


Abstract
The automated transcript discovery and quantification of high-throughput RNA sequencing (RNA-seq) data are important tasks of next-generation sequencing (NGS) research. However, these tasks are challenging due to the uncertainties that arise in the inference of complete splicing isoform variants from partially observed short reads. Here, we address this problem by explicitly reducing the inherent uncertainties in a biological system caused by missing information. In our approach, the RNA-seq procedure for transforming transcripts into short reads is considered an information transmission process. Consequently, the data uncertainties are substantially reduced by exploiting the information transduction capacity of information theory. The experimental results obtained from the analyses of simulated datasets and RNA-seq datasets from cell lines and tissues demonstrate the advantages of our method over state-of-the-art competitors. Our algorithm is an open-source implementation of MaxInfo.
INTRODUCTION
Due to the ever-increasing development of next-generation sequencing (NGS) technology in genome biology (1–4), powerful quantitative methods are needed to depict inherent gene regulation and the transcriptome landscape from high-throughput RNA sequencing (RNA-seq) data (5–7). At the RNA level, isoform identification and abundance estimation are two important approaches for evaluating heterogeneous transcriptional functions, and their use in NGS studies can reveal the underlying mechanisms of disease and lead to novel insights. Transcript (isoform) assembly is performed to structurally recover the splicing isoform variants of expressed genes from a large quantity of short sequencing reads. Abundance estimations (transcript quantification) quantitatively evaluate the expression levels of the discovered isoforms. However, the only available data for de novo assembly in these two inference tasks are incomplete sequencing results of isoform fragments. Obtaining a complete understanding from limited observations is essentially an ill-posed mathematical problem, and significant uncertainties arise as a result of missing information.
Conventional transcript discovery and quantification methods employ parametric statistical models established from various perspectives, e.g. probabilistic generative models (8–11) and linear regressions (12–14). Although their mathematical formulations broadly differ, the inherent concepts fall into similar data-fitting categories. The process for the transformation of transcripts to RNA-seq reads introduces high-level uncertainties caused by missing information and data ambiguities. For example, the indetermination of transcript components, the multiple mapping of short RNA-seq reads to isoforms and non-uniform read distributions over the isoforms (15–17) are all unknown factors that are difficult to control. When the data-fitting process involves too many uncertainties, the estimated isoforms may be inaccurate and exhibit great differences from the true isoforms (18–21).
Some data-fitting approaches rely on additional information to reduce data uncertainties and might require partial or full genome annotations for transcript assembly. SLIDE (14) utilizes gene annotations to locate subexons. Although iReckon (9) is more advanced, it still requires the start and end sites of transcripts. While genome annotations are available for certain species, novel gene splicing events are continually being discovered, and the annotation process has not been completed (13,14,22). Various annotation-free methods, such as Cufflinks (11), RSEM (23) and IsoLasso (13), are also available. However, the accuracies of these methods are still relatively low, and methods with better performance are desired. In addition, transcripts identified by different methods exhibit great diversity, and this diversity has been observed even among transcripts identified by methods based on similar mathematical assumptions (24). Therefore, more accurate and general approaches for annotation-free transcript inferences are highly desired.
Rather than exploiting the aforementioned data-fitting strategy, a more reasonable method that directly targets the uncertainties in the system is useful. Here we introduce a maximal information transduction pursuit (MaxInfo) approach for the simultaneous identification and quantification of isoforms based on information coding theory. In this approach, the isoforms and reads are regarded as the ‘signal sources’ and ‘short codes’ of an information transmission channel, respectively. The uncertainties in the channel are then reduced by maximizing the transduction capability of the information system. Transduction capacity (a.k.a. channel capacity) is conventionally quantified by mutual information, which is a formal term defined in information theory (25,26). Intuitively, this transduction capacity well depicts how much information about the isoform is well encoded on short reads after the RNA-seq process. Experiments based on simulated datasets demonstrated that MaxInfo consistently outperforms state-of-the-art methods for transcript and gene prediction. Moreover, MaxInfo showed to exhibit good performance in experiments based on six different datasets of human and Drosophila melanogaster tissues. MaxInfo is also flexible and can be run with reference annotations. The open-source software MaxInfo is available at http://maxinfo.sourceforge.net.
