

Abstract
Gimeracil or 5-chloro-2,4-dihydroxypyridine (CDHP) enhances the antitumor effects of 5-fluorouracil (5-FU) by inhibiting dihydropyrimidine dehydrogenase (DPD), which is involved in the degradation of 5-FU. CDHP, as part of a combination therapy, was also reported to exert a radiosensitizing effect. Therefore, CDHP may have underlying mechanisms of action other than DPD inhibition. The focus of the present study was to investigate the antitumor effects of CDHP and cisplatin (CDDP) combination treatment in vitro and in vivo against oral squamous cell carcinoma (OSCC) tumors. The inhibitory growth effects of CDHP and/or CDDP treatment on SAS and HSC2 cells were examined using an MTT assay. The expression levels of DNA double strand break repair proteins, including Ku70, DNA-dependent-protein kinase catalytic subunit (DNA-PKcs), Rad50 and Rad51 in CDHP and/or CDDP-treated cells were detected using western blotting. Nude mice with SAS or HSC2 tumors were treated with CDHP (administered orally 7 times/week) and/or CDDP (administered by intraperitoneal injection once/week) for 2 weeks. Combined treatment of CDHP and CDDP significantly suppressed the growth of SAS and HSC2 cells in vitro and that of tumors in vivo compared with the effects caused by single drug only or control treatments. Western blotting demonstrated that the expression levels of Ku70, DNA-PKcs, Rad50 and Rad51 were downregulated in cells treated with CDHP and CDDP combination treatment. Immunohistochemistry also identified that the expression of DNA double strand break repair proteins was downregulated in tumors treated with CDHP and CDDP combination treatment compared with that of tumors treated with CDDP alone or control. The results of the current study suggest that CDHP may be responsible for enhancing the antitumor effects of CDDP by suppressing the DNA double strand break repair system. Therefore, the combination of CDHP and CDDP may be a potential effective option for OSCC treatment.
Keywords: cisplatin, gimeracil, antitumor effect, combined therapy, oral squamous cell carcinoma
Introduction
Surgery, radiotherapy or combined therapies (surgery and radiation) have been used for curative treatment of oral squamous cell carcinoma (OSCC). However, multidrug chemotherapy or chemo-radiotherapy is primarily used for patients with advanced OSCC to preserve oral functions. In multidrug chemotherapy, cisplatin (CDDP) in combination with 5-fluorouracil (5-FU) is typically used for the treatment of head and neck cancer, including OSCC (1). However, combined chemotherapy of CDDP and the combination tablet of tegafur/5-chloro-2,4-dihydroxypyridine (CDHP)/oteracil potassium (S-1) is used instead of CDDP and 5-FU combined chemotherapy to reduce the length of time of hospital admission or to avoid continuous infusion therapy (2,3).
S-1 is an anticancer drug that is orally administered. It is a combination of tegafur, CDHP and oteracil potassium in a 1:0.4:1 molar concentration ratio (4). S-1 therapy in nude rats was associated with an increased and prolonged concentration of 5-FU in plasma and tumor tissues compared with that of a combination of tegafur/uracil (5). Amongst the components of S-1, CDHP functions to maintain efficacious concentrations of 5-FU in plasma and tumor tissues by inhibiting dihydropyrimidine dehydrogenase (DPD), which is involved in the degradation of 5-FU (6). The antitumor effects of S-1 have been demonstrated in a variety of solid tumors, and the response rates for head and neck cancer (7) and oral cancer (8) in phase II studies conducted in Japan were 29.0 and 41.5%, respectively. However, S-1 single-agent therapy may be inadequate against advanced or recurrent tumors. In those cases, systemic chemotherapy may be considered. Therefore, combined chemotherapy of CDDP and S-1 has received increased interest in recent years (9–13).
It has been reported that CDDP reduces the expression level of 5-FU-resistant factors, including thymidylate synthase, DPD and multidrug resistance-associated protein. In addition, low-dose CDDP may also enhance the inhibition of DNA synthesis following the combined therapy of CDDP and 5-FU (14,15). However, the novel underlying mechanisms of CDDP and S-1 combination chemotherapy may occur as S-1 has three components (tegafur, CDHP and oteracil potassium) and CDDP in combination with any of these components may be effective in suppressing tumor growth. CDHP enhanced the efficacy of radiotherapy by inhibiting DNA repair pathways (16–18) and promoting the anticancer effects of a number of drugs (19). Therefore, CDHP may have the ability to increase the efficacy of CDDP.
