScTrm10 E111A (pKa = 8.0 ± 0.1); hTRMT10A WT (pKa = 7.9 ± 0.2); hTRMT10A D210N (pKa = 7.3 ± 0.1).
To test whether the conserved carboxylates impact the observed pH-dependence of Trm10, pH-rate profiles were similarly determined for each of the single variant enzymes (Table 1). Enzyme concentrations were used for the variants that were saturating according to the wild-type analysis (≥2 μM), which is further supported by the similar KD,tRNA also measured by FA for these variant enzymes compared to WT (Table 1, Supplementary Figure S5). Although the magnitude of the observed rates differed as expected for each variant based on the fold-defects measured earlier, the overall shape of the pH-rate profiles was in each case similar to wild-type and best fit by the single ionization equation (Figure 6). Interestingly, however, the measured pKa values for variants at either D210 or D100 were all shifted in the acidic direction by ∼0.5–1 pH unit (Figure 6). In contrast, the E111A variant exhibited a nearly identical pH dependence to the wild-type enzyme (Figure 6B).
ScTrm10 does not utilize a catalytic metal ion-dependent mechanism
Recently, a detailed mechanistic study with a different SPOUT family m1G methyltransferase (TrmD) suggested an essential role for a metal ion (likely Mg2+) in catalysis (31). This metal likely promotes m1G formation by TrmD through stabilization of the developing negative charge on the C6 carbonyl via a direct interaction that was supported by metal rescue studies (31). Since the role of metal ions in Trm10 has not yet been investigated, we first tested the ability of EDTA to inhibit ScTrm10 activity and saw that unlike for TrmD, there was virtually no detectable effect on the extent of enzyme activity at concentrations of EDTA up to 0.5 mM (in large excess over the 4 nM ScTrm10 in the assay), suggesting that the two SPOUT m1G methyltransferases do not share this aspect of catalysis (Figure 7A). However, since the standard Trm10 purification conditions contain Mg2+ in the buffers (16,17), we re-purified ScTrm10 in buffer lacking any added metal and measured the rate of m1G9 catalysis with this enzyme in the absence and presence of Mg2+. Activity was readily detected in time courses using the enzyme purified under metal-free conditions, although addition of 5 mM MgCl2 to the assays resulted in an ∼10-fold increase in the observed rate, likely due to the optimal folding of and interaction with the tRNA substrate in the presence of Mg2+, as is frequently observed with enzymes that act on RNA (Figure 7B). Nonetheless, the relatively small magnitude of this effect on the overall rate (compared to the >300-fold effect exerted by Mg2+ on TrmD and its complete loss of activity in the presence of EDTA) argues against catalytically important roles for metal ions in m1G9 methylation by Trm10.
Figure 7.

Divalent metal ions are not essential for Trm10 catalytic activity. (A) ScTrm10 methylation activity on tRNAGly was measured in the presence of varied concentration of EDTA in the assay. The percent conversion of the labeled G9 to m1G9 catalyzed by 4 nM ScTrm10 was measured as described in methods and plotted versus [EDTA]. (B) Time courses of m1G9 methylation activity catalyzed by ScTrm10 purified under metal-free conditions on G9-tRNAPhe. Percent product formed under these single turnover conditions was plotted and fit to Equation (2) to yield the indicated kobs (shown in parentheses) for catalysis in the absence and presence of Mg2+ in the reactions; NE is no enzyme control.
DISCUSSION
The m1G tRNA modification has now been associated with three distinct families of methyltransferases acting at two tRNA positions, G37 and G9. Trm10 is so far uniquely observed to catalyze the methylation at position 9, while the two other enzymes (TrmD in Bacteria and Trm5 in Archaea and Eukarya) act at G37 (16,32,33). The two m1G37 enzymes themselves comprise different and evolutionarily unrelated classes of methyltransferases (Trm5 is a Class I Rossman-fold containing enzyme and TrmD is a SPOUT methyltransferase) and, perhaps not surprisingly in light of their distinct origins, use dissimilar mechanisms for catalysis (31,34–36). Here, we investigated the mechanism of m1G9 formation by two different eukaryotic Trm10 enzymes and demonstrate through biochemical and kinetic studies that Trm10 represents yet another distinct family in terms of its mechanism of methylation, since it does not share many features that are associated with either Trm5 or its other SPOUT relative TrmD. Here we showed that a single general base analogous to the aspartate/glutamate residues that are highly conserved and catalytically important in other methyltransferases is not an obligate feature of the mechanism of Trm10. This conclusion is supported by first, our observation that the activity of variants altered at any of the three highly conserved candidate residues of Trm10 are only modestly affected in activity (Figure 3, Table 1), and second, because the similar shape of the pH-rate profiles of these same variants suggests that the basic ionization required for catalysis (discussed further below) is also preserved upon alteration of these residues (Figure 6). Moreover, unlike TrmD, Trm10 does not utilize a catalytically-important metal ion to stabilize a negatively charged O6 that possibly forms during the course of the methylation reaction (Figure 7). This conclusion is supported by the observation of substantial activity associated with enzyme purified in the absence of metal or treated with high concentrations of the chelator EDTA. Taken together, these results underscore the lack of a unified mechanism for N-1 methyltransferases, even among related members of the larger SPOUT superfamily, and raise additional questions about the catalytic features employed by Trm10.