MATERIALS AND METHODS
MaxInfo dissects RNA-seq processes based on information transduction
In Shannon's information theoretic configuration, a fundamental information transduction system (27) is always composed of three parts: the information source, the coding channel and the receiver terminal. Briefly, the information source continually sends signals to the coding channel, where the signals are coded into short codes that accumulate in the receiver terminal. However, information loss between the original signal and the short codes may occur due to channel noise and the shortening process. A basic pursuit in information science is to identify the signals that exhibit minimal information loss after passing through a particular coding channel (27,28). Therefore, once the measurements (short codes) reach the receiver, the property of the signals from the information source can be characterized. This process has been described in the context of the transduction capacity problem in information theory (27–29).
The natural relationships between the RNA-seq process and the aforementioned information transmission system are of interest. As shown in Figure 1A, DNA can be viewed as an information source that sends various transcripts (signals) with different probabilities (abundances) through an RNA-seq channel (coding channel) to code the transcripts as short reads (codes). A reduction of uncertainties and errors in the biological signaling process is achieved by maximizing the information transmitted through the RNA-seq channel. Mathematically this process is modeled through the joint identification of transcripts T and their generative probabilities (abundances) 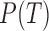 at exhibit maximal mutual information with the reads R at the receiver, i.e.
at exhibit maximal mutual information with the reads R at the receiver, i.e.  . This formulation is the well-known channel capacity (27) pursuit in information science. Intuitively, mutual information (25,30) measures the mutual dependence of putative isoforms and sequencing reads, which ideally depicts the level of uncertainty regarding the remaining transcripts by observing the reads and vice versa.
. This formulation is the well-known channel capacity (27) pursuit in information science. Intuitively, mutual information (25,30) measures the mutual dependence of putative isoforms and sequencing reads, which ideally depicts the level of uncertainty regarding the remaining transcripts by observing the reads and vice versa.
Figure 1.

Overview of MaxInfo. (A) Dissect RNA-seq procedures from the perspective of information transduction. The RNA-seq procedures construct a coding channel that transmits the information from the source to the receiver. On both terminals of the channel, isoforms are the signal source and short reads are the encoded codes. (B) Algorithmic gene prediction and candidate isoform reconstruction. For illustration purposes, two genes (A and B) are located on the genome and determined by the read distribution. Within gene A, eight subexons are identified and used as nodes to construct the directed graph. A pair of source (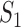 ) and sink (
) and sink (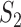 ) nodes are added to the graph to identify the start/end exon of a putative isoform. (C) Information transduction capacity model.
) nodes are added to the graph to identify the start/end exon of a putative isoform. (C) Information transduction capacity model. 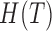 and
and 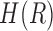 represent the entropies of transcripts and reads, respectively.
represent the entropies of transcripts and reads, respectively.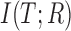 is the mutual information and used to measure the information content shared by the transcripts and the reads. A probabilistic graphical model (in the rectangle) is incorporated to depict the read generation procedures from transcripts (T) to RNA-seq data (R).
is the mutual information and used to measure the information content shared by the transcripts and the reads. A probabilistic graphical model (in the rectangle) is incorporated to depict the read generation procedures from transcripts (T) to RNA-seq data (R). 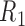 ,
,  indicate a pair of reads (paired-end). In the graphical model, S and L represent the starting position along the transcript and the length of the fragment, respectively, and Q describes the match quality of the read alignment.
indicate a pair of reads (paired-end). In the graphical model, S and L represent the starting position along the transcript and the length of the fragment, respectively, and Q describes the match quality of the read alignment.
Inspired by the information theoretic model, we propose a maximal information transduction estimation approach, MaxInfo, for transcriptome analyses using RNA-seq. In this method, sequencing reads are first aligned to the reference genome (Figure 1B), and MaxInfo then begins the initial gene and transcription start/end site predictions. After generating the predictions, a directed graph is built for path selection, and the paths connecting the source node (the node with only outgoing edges) and the sink node (the node with only incoming edges) are regarded as possible isoforms. Consequently, MaxInfo accomplishes simultaneous isoform identifications and abundance estimations based on maximal information transduction capacity (Figure 1C). The uncertainties and errors generated in all the previous steps, such as the gene prediction step, are reduced and refined in this step. In addition, by considering the generative mechanism of RNA-seq data, a probabilistic likelihood term is incorporated into the mutual information term to improve the estimation accuracy.
Initial gene prediction and coarse isoform selection
When implemented in the de novo assembly mode, MaxInfo performs gene predictions directly from the read distribution and detected junctions. Gene prediction processes generally consist of three steps: subexon discovery, gene boundary determinations and transcription start/end sites predictions. Reads are initially aligned to the genome with junction-sensitive tools in TopHat 2, and the junctions spanned by split reads are considered potential splice sites. Suspicious splice sites with weak read support are excluded to reduce assembly errors. Two types of expressed segments are marked as putative subexons: the region between the adjacent 3′ end and 5′ end splice sites and the region between alternative splice sites (both 5′ ends or both 3′ ends).