The present study investigated whether CDHP enhances the antitumor effects of CDDP in OSCC cells, as well as the underlying mechanism of CDDP and CDHP combination therapy, with particular emphasis on its effect on the DNA double strand break repair system.
Materials and methods
Cell lines and cell culture
The SAS and HSC2 OSCC cell lines were obtained from the Cell Bank (RIKEN BioResource Center, Tsukuba, Japan). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) supplemented with 10% fetal bovine serum (FBS), 100 µg/ml streptomycin and 100 U/ml penicillin (all from Thermo Fisher Scientific, Inc., Waltham, MA, USA) in a humidified atmosphere containing 5% CO2 at 37°C.
In vitro cell growth assay
The HSC2 and SAS cells were seeded at a density of 5×103 cells/well on 96-well plates (BD Biosciences, Franklin lakes, NJ, USA) in DMEM supplemented with 10% FBS. After 24 h, the cells were cultured in the same medium without any treatment (untreated control) or treated with CDDP (5–100 µg/ml), CDHP (0.1–5 µg/ml) or a combination (CDDP 1 µg/ml and CDHP 10 µg/ml) of the two for 0, 24, 48 or 72 h. Subsequently, MTT was added to each well (25 µl/well) and incubated for 4 h. The blue dye absorbed by the cells was dissolved in dimethyl sulfoxide (100 µl/well) and the absorbance was measured using a spectrophotometer (Bio-Rad Laboratories, Inc., Hercules, CA, USA) at a wavelength of 490 nm. All assays were performed in triplicate.
Western blot analysis
Following treatment for 48 h with CDHP (10 µg/ml), CDDP (1 µg/ml), or the combination of the two or without any treatment (untreated control), the SAS and HSC2 cells were collected and lysed with radioimmunoprecipitation buffer (Wako Pure Chemical Industries, Ltd., Osaka, Japan). The quantity of protein from the whole cell lysates was quantified using NanoDrop™ 1000 spectrophotometer (Thermo Fisher Scientific, Inc.). A total of 20 µl of protein sample containing 50 µg/ml protein was loaded into each well of NuPAGE™ 4–12% Bis-Tris gel (Thermo Fisher Scientific, Inc.) and separated using electrophoresis (200 V, 110–125 mA gel, 40 min), then transferred to a polyvinylidene fluoride membrane using iBlot® gel transfer stacks (Thermo Fisher Scientific, Inc.). A blocking solution and a primary antibody dilution solution were made from WesternBreeze® Blocker/Diluent part A and B (no. WB7050; Thermo Fisher Scientific, Inc.) according to manufacturer's protocol. Following blocking the membranes at room temperature for 30 min, the membranes were incubated with anti-Ku70 mouse monoclonal antibody (dilution, 1:250; no. sc-5309; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-Ku80 rabbit polyclonal antibody (dilution, 1:500; no. 2753; Cell Signaling Technology, Danvers, MA, USA), anti-DNA-dependent-protein kinase catalytic subunit (DNA-PKcs) mouse monoclonal antibody (dilution, 1:500; no. sc-9051; Santa Cruz Biotechnology, Inc.), anti-Rad50 mouse monoclonal antibody (dilution, 1:200; no. sc-56209; Santa Cruz Biotechnology, Inc.), anti-X-ray repair cross complementing protein 4 (XRCC4) rabbit polyclonal antibody (dilution, 1:500; no. ab12069; Abcam, Cambridge, UK), anti-XRCC4-like factor (XLF) rabbit polyclonal antibody (dilution, 1:250; no. ab33499; Abcam), anti-Rad51 rabbit polyclonal antibody (dilution, 1:500; no. sc-8349; Santa Cruz Biotechnology, Inc.), or anti-α-tubulin mouse monoclonal antibody (dilution, 1:500; no. sc-5286; Santa Cruz Biotechnology, Inc.) at 4°C overnight. Then the membranes were washed using 1X WesternBreeze® wash solution (Thermo Fisher Scientific, Inc.) 3 times at room temperature (5 min/wash), followed by incubation with Novex® alkaline-phosphatase conjugated goat anti-rabbit (no. WP2007; Thermo Fisher Scientific, Inc.) or goat anti-mouse immunoglobulin G secondary antibodies (no. WP20006; Thermo Fisher Scientific, Inc.) at room temperature for 30 min. Following washing of the membranes 3 times with the wash solution at room temperature, protein bands were detected upon incubation of the membranes with Novex® AP Chromogenic substrate (no. WP20001; Thermo Fisher Scientific, Inc.) at room temperature for 5–15 min.