The requirement for a general base to abstract a proton from the target N1 position of G9 was originally suggested based on an observation of increased m1G methylation with reaction pH and the idea that this might be due to an enzyme side chain capable of deprotonating the N1 atom during methylation (24). Two different groups identified D210 as a probable general base due to its universal conservation and what appeared to be a total loss of activity associated with alterations at this position in three different enzymes (ScTrm10, SpTrm10 and a human Trm10 ortholog TRMT10C) (20,25). However, limitations of the single time point enzyme assays used to evaluate activities of each of these D210 variant enzymes combined with relatively low concentrations of SAM used in those assays likely resulted in this misleading observation. Using more detailed kinetic assays and multiple substitutions at this position, here we demonstrated that D210 is not a catalytically critical residue, since the effects of its alteration are much less severe than the several hundred to ∼103-fold defects in the observed rate upon alteration of the relevant general base residues in Trm5 and TrmD, respectively (30,31,34). We note that although rates were not reported in a similar analysis of the residue corresponding to D210 in an archaeal m1A9 methyltransferase, the general effect on activity at high concentrations of enzyme appears to be similar in magnitude to the effects observed here (26). Moreover, alteration of either of two other highly conserved acidic residues (D100 and E111) resulted in similarly modest effects on the observed rate (Figure 3, Table 1), suggesting that none of these residues is likely to function as a single obligate general base during catalysis.
Interestingly, we observed greater than additive effects of combined alterations at the conserved D/E residues. Three possibilities (not necessarily mutually exclusive) seem most consistent with these results. First, the Trm10 active site may allow promiscuity in terms of the identity of a residue that accepts the N1 proton from G9, and the modest effects on activity with single variants could be due to more or less redundant roles for these conserved residues. We consider this the least likely possibility due to the extreme conservation of all three of these residues (D100, E111 and D210) in all enzymes identified to date (only a small number of which are shown in Figure 2). Second, although the observation that the double and triple variants exhibit a similar tRNA affinity to wild-type Trm10 suggests no gross structural defects, it is possible that these three residues play an interdependent role in maintaining structure at the active site in the context of the bound tRNA. This could result in the observed non-additive catalytic defects associated with loss of more than one of these side chains. Finally, the mechanism of N1 methylation by Trm10 may instead be the result of a specific base mediated process where the N1 proton is accepted by bulk solvent. An intriguing possibility is that the carboxylate-containing residues could be involved indirectly in catalysis by helping to organize a solvent network to facilitate this process, and that disruption of this network requires loss of more than one of these residues. This type of mechanism has been documented through extensive structural characterization of arylalkylamine N-acetyltransferases, where two histidines originally thought to serve as general base residues exhibited similarly modest effects on catalysis when altered, but were later shown to form part of a ‘proton wire’ that facilitates deprotonation of a substrate amine to activate it for attack on acetyl coenzyme A (37–39). Further structural and biochemical characterization of Trm10 will be essential to evaluate these and other hypotheses regarding the role(s) of these absolutely conserved residues, as well as the roles of others, such as the invariant Q118 residue that is located in a possible RNA substrate binding pocket visible in both fungal structures (25).