After assembling the subexons, MaxInfo determines the gene boundaries and allocates subexons into different gene loci. Gene loci are initially identified with respect to their orientation information. If junctions reported by TopHat 2 are marked with different orientations, they should belong to different genes. In a local genetic region, MaxInfo uses high-quality subexons to estimate the probabilistic distribution of subexon lengths, and extremely long subexons are noted as suspicious segments that likely span two gene loci. Within these suspicious segments, MaxInfo locates gene boundaries in positions that present an obvious discontinuity of read distribution.
With the predicted gene structures, a directed graph is built to interpret the splicing variants of the gene. On the graph, nodes represent subexons obtained via de novo assembly, and two nodes are linked by a directed edge if their corresponding subexons are adjacent or spanned by split reads. A pair of source/sink nodes is added to the graph to connect the potential transcription start/end sites of a putative isoform. Entire possible isoforms can be enumerated through path searches over the graph. To reduce the size of the candidate isoform set, we employed the concept of ‘flow’ on the directed graph for isoform selection. Specifically, flow on an edge is defined as the number of reads spanning the linked nodes (subexons). Each path p is evaluated according to the flow cost 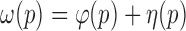 along with the balance cost
along with the balance cost  d connectivity cost
d connectivity cost 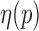 . The balance cost measures the possibility of the candidate isoform with considered beginning and ending exons, while the connectivity cost measures the probability of the putative isoform of a certain length (Supplementary Note 1). One path is discarded if its cost exceeds the threshold. We should note that the isoforms selected in this step are only preliminary coarse candidates for subsequent precise selection by the information theoretic model.
. The balance cost measures the possibility of the candidate isoform with considered beginning and ending exons, while the connectivity cost measures the probability of the putative isoform of a certain length (Supplementary Note 1). One path is discarded if its cost exceeds the threshold. We should note that the isoforms selected in this step are only preliminary coarse candidates for subsequent precise selection by the information theoretic model.
Information theoretic model
MaxInfo exploits the principle of information transduction to simultaneously conduct isoform discovery and abundance estimation from the coarse isoform set. In this process, the RNA-seq processing technique is analogous to a noisy information channel that encodes the signal source (isoform) into short codes (RNA-seq reads) (Figure 1A). In this context, reducing uncertainties during isoform discovery is subject to an information theoretic optimization by maximizing the information transmitted through the channel. Mathematically, the channel capacity is defined as follows:
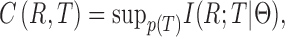 |
(1) |
where, 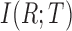 is the mutual information between the reads and isoforms,
is the mutual information between the reads and isoforms, 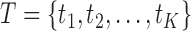 is the set of isoform candidates and
is the set of isoform candidates and  represents the observed RNA-seq (paired-end or single-end) reads.
represents the observed RNA-seq (paired-end or single-end) reads.  represents the generative probability of the isoform, which is related to the abundance. For clarity, we define
represents the generative probability of the isoform, which is related to the abundance. For clarity, we define  and
and 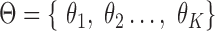 . It can be seamlessly converted to a formal abundance measurement, e.g. fragments per kb per million reads (FPKM).
. It can be seamlessly converted to a formal abundance measurement, e.g. fragments per kb per million reads (FPKM).
Channel capacity defines the maximum amount of information that can be transmitted throughout a noisy channel, and its objective function is quantitatively defined in the probabilistic form with the parameter set 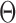 :
:
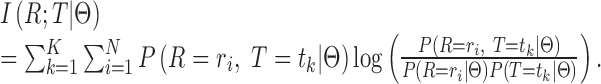 |
(2) |
Intuitively, the mutual information term can be explained as the level of uncertainty on the isoforms that is reduced after observing the RNA-seq data. Mathematically, this value should be maximized to reduce the uncertainty. Using Equation (2), we attempt to solve  and
and 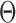 based on the sequenced reads
based on the sequenced reads  .
.