In the second western blot experiment, SAS cells were cultured for 24, 48 or 72 h without any treatment (untreated control) or with CDHP 10 µg/ml. Protein samples were prepared and western blotting was performed using the same protocol as aforementioned, except the primary antibodies were anti-proliferating cell nuclear antigen (PCNA) mouse monoclonal antibody (dilution, 1:500; no. sc-25280; Santa Cruz Biotechnology, Inc.), anti-phosphorylated-Akt (p-Akt) rabbit polyclonal antibody (dilution, 1:250; no. sc-16646; Santa Cruz Biotechnology, Inc.) and anti-α-tubulin mouse monoclonal antibody (dilution, 1:500; no. sc-5286; Santa Cruz Biotechnology, Inc.). Anti-α-tubulin monoclonal antibody was used as an internal control for the normalization of western blot analysis. All western blot experiments were repeated ≥3 times and demonstrated similar results. Quantification of protein bands was performed using ImageJ v1.51 h software available at http://rsb.info.nih.gov/ij/, and the fold change of expression of each protein of interest was calculated relative to the internal control.
Nude mice and tumor inoculations
In total 20 female CAnN. Cg-Foxnlnu/CrlCrlj athymic nude mice (CLEA Japan, Inc., Tokyo, Japan) were purchased at 4 weeks of age and were maintained in a pathogen-free environment at 20–25°C with a 12 h light/dark cycle. Sterile water and food was available to the mice at all times. In addition, all manipulations were performed inside an aseptic laminar flow hood. Untreated SAS and HSC2 cells were used for the xenograft model in the nude mice, and 1×106 cells were suspended in 0.1 ml of serum-free medium and subcutaneously injected into 5-week-old nude mice (average weight, 20 g). Tumors grew for 10 days prior to treatment. The mice were divided into four groups of 5 mice, with similar mean tumor volumes (100–150 mm3). The humane end point of the experiment was decided as maximum tumor size: 1,500 mm3 and <50% loss of body weight. The mice were treated for 2 weeks and were sacrificed using an overdose of Somnopentyl (sodium pentobarbital, 200 mg/kg; Merck & Co., Inc., Whitehouse Station, NJ, USA) at the end of the experiment. Maximum efforts were taken to minimize the suffering of the animals during the experiment. All animal procedures were approved by the Guidelines for Animal Experimentation of Yamaguchi University (approval no. 55-021). All in vivo experiments were repeated ≥3 times.
Reagents and in vivo treatment protocol
CDDP and CDHP were obtained from Sigma-Aldrich (Merck KGaA) and Taiho Pharmaceutical Co., Ltd. (Tokyo, Japan), respectively. CDHP was suspended in sterile 0.5% sodium hydroxypropylmethylcellulose (HPMC; DKS Co., Ltd., Kyoto, Japan) in sterile conditions at 1.0 mg/ml and subsequently homogenized. Mice in the untreated control group were administered orally an equal volume of 0.5% HPMC for 2 weeks (7 times/week) and saline (200 µl) intraperitoneally for 2 weeks (once/week). Mice in the CDHP groups were administered orally via a gastric tube CDHP (2.0 mg/kg) for 2 weeks (7 times/week) and saline (200 µl) intraperitoneally for 2 weeks (once/week). Mice in the CDDP group were administered orally an equal volume of 0.5% HPMC for 2 weeks (7 times/week) and injected with CDDP (6.0 mg/kg/day) intraperitoneally for 2 weeks (once/week). Mice in the CDDP and CDHP combination therapy group (CDDP+CDHP) received CDHP (7 times/week) and CDDP (once/week) at the same doses described above for 2 weeks. The sizes of these tumors were measured every 2 days and the tumor volumes were calculated. At 14 days, mice were sacrificed and the tumors were dissected out, fixed at room temperature for 24 h in 10% neutral-buffered formalin (Mildform® 10N; Wako Pure Chemical Industries, Ltd.) and embedded in paraffin for further study.