If Trm10 does not employ an enzymatic general base mechanism, this raises the question as to the identity of the ionizing residue associated with the observed pKa of ∼8 for the wild-type enzymes. Here, a strong possibility is that the results are consistent with those observed for both Trm5 and TrmD in which the catalytic pKa reflects the ionization of the N1 atom of the target guanine itself, possibly promoted by an induced fit process that occurs upon formation of the catalytic ES complex (33–35). Whether the observed maximal [Trm10]-independent rate corresponds to the actual chemical step of N1 deprotonation, or to a previous slower step such as a conformational rearrangement to form the ES complex, remains to be determined by further kinetic analysis. Nonetheless, the acid shift of the catalytic pKa by ∼ 1 unit observed upon alteration of D210 and D100 is intriguing, since this is in the opposite direction from the shift expected toward the solution pKa of ∼9.5 for the N1 proton. If these residues were involved in organization of the active site to promote deprotonation of the target N1 atom, their loss could be expected to result in a basic shift toward the non-enzyme associated pKa. We note that the acidic pKa shift is not correlated with the overall electrostatic character of the active site, since replacement of D210 with either uncharged A or positively charged K causes the same acid-shift in the observed pKa. The existing structures do not provide insight into the possible molecular basis of this effect, since the three carboxylates are not located close enough to interact in the absence of bound tRNA, and the precise nature of their interaction with G9 (if any) is not known (Figure 8). Fully unraveling the nature of the active site in Trm10 is an important future direction that will require additional structural and biochemical analysis, especially of complexes that contain tRNA substrate.
Figure 8.

Locations of conserved carboxylate residues in SPOUT methyltransferases. (A) Crystal structure of Saccharomyces cerevisiae Trm10 (PDB:4JWJ) (25) showing the positions of D210, D100 and E111. The S-adenosyl homocysteine is shown in green. (B) Crystal structure of Haemophilus influenzae TrmD (PDB: 4YVI) (40) showing the active site composed of residues from two subunits. The SAM analog (sinefungin in green) is bound to subunit A (opaque) and the catalytic residue D169* of subunit B (transparent) is shown in red.
In addition to its previously proposed role as a general base, D210 had also been implicated in a role in SAM binding based on its location within hydrogen bonding distance of the homocysteine amino group in the SAH-bound structures. Consistent with this role, D210 variants exhibited a significant dependence on the concentration of SAM in the assays, with the D210K alteration exhibiting similar SAM-dependence to the variants with alterations at the two conserved G residues that are hallmarks of the SPOUT SAM binding motif (Figure 4). Nonetheless, the readily detectable activity of these variants combined with the moderate effect on SAM-binding by ITC supports the idea that D210 is involved in, but not critical for SAM binding. These results with the yeast and human Trm10 D210 variants agree with those observed for the m1A9 catalyzing Sulfolobus acidocaldarius Trm10 (SaTrm10) where altering the residue corresponding to D210 similarly does not abolish SAM binding (26). These data add support to the crystallographically observed SAM-binding site and suggest that any conformational changes associated with catalysis, such as a rate-determining conformational change that facilitates loss of the N1 proton from G9, are more likely to involve the interaction with tRNA than with the SAM methyl donor. In any case, a rearrangement of the active site in the presence of the substrate tRNA appears likely and it is clear that Trm10 exhibits significant deviation from mechanisms exhibited by other members of the SPOUT family. An active site comprised of multiple catalytic residues could serve to compensate for Trm10’s monomeric quaternary structure compared to the dimeric structure employed by TrmD in which residues from the two subunits form its active site, with the catalytic aspartate general base arising from subunit B and the methyl group is donated from SAM bound by subunit A (30,40) (Figure 6B). These and other intriguing issues await further mechanistic and structural characterization.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jeremy C. Henderson for construction of several Trm10 variant plasmids used in this work.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM087543 to J.E.J]. Funding for open access charge: National Institutes of Health [GM087543].
Conflict of interest statement. None declared.