In addition to modeling the uncertainty from the isoforms to the reads, another issue that should be addressed is the likelihood of read generation, i.e. 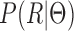 . In detail, this term depicts the probability of generating RNA-seq data that explain how the estimated model fits the data. This general objective has also been adopted by other data-fitting approaches (31). Using this term alone for model estimation is not reasonable because
. In detail, this term depicts the probability of generating RNA-seq data that explain how the estimated model fits the data. This general objective has also been adopted by other data-fitting approaches (31). Using this term alone for model estimation is not reasonable because 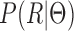 tends to use complex model structures (e.g. many isoforms) to over-fit the observed RNA-seq data. However, when considering mutual information term, the uncertainty regarding the read source of the isoform will be high if complex isoforms are used. Therefore, the optimization process will force to obtain simple model structures in order to minimize the uncertainty. Accordingly, the objective of the MaxInfo method is as follows:
tends to use complex model structures (e.g. many isoforms) to over-fit the observed RNA-seq data. However, when considering mutual information term, the uncertainty regarding the read source of the isoform will be high if complex isoforms are used. Therefore, the optimization process will force to obtain simple model structures in order to minimize the uncertainty. Accordingly, the objective of the MaxInfo method is as follows:
 |
(3) |
where,  is the log-likelihood term. The optimization objective function involves two additive terms: log-likelihood and mutual information.
is the log-likelihood term. The optimization objective function involves two additive terms: log-likelihood and mutual information.  balances the relative importance of these two terms. In our approach, we selected the value based on simulation studies and then varied it over a relatively wide range (from 0.001 to 1000) and observed the corresponding performances of MaxInfo under these different parameters. We found that different parameter settings over a flexible range (from 0.1 to 100) all yielded reasonable recoveries (Supplementary Note 3).
balances the relative importance of these two terms. In our approach, we selected the value based on simulation studies and then varied it over a relatively wide range (from 0.001 to 1000) and observed the corresponding performances of MaxInfo under these different parameters. We found that different parameter settings over a flexible range (from 0.1 to 100) all yielded reasonable recoveries (Supplementary Note 3).
The two terms in (3) require explicit probabilistic definitions. Here, we model the read generation mechanism using a graphical model (Figure 1C) that considers the starting position of fragmentation, the length of the selected fragments and the read matching quality (Supplementary Note 2). The above model is solved within an Expectation-Maximization (EM) framework (32–34) to iteratively and alternatively update the latent variables  and the model parameters (
and the model parameters ( ) (Supplementary Note 3).
) (Supplementary Note 3).
Forward selection for isoform set refinement
Mathematically, the optimization in (1) is not convex and could guarantee only the local optimum. Therefore, for complex gene structures, the EM solution is implemented multiple times with varying initializations. If the identified isoforms of different runs are not consistent, a sequential forward isoform selection procedure is adopted. Before the sequential selection, we assign a confidence score to each isoform, and the isoforms with low confidence are excluded.
In the forward selection,  and
and  are defined as the selected and unselected isoform set after the kth selection, respectively. Then, isoform
are defined as the selected and unselected isoform set after the kth selection, respectively. Then, isoform 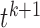 is selected in the
is selected in the 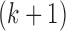 th selection with the function
th selection with the function
 |
(4) |
The sequential forward isoform selection is stopped when the objective function reaches the maximum.
Implementation with genome annotation
MaxInfo also works when the genome annotations are known. In the software, MaxInfo combines the initial gene and isoform predictions with the provided authentic annotations. If an initially predicted gene locus is consistent with an annotated gene, MaxInfo utilizes the annotation to refine the assembly of isoforms. If the gene locus is not covered by any annotated gene, MaxInfo reconstructs isoforms in the locus similar to that in the de novo assembly mode. The expressed annotated isoforms and the novel discovered isoforms are collectively used for the investigation.
RESULTS
Evaluating the identification performance of MaxInfo on simulation datasets
We evaluated the performance of MaxInfo in identifying isoforms from simulated data generated with true transcripts. Based on the known transcripts in chromosome 1 (Chr. 1) of the UCSC Known Genes annotation, we simulated 15 million 75-bp pair-ended RNA-seq reads for an in silico study using Flux Simulator software (35) (Supplementary Notes). We then mapped the generated reads to the UCSC hg19 reference genome using TopHat 2. For evaluation purposes, MaxInfo was compared with the other leading methods, including SLIDE, iReckon, IsoLasso and Cufflinks. It is worth noting that the two latter methods are automated methods that do not require genome annotations to guide the assembly, whereas SLIDE and iReckon require gene or transcript annotations. In this study, we configured iReckon to handle unannotated cases by providing it with only minimal annotations based on the boundaries of reference transcripts, while for SLIDE we provided full annotations.