Immunohistochemistry
The avidin-biotin complex immunohistochemical technique was used to detect DNA double strand break repair proteins in tissue specimens. Paraffin-embedded tissue sections (4-µm thick) were deparaffinized in 100% xylene (Wako Pure Chemical Industries, Ltd.) for 10 min and rehydrated through graded (100–70% v/v) ethyl alcohol (Muto Pure Chemicals Co., Ltd., Tokyo, Japan) for 5 min/each step at room temperature. Slides were quenched with a 0.3% hydrogen peroxide/methanol mixture for 30 min at room temperature. Sections were washed and pre-incubated with Dako REAL™ Peroxidase-Blocking solution (no. S2023; Agilent Technologies, Inc., Santa Clara, CA, USA) for 30 min at room temperature; followed by incubation with anti-Ku70 mouse monoclonal antibody (dilution, 1:50; no. sc-5309; Santa Cruz Biotechnology, Inc.), anti-Ku80 rabbit polyclonal antibody (dilution, 1:100; no. 2753; Cell Signaling Technology), anti-DNA-PKcs mouse monoclonal antibody (dilution, 1:100; no. sc-9051; Santa Cruz Biotechnology, Inc.), anti-Rad50 mouse monoclonal antibody, anti-XRCC4 rabbit polyclonal antibody (dilution, 1:50; no. ab12069; Abcam), anti-XLF rabbit polyclonal antibody (dilution, 1:100; no. ab33499; Abcam), anti-Rad51 rabbit polyclonal antibody (dilution, 1:100; no. sc-8349; Santa Cruz Biotechnology, Inc.) or PCNA mouse monoclonal antibody (dilution, 1:100; no. sc-25280; Santa Cruz Biotechnology, Inc.) for 8 h at 4°C. Following washing of the tissue sections in PBS for 10 min at room temperature, the antibodies were detected using the Dako REAL™ EnVision™ Detection system (K5007; Agilent Technologies) according to the manufacturer's protocol. Tissues were washed in PBS for 5 min and H2O for 5 min in room temperature, and counterstained with hematoxylin (Muto Pure Chemicals Co., Tokyo, Japan) in room temperature for 1 min. The tissue sections were subsequently dehydrated in (70–100% v/v) graded ethyl alcohol (Muto Pure Chemicals Co., Ltd.), then removed using xylene (Wako Pure Chemical Industries Ltd.) for 10 min in room temperature and mounted with glass coverslips using DPX mounting medium (Sigma-Aldrich; Merck KGaA). In total, >1,000 cells were counted under an optical microscope (BX51; Olympus Corporation, Tokyo, Japan) in ≥3 random fields in each section at ×200 magnification. The number of positive cells was divided by the total number of counted cells, and each labeling index was expressed as a percentage.
Statistical analysis
Statistical analyses were performed using StatView software version 5.0 J (SAS Institute, Inc., Cary, NC, USA). The statistical significance of the in vivo and in vitro results was determined using the Mann-Whitney U test. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of CDHP and CDDP on cell growth inhibition of OSCC cell lines in vitro
The growth inhibitory effect of CDHP and/or CDDP on SAS and HSC2 cells was analyzed using the MTT assay. Cells were treated with various concentrations of CDHP (5, 10, 50 and 100 µg/ml) or CDDP (0.1, 0.5, 1.0 and 2.0 µg/ml) alone for 24, 48 or 72 h. CDHP did not inhibit the growth of SAS or HSC2 cells (Fig. 1A), while CDDP inhibited the growth of SAS (0.5–5.0 µg/ml CDDP) and HSC2 (1.0–5.0 µg/ml CDDP) cells in a dose-dependent manner (Fig. 1B). Furthermore, the combination treatment of CDHP (10 µg/ml) and CDDP (1.0 µg/ml) significantly inhibited the growth of SAS or HSC2 cells compared with the effect caused by CDHP or CDDP alone and that observed in the untreated control (Fig. 1C).
Figure 1.