REFERENCES
- 1. Phizicky E.M., Alfonzo J.D.. Do all modifications benefit all tRNAs?. FEBS Lett. 2010; 584:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jackman J.E., Alfonzo J.D.. Transfer RNA modifications: nature's combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA. 2013; 4:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M. et al. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 2013; 41:D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jühling F., Mörl M., Hartmann R.K., Sprinzl M., Stadler P.F., Pütz J.. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009; 37:D159–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Machnicka M.A., Olchowik A., Grosjean H., Bujnicki J.M.. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014; 11:1619–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Urbonavičius J., Qian Q., Durand J.M.B., Hagervall T.G., Björk G.R.. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J. 2001; 20:4863–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yarian C., Townsend H., Czestkowski W., Sochacka E., Malkiewicz A.J., Guenther R., Miskiewicz A., Agris P.F.. Accurate translation of the genetic code depends on tRNA modified nucleosides. J. Biol. Chem. 2002; 277:16391–16395. [DOI] [PubMed] [Google Scholar]
- 8. Freude K., Hoffmann K., Jensen L.-R., Delatycki M.B., Portes V. Des, Moser B., Hamel B., Van Bokhoven H., Moraine C., Fryns J.-P. et al. Report mutations in the FTSJ1 gene coding for a novel S-adenosylmethionine—binding protein cause nonsyndromic X-linked mental retardation. Am. J. Hum. Genet. 2004; 75:305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alazami A.M., Hijazi H., Al-Dosari M.S., Shaheen R., Hashem A., Aldahmesh M.A., Mohamed J.Y., Kentab A., Salih M.A., Awaji A. et al. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J. Med. Genet. 2013; 50:425–430. [DOI] [PubMed] [Google Scholar]
- 10. Igoillo-Esteve M., Genin A., Lambert N., Désir J., Pirson I., Abdulkarim B., Simonis N., Drielsma A., Marselli L., Marchetti P. et al. tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet. 2013; 9:e1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gillis D., Krishnamohan A., Yaacov B., Shaag A., Jackman J.E., Elpeleg O.. TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J. Med. Genet. 2014; 51:581–586. [DOI] [PubMed] [Google Scholar]
- 12. Abbasi-Moheb L., Mertel S., Gonsior M., Nouri-Vahid L., Kahrizi K., Cirak S., Wieczorek D., Motazacker M.M., Esmaeeli-Nieh S., Cremer K. et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012; 90:847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khan M.A., Rafiq M.A., Noor A., Hussain S., Flores J. V, Rupp V., Vincent A.K., Malli R., Ali G., Khan F.S. et al. Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012; 90:856–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Torres A.G., Batlle E., Ribas De Pouplana L.. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014; 20:306–314. [DOI] [PubMed] [Google Scholar]
- 15. Towns W.L., Begley T.J.. Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: activities, predications, and potential roles in human health. DNA Cell Biol. 2012; 31:434–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jackman J.E., Montange R.K., Malik H.S., Phizicky E.M.. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA. 2003; 9:574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swinehart W.E., Henderson J.C., Jackman J.E.. Unexpected expansion of tRNA substrate recognition by the yeast m1G9 methyltransferase Trm10. RNA. 2013; 19:1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gustavsson M., Ronne H.. Evidence that tRNA modifying enzymes are important in vivo targets for 5-fluorouracil in yeast. RNA. 2008; 14:666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holzmann J., Frank P., Löffler E., Bennett K.L., Gerner C., Rossmanith W.. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008; 135:462–474. [DOI] [PubMed] [Google Scholar]
- 20. Vilardo E., Nachbagauer C., Buzet A., Taschner A., Holzmann J., Rossmanith W.. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012; 40:11583–11593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zung A., Kori M., Burundukov E., Ben-Yosef T., Tatoor Y., Granot E.. Homozygous deletion of TRMT10A as part of a contiguous gene deletion in a syndrome of failure to thrive, delayed puberty, intellectual disability and diabetes mellitus. Am. J. Med. Genet. A. 2015; 167:3167–3173. [DOI] [PubMed] [Google Scholar]