To assess the accuracy of the assembly results in relation to the known true transcripts, we followed the representative study Cufflinks and employed precision and recall as criteria. We considered each true transcript as correctly discovered only if all of the exons inside the transcript were correctly identified. We considered exons located at the transcript boundaries as correctly identified if they fell within 100 bp of the true positions in the known transcripts. For extremely long transcripts (>20 exons), if 90% of the exons were correctly identified, the prediction was considered correct.
The precision and recall (Figure 2A) performances were reported for the overall isoforms. The corresponding F-1 scores of different methods are summarized in Supplementary Figure S1. With respect to both criteria, MaxInfo achieved the best overall discovery accuracy. The results of all competitors fluctuated across different isoform numbers, whereas MaxInfo presented stable performances.
Figure 2.

Performance of MaxInfo based on a simulation study of the human genome. (A) Precision and recall of MaxInfo with respect to isoform discovery compared with other state-of-the-art methods. (B) Box plot of the precision and recall for 100 replicated experiments on human datasets. The upper and lower whiskers indicate the 10th and 90th percentiles, respectively. (C) Significance tests under the hypothesis that the accuracy attained by MaxInfo is worse than that of each competitor. The rank sum test was performed based on 100 replicated experiments on human datasets. (D) Analysis of the abundance estimation accuracy for the isoforms. Spearman's correlations were calculated between the true abundances and predicted abundances in FPKM. (E) Fractions of isoforms whose abundances were correctly estimated under different error tolerance thresholds.
We also calculated the precision and recall performances of each method in terms of gene discovery. In the simulation, we regarded a multi-transcript gene as correctly identified if at least one transcript generated from it was discovered. For the discovery accuracy (Supplementary Figure S2), MaxInfo attained better precision than all other methods. In addition, MaxInfo also outperformed the other methods in recall. The precision and recall results were combined to determine the F-1 score, and we observed that MaxInfo achieved the highest score among all five benchmark methods (Supplementary Figure S3). In the simulation process using the Chr. 1 dataset, the computational costs for each method were recorded for comparison (Supplementary Figure S4). The speed of MaxInfo was similar to that of Cufflinks and IsoLasso and was better than that of iReckon and SLIDE.
To better compare the robustness and consistency of the methods, we performed statistical analyses of the prediction accuracies. We generated 100 different datasets with the Flux Simulator, and each of the datasets contained 1 million 75-bp paired-end reads. In addition, the datasets were simulated using 100 randomly selected genes from Chr. 1 as a reference, and the aforementioned evaluation strategy was employed. The precision and recall distributions of the 100 different datasets determined by the different methods are shown in Figure 2B. The MaxInfo results exhibited smaller variances and higher median accuracies than the other methods. We also performed a rank-sum test (Figure 2C) and a one-sided paired t-test (Supplementary Figure S5) to compare the results of MaxInfo with the results of the other methods. The null hypothesis for the test was that the accuracy of MaxInfo was not better than that of the competing methods (SLIDE, iReckon, IsoLasso and Cufflinks). The P-values suggested that the precision of MaxInfo was significantly better than that of the other methods.
To evaluate the performance of MaxInfo for the analysis of different species, we also performed a simulation study using Chr. 3R of D. melanogaster, a chromosome that contains 880 genes and 2327 isoforms. A set of 5 million 75-bp paired-end reads was generated for this in silico study, and these D. melanogaster data were utilized to determine the transcript accuracies and gene accuracies (Supplementary Figure S6). Most of the comparisons performed revealed that MaxInfo also outperformed the other approaches.
Evaluating the quantification performance of MaxInfo using simulated datasets
We also evaluated the accuracy of MaxInfo in estimating isoform abundance. Using the simulated dataset, Flux Simulator provided the exact number of fragments generated from each isoform. We calculated the abundance in the form of FPKM for the evaluation and considered only comparisons between the correctly identified transcripts and the corresponding true transcripts. When more than one of the predicted transcripts was matched to the same true isoform, we merged these transcripts into one by adding the fragments to each together.
The abundance estimations were first evaluated by calculating the Spearman’s rank correlation coefficients between the true transcripts and the assembled transcripts from each method (Figure 2D). All methods showed positive correlations between the two abundance results, and the results of MaxInfo were generally better than others. We also calculated the Pearson’s correlation coefficients for the predicted transcripts using the log-transformed abundances (Supplementary Figure S7).