Effects of CDHP and CDDP on cell growth inhibition in vitro. Inhibition of cell growth was evaluated using an MTT assay. (A) CDHP did not inhibit the growth of SAS or HSC2 cells. (B) CDDP inhibited the growth of SAS and HSC2 cells in a dose-dependent manner. (C) CDDP and CDHP combined treatment significantly inhibited the growth of SAS and HSC4 cells compared with that of single agent treatment or that observed in the untreated control. Error bars represent the standard deviation of the mean of six independent experiments. *P<0.01 compared with the control (Mann-Whitney's U test). #P<0.01 compared with CDDP alone (Mann-Whitney's U test). OD, optical density; CDHP, 5-chloro-2,4-dihydroxypyridine; CDDP, cisplatin.
Effects of CDHP and CDDP on the expression of DNA double strand break repair proteins in vitro
To clarify the underlying mechanisms of the growth inhibitory effect of CDHP and CDDP combination treatment, the expression of DNA double strand break repair proteins (Ku70, Ku80, DNA-PKcs, Rad50, Rad51 XRCC4 and XLF) was examined in cells using western blotting. Combined treatment with CDHP (10 µg/ml) and CDDP (1.0 µg/ml) reduced the expression of DNA double strand break repair proteins (Ku70, Ku80, DNA-PKcs, Rad50 XRCC4, XLF and Rad51) in OSCC cells (Fig. 2). In addition, CDHP treatment for 24, 48 or 72 h had small affect on the expression of PCNA (cell proliferative ability), but only marginally reduced the expression of p-Akt (survival signal; Fig. 3).
Figure 2.
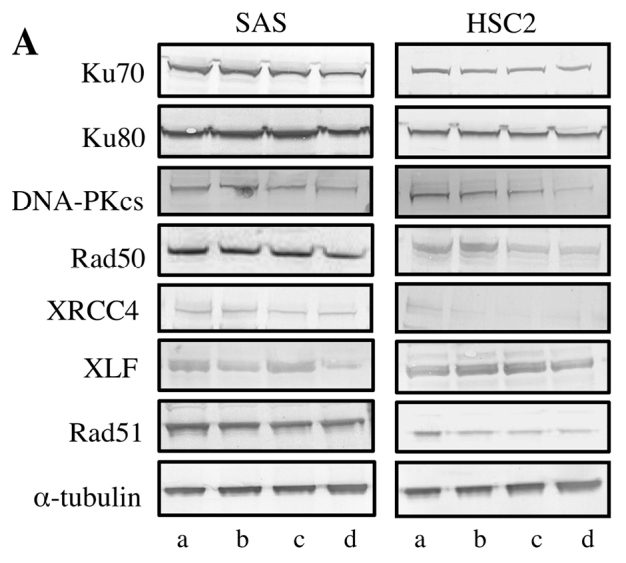


Effects of CDHP and CDDP on the expression of DNA double strand break repair proteins in vitro. (A) Western blotting was performed to detect the expression levels of DNA double strand break repair proteins in SAS and HSC2 cells following CDDP and/or CDHP treatment. (B and C) CDDP and CDHP combination treatment reduced the expression of Ku70, Ku80, DNA-PKcs, Rad50, XRCC, XLF and Rad51 compared with that of CDDP or CDHP treatment alone, or the expression observed in the untreated control. Error bars represent the standard deviation of the mean of 3 independent experiments. *P<0.05 compared with the control (Mann-Whitney's U test). a, Untreated Control; b, CDHP 10 µg/ml; c, CDDP 1.0 µg/ml; d, CDDP+CDHP; CDHP, 5-chloro-2,4-dihydroxypyridine; CDDP, cisplatin; DNA-PKcs, DNA-dependent-protein kinase catalytic subunit; XRCC4, X-ray repair cross complementing protein 4; XLF, XRCC4-like factor.
Figure 3.

Effects of CDHP on cell-growth ability and survival signals. Western blotting was performed to investigate the effect of CDHP on SAS cell growth ability (PCNA) and survival signals (p-Akt). CDHP treatment had minor effect on the expression of PCNA, but reduced the expression of p-Akt. CDHP, 5-chloro-2,4-dihydroxypyridine; PCNA, proliferating cell nuclear antigen; p, phosphorylated. *P<0.01 compared with the control (Mann-Whitney's U test).