- 22. Yew T.W., McCreight L., Colclough K., Ellard S., Pearson E.R.. tRNA methyltransferase homologue gene TRMT10A mutation in young adult-onset diabetes with intellectual disability, microcephaly and epilepsy. Diabet. Med. 2015; doi:10.1111/dme.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Narayanan M., Ramsey K., Grebe T., Schrauwen I., Szelinger S., Huentelman M., Craig D., Narayanan V.. Case Report: Compound heterozygous nonsense mutations in TRMT10A are associated with microcephaly, delayed development, and periventricular white matter hyperintensities. F1000Research. 2015; 4:912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kempenaers M., Roovers M., Oudjama Y., Tkaczuk K.L., Bujnicki J.M., Droogmans L.. New archaeal methyltransferases forming 1-methyladenosine or 1-methyladenosine and 1-methylguanosine at position 9 of tRNA. Nucleic Acids Res. 2010; 38:6533–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shao Z., Yan W., Peng J., Zuo X., Zou Y., Li F., Gong D., Ma R., Wu J., Shi Y. et al. Crystal structure of tRNA m1G9 methyltransferase Trm10: insight into the catalytic mechanism and recognition of tRNA substrate. Nucleic Acids Res. 2014; 42:509–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Laer B., Roovers M., Wauters L., Kasprzak J.M., Dyzma M., Deyaert E., Kumar Singh R., Feller A., Bujnicki J.M., Droogmans L. et al. Structural and functional insights into tRNA binding and adenosine N1-methylation by an archaeal Trm10 homologue. Nucleic Acids Res. 2016; 44:940–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tkaczuk K.L., Dunin-Horkawicz S., Purta E., Bujnicki J.M.. Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinformatics. 2007; 8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lv F., Zhang T., Zhou Z., Gao S., Wong C.C., Zhou J.-Q., Ding J.. Structural basis for Sfm1 functioning as a protein arginine methyltransferase. Cell Discov. 2015; 1:15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Watanabe K., Nureki O., Fukai S., Ishii R., Okamoto H., Yokoyama S., Endo Y., Hori H.. Roles of conserved amino acid sequence motifs in the SpoU (TrmH) RNA methyltransferase family. J. Biol. Chem. 2005; 280:10368–10377. [DOI] [PubMed] [Google Scholar]
- 30. Elkins P.A., Watts J.M., Zalacain M., Van Thiel A., Vitazka P.R., Redlak M., Andraos-Selim C., Rastinejad F., Holmes W.M.. Insights into catalysis by a knotted TrmD tRNA methyltransferase. J. Mol. Biol. 2003; 333:931–949. [DOI] [PubMed] [Google Scholar]
- 31. Sakaguchi R., Lahoud G., Christian T., Gamper H., Hou Y.-M.. A divalent metal ion-dependent N(1)-methyl transfer to G37-tRNA. Chem. Biol. 2014; 21:1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Byström A.S., Björk G.R.. Chromosomal location and cloning of the gene (trmD) responsible for the synthesis of tRNA (m1G) methyltransferase in Escherichia coli K-12. MGG Mol. Gen. Genet. 1982; 188:440–446. [DOI] [PubMed] [Google Scholar]
- 33. Brulé H., Elliott M., Redlak M., Zehner Z.E., Holmes W.M.. Isolation and characterization of the human tRNA-(N1G37) methyltransferase (TRM5) and comparison to the Escherichia coli TrmD protein. Biochemistry. 2004; 43:9243–9255. [DOI] [PubMed] [Google Scholar]
- 34. Christian T., Gamper H., Hou Y.. Conservation of structure and mechanism by Trm5 enzymes. RNA. 2013; doi:10.1261/rna.039503.113.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Christian T., Lahoud G., Liu C., Hoffmann K., Perona J.J., Hou Y.-M.. Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. RNA. 2010; 16:2484–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Christian T., Hou Y.-M.. Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J. Mol. Biol. 2007; 373:623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klein D.C. Arylalkylamine N-acetyltransferase: ‘The timezyme’. J. Biol. Chem. 2007; 282:4233–4237. [DOI] [PubMed] [Google Scholar]
- 38. Dempsey D.R., Carpenter A.-M., Ospina S.R., Merkler D.J.. Probing the chemical mechanism and critical regulatory amino acid residues of Drosophila melanogaster arylalkylamine N-acyltransferase like 2. Insect Biochem. Mol. Biol. 2015; 66:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hickman A.B., Namboodiri M.A., Klein D.C., Dyda F., Nakatani Y., Burley S.K., Zatz M., Iuvone P.M., Rodriguez I.R., Begay V. et al. The structural basis of ordered substrate binding by serotonin N-acetyltransferase: enzyme complex at 1.8 A resolution with a bisubstrate analog. Cell. 1999; 97:361–369. [DOI] [PubMed] [Google Scholar]
- 40. Ito T., Masuda I., Yoshida K., Goto-Ito S., Sekine S., Suh S.W., Hou Y.-M., Yokoyama S.. Structural basis for methyl-donor-dependent and sequence-specific binding to tRNA substrates by knotted methyltransferase TrmD. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E4197–E4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., Mcwilliam H., Remmert M., Sö Ding J. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011; 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R.. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010; 38:W695–W699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McWilliam H., Li W., Uludag M., Squizzato S., Park Y.M., Buso N., Cowley A.P., Lopez R.. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013; 41:W597–W600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.