To analyze the detailed abundance errors, we calculated the same ratio between the true and predicted abundance estimation, as described for iReckon. The evaluation strategy yielded the abundance accuracies under different error tolerance rates. In detail, defining the error tolerance rate as γ, if the difference between an estimated abundance and the true abundance is less than γ, it is regarded as correctly estimated. For each correctly predicted isoform, we conducted this comparison of abundance and plotted the overall proportion of correctly predicted isoforms (Figure 2E). Under the same error tolerance rate, MaxInfo consistently generated the highest fractions of correct estimates, indicating its high accuracy for abundance estimation. The abundance estimation results for D. melanogaster were also summarized (Supplementary Table S1).
Performance of MaxInfo with genome annotation
The aforementioned evaluations verified that MaxInfo generally outperformed other state-of-the-art methods when implemented without genome annotations. However, the performance can be further improved by providing reference annotations. The released software can be flexibly adjusted between unannotated and annotated modes depending on the type of data source that is provided.
In this study, SLIDE and iReckon are methods that require annotation, and Cufflinks, MaxInfo can work both in annotation and de novo modes. We used these four methods for analysis and conducted experiments on the human Chr. 1 dataset generated by Flux Simulator. We first provided the known transcripts from the UCSC Known Genes annotation as a reference. With such high-quality annotations, the accuracy of all methods was greatly improved (Supplementary Figure S8a) compared with the de novo results. MaxInfo exhibited the best performance under this condition.
We also provided the assembled transcripts of MaxInfo to other methods as a reference. In this case, we evaluated the difference between the transcripts from MaxInfo and the transcripts assembled by other methods. The results indicated that the performance of Cufflinks could also be improved using MaxInfo transcripts as a reference (Supplementary Figure S8b).
Application of MaxInfo to real datasets
To test the performance of MaxInfo in practice, we applied the method to three human RNA-seq datasets: H1 human embryonic stem cells (H1-hESC), neurons derived from H1 embryonic stem cells (H1-Derived Neurons) and human pulmonary alveolar epithelial cells (HPAEpiC) (Supplementary Note 9). We first mapped the reads to the Homo sapiens (hg19) genome using TopHat 2. Compared with the simulated data, the real sequence data contained more practical factors that were hardly covered by the simulation. One known problem associated with evaluating real sequence data is the lack of ground truths to define the expressed transcripts. Alternatively, we evaluated the assembled isoforms with known genes in the RefSeq, Ensembl or UCSC Known Genes annotation datasets. For transcript accuracy, we compared the assembled transcripts with known transcripts following the aforementioned transcript matching criteria. In our evaluation, a known gene was considered to be identified if at least one of its known isoforms was correctly predicted.
For isoform discovery over all the datasets across different tissues, MaxInfo could identify more than 20% of the known human transcripts, regardless of the cell type (Figure 3A and Supplementary Table S2). In the precision evaluation, the scores of MaxInfo for the different datasets were also better than those of the other methods (Figure 3A). MaxInfo also showed consistent advantages in gene prediction and achieved >30% accuracy in recall and ∼40% accuracy in precision (Figure 3B).
Figure 3.

Performance of MaxInfo for isoform discovery using unannotated genes in human datasets. (A) Overall performance evaluation for isoform discovery over three RNA-seq datasets: H1-hESC, H1-Derived Neurons and HPAEpiC. (B) Overall performance evaluation for gene prediction. (C) Distribution of the predicted abundance for each dataset. Abundance was quantified in FPKM with log-transformations.
To provide a comprehensive perspective of the recovered isoforms, we analyzed the number of correct transcripts and genes that were consistently identified in all the three datasets. Although the reads data were sequenced from different bio-samples, their basic transcriptomes shared homogeneity. If a method produced diverse predictions on these inherently related biological data, then it was regarded as data source sensitive. MaxInfo exhibited increased consistency compared with the other methods (Supplementary Figure S9). We also assessed the performances of MaxInfo on cancer cell (human colorectal cell line HT-29) and normal tissue (human sigmoid colon) datasets (Supplementary Table S3). When run on these two human samples, MaxInfo showed similar performances with known annotations.
For the evaluation of estimated abundances, there was no abundance ground truth for comparisons. Alternatively, we only plotted the distributions of the estimated abundances for each method (Figure 3C). SLIDE tended to be more tightly distributed than the other methods. MaxInfo had a similar distribution center as IsoLasso but predicted more transcripts with low abundances (long tail in the left of the distribution). For the different datasets, the estimated abundances using different assemblers varied. For example, the centers of abundances in H1-hESC and HPAEpiC were close to zero but were much closer to −1 in H1-Derived Neurons.
Experiments were also conducted on three D. melanogaster datasets from the Gene Expression Omnibus (GEO): head, testis and ovary. Each dataset was processed using TopHat 2 and analyzed using all the methods as previously described. We report the results for transcript prediction (Supplementary Table S4) and gene discovery (Supplementary Table S5). All methods tended to achieve better accuracies for the D. melanogaster datasets than the human datasets. Compared with its competitors, MaxInfo also exhibited an enhanced ability to identify transcripts (>30% accuracy) and genes (40–50% accuracy).
DISCUSSION
In this study, we proposed the information theoretic model MaxInfo, which is capable of simultaneously performing isoform discovery and abundance estimation from high-throughput RNA-seq data. The novelty of the method is the formulation of the RNA-seq process via channel coding theory. According to a review of transcript reconstruction methods (24), the transcript identification results were shown to vary greatly among different assemblers, even if the methods were developed based on similar mathematical assumptions. MaxInfo tackles the isoform discovery problem from an information theory perspective, introducing a novel mathematical framework to this field. We thus believe that MaxInfo could illustrate this challenging problem from a fresh new perspective compared with the existing assembly methods. To evaluate the performance of MaxInfo, we experimentally compared it with four benchmark NGS technologies: SLIDE, iReckon, IsoLasso and Cufflinks.
Using simulated human and D. melanogaster datasets, we achieved transcript assembly accuracies of >60% and ∼70%, respectively. However, we noted that the predictive accuracy for transcripts from real sequence data was considerably reduced compared with that for transcripts from simulated data because the accuracy scores were calculated according to real data for which the evaluation ground truth was not known. Therefore, we followed previously published evaluation strategy and used the human genome annotations as a ‘surrogate’ ground truth. The human genome annotations offer only partial information regarding the ‘true’ ground truth. For instance, although various isoforms recovered by computational methods might be correct, they have not been annotated in the human genome annotations. Such recoveries are regarded as false negatives when using known annotations as true transcripts. This ‘partial ground truth’ problem with real datasets decreases the calculated accuracy. The best evaluation includes validating the discoveries with functional studies and in vivo experiments. However, accurately identifying tens of thousands of transcripts is difficult with the current experimental technology.
Although the results demonstrated that MaxInfo presents improved assembly performance, we should note that there is room for further enhancement. For example, instead of using the uniform distribution to model the starting position of the fragments, non-uniform assumptions can be considered in the RNA-seq process. Alternative error models of alignment quality evaluations could also be used in the MaxInfo framework. A super-read strategy can also be employed to extend the length of short reads into super-reads. When the super-reads are mapped to the reference genome, less ambiguity and better accuracy could be expected. MaxInfo framework is flexible and can be integrated with these advanced modifications for potential performance improvements. However, such modifications will also increase the complexity of our model. Therefore, we prefer to adopt the simplest implementation of MaxInfo because our evaluations demonstrate the simple and natural choices lead to improved performance.
AVAILABILITY
The MaxInfo software package is available at http://maxinfo.sourceforge.net for public usage, and the source code is included.
ACCESSION NUMBERS
The RNA-seq datasets are downloaded from the Gene Expression Omnibus (GEO) under the access number SRR317039 (H1 human embryonic stem cells), SRR3192700 (neurons derived from H1 embryonic stem cells), SRR3192690 (human pulmonary alveolar epithelial cells), SRR3659025 (head of D. melanogaster), SRR3663888 (testis of D. melanogaster), SRR3664030 (ovary of D. melanogaster), GSE78684 (human colorectal cell lines HT-29), and GSE88557 (human sigmoid colon).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Shanshan Liu in University of California, San Francisco for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China (NSFC) [61327902, 61531014]; Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (Public Health and Preventive Medicine). Funding for open access charge: Tsinghua University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Holt K.E., Parkhill J., Mazzoni C.J., Roumagnac P., Weill F.-X., Goodhead I., Rance R., Baker S., Maskell D.J., Wain J.. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet. 2008; 40:987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.-J., Makhijani V., Roth G.T.. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008; 452:872–876. [DOI] [PubMed] [Google Scholar]
- 3. Marioni J.C., Mason C.E., Mane S.M., Stephens M., Gilad Y.. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008; 18:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Z., Gerstein M., Snyder M.. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jaitin D.A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I., Mildner A., Cohen N., Jung S., Tanay A.. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014; 343:776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katz Y., Wang E.T., Airoldi E.M., Burge C.B.. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods. 2010; 7:1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B.. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xing Y., Yu T., Wu Y.N., Roy M., Kim J., Lee C.. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006; 34:3150–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mezlini A.M., Smith E.J., Fiume M., Buske O., Savich G.L., Shah S., Aparicio S., Chiang D.Y., Goldenberg A., Brudno M.. iReckon: simultaneous isoform discovery and abundance estimation from RNA-seq data. Genome Res. 2013; 23:519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rossell D., Attolini C.S.-O., Kroiss M., Stöcker A.. Quantifying alternative splicing from paired-end RNA-sequencing data. Annl. Appl. Stat. 2014; 8:309–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., Van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bernard E., Jacob L., Mairal J., Vert J.-P.. Efficient RNA isoform identification and quantification from RNA-Seq data with network flows. Bioinformatics. 2014; 30:2447–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li W., Feng J., Jiang T.. IsoLasso: a LASSO regression approach to RNA-Seq based transcriptome assembly. J. Comput. Biol. 2011; 18:1693–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J.J., Jiang C.-R., Brown J.B., Huang H., Bickel P.J.. Sparse linear modeling of next-generation mRNA sequencing (RNA-Seq) data for isoform discovery and abundance estimation. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:19867–19872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li B., Ruotti V., Stewart R.M., Thomson J.A., Dewey C.N.. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010; 26:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roberts A., Trapnell C., Donaghey J., Rinn J.L., Pachter L.. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011; 12:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones D.C., Ruzzo W.L., Peng X., Katze M.G.. A new approach to bias correction in RNA-Seq. Bioinformatics. 2012; 28:921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hocking R.R., Speed F., Lynn M.. A class of biased estimators in linear regression. Technometrics. 1976; 18:425–437. [Google Scholar]
- 19. Whitehead J. On the bias of maximum likelihood estimation following a sequential test. Biometrika. 1986; 73:573–581. [Google Scholar]
- 20. Li W., Jiang T.. Transcriptome assembly and isoform expression level estimation from biased RNA-Seq reads. Bioinformatics. 2012; 28:2914–2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993; 80:27–38. [Google Scholar]
- 22. Roberts A., Schaeffer L., Pachter L.. Updating RNA-Seq analyses after re-annotation. Bioinformatics. 2013; 29:1631–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Steijger T., Abril J.F., Engström P.G., Kokocinski F., Hubbard T.J., Guigó R., Harrow J., Bertone P., Consortium R.. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods. 2013; 10:1177–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cover T.M., Thomas J.A.. Elements of lnformation Theory. 1991; NY: A Wiley—Interscience Publication. [Google Scholar]
- 26. Deng Y., Bao F., Deng X., Wang R., Kong Y., Dai Q.. Deep and structured robust information theoretic learning for image analysis. IEEE Trans. Image Process. 2016; 25:4209–4221. [DOI] [PubMed] [Google Scholar]
- 27. Lesurf J.C.G. Information and Measurement. 2001; NY: CRC press. [Google Scholar]
- 28. Reza F.M. An Introduction to Information Theory. 1961; MA: Courier Corporation. [Google Scholar]
- 29. Cheong R., Rhee A., Wang C.J., Nemenman I., Levchenko A.. Information transduction capacity of noisy biochemical signaling networks. Science. 2011; 334:354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Astola J., Virtanen I.. Entropy Correlation Coefficient, a Measure of Statistical Dependence for Categorized Data. 1981; Helsinki: Lappeenrannan teknillinen korkeakoulu. [Google Scholar]
- 31. Saliba A.-E., Westermann A.J., Gorski S.A., Vogel J.. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014; 42:8845–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moon T.K. The expectation-maximization algorithm. IEEE Signal Process. Mag. 1996; 13:47–60. [Google Scholar]
- 33. Deng Y., Dai Q., Liu R., Zhang Z., Hu S.. Low-rank structure learning via nonconvex heuristic recovery. IEEE Trans. Neural Netw. Learn. Syst. 2013; 24:383–396. [DOI] [PubMed] [Google Scholar]
- 34. Bao F., Deng Y., Du M., Ren Z., Zhang Q., Zhao Y., Suo J., Zhang Z., Wang M., Dai Q.. Probabilistic natural mapping of gene-level tests for genome-wide association studies. Brief. Bioinform. 2017; bbx002. [DOI] [PubMed] [Google Scholar]
- 35. Griebel T., Zacher B., Ribeca P., Raineri E., Lacroix V., Guigó R., Sammeth M.. Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res. 2012; 40:10073–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.