Effects of CDHP and CDDP on tumor growth inhibition in vivo
Nude mice with SAS or HSC2 tumor xenografts were used to examine the antitumor activity of CDHP and CDDP single/combination treatment. The in vivo treatment protocol is presented in Fig. 4A. Growth inhibition was not observed in CDHP-treated tumors compared with that in HPMC-treated tumors (Fig. 4B). However, significant growth inhibition was observed in CDDP-treated tumors and CDHP+CDDP-treated tumors (P<0.01; Fig. 4B). In addition, CDHP+CDDP treatment significantly inhibited tumor growth compared with that of CDDP alone (P<0.01). No significant weight loss was observed in mice treated with CDHP and/or CDDP during the experimental period in any treatment group (Fig. 4C).
Figure 4.

Effects of CDHP and/or CDDP on tumor growth inhibition in vivo. (A) Control, CDHP and CDDP treatment protocol. There were four experimental groups of mice: Control group, administered orally 0.5% HPMC (200 µl, 7 times/week) and saline (200 µl, once/week) by IP for 2 weeks; CDHP group, administered CDHP (2.0 mg/kg, PO, 7 times/week) and saline (200 µl IP, once/week) for 2 weeks; CDDP group, administered 0.5% HPMC (200 µl, PO, 7 times/week) and with CDDP (6.0 mg/kg/day, IP, once/week) for 2 weeks; and CDDP+CDHP combination therapy group, which received CDHP (2.0 mg/kg, PO, 7 times/week) and CDDP (6.0 mg/kg/day, IP, once/week) for 2 weeks. (B) CDHP+CDDP treatment significantly inhibited SAS and HSC2 tumor growth compared with that of CDDP treatment alone. CDHP or HPMC alone did not inhibit tumor growth. (C) CDHP and/or CDDP did not cause significant weight loss in any of the treatment groups compared with that of the untreated control. Error bars represent the standard error of the mean from the results of 5 mice (n=5). *P<0.01 compared with the control (Mann-Whitney's U test). #P<0.01 compared with CDDP alone (Mann-Whitney's U test). CDHP, 5-chloro-2,4-dihydroxypyridine; CDDP, cisplatin; HPMC, hydroxypropylmethylcellulose; PO, per os; IP, intraperitoneal injection.
Effects of CDHP and CDDP on the expression of DNA double strand break repair proteins in vivo
The expression of DNA double strand break repair proteins in mice tumors was examined using immunohistochemistry. The expression of DNA double strand break repair proteins (Ku70, Ku80, DNA-PKcs, Rad50 and Rad51) was decreased in CDHP-treated tumors and markedly reduced in CDHP+CDDP-treated tumors compared with that in CDDP-treated or untreated tumors (w. 5). Additionally, the expression of PCNA was decreased in CDDP- or CDHP+CDDP-treated tumors compared with that in untreated or CDHP-treated tumors (Fig. 5).
Figure 5.


DNA double strand break repair protein expression levels in vivo. Immunohistochemical staining was performed to investigate the underlying mechanisms of the antitumor effects of CDDP and CDHP combination therapy. CDDP and CDHP combination therapy reduced the expression of Ku70, Ku80, DNA-PKcs, Rad50 and Rad51 in HSC2 tumors. Percentage of positively stained cells is described in each figure. Magnification, ×200. CDHP, 5-chloro-2,4-dihydroxypyridine; CDDP, cisplatin; DNA-PKcs, DNA-dependent-protein kinase catalytic subunit; XRCC4, X-ray repair cross complementing protein 4; XLF, XRCC4-like factor; PCNA, proliferating cell nuclear antigen.
Discussion
In the current study, the efficacy of CDHP in combination with CDDP on OSCC in vitro and in vivo has been demonstrated. In addition, these results suggest that CDHP may enhance the effects of CDDP in OSCC via the downregulation of DNA double strand break repair proteins.
As CDHP is a competitive inhibitor of DPD, which degrades 5-FU, CDHP increases the blood level of 5-FU and therefore enhances the effect of 5-FU on cancer cells (4). Notably, Takagi et al (16) and Sakata et al (17) reported that CDHP may enhance the efficacy of radiotherapy via the suppression of homologous recombination (HR)-mediated DNA repair pathways. This was also observed with similar results in a previous study (18). In addition, it has been reported that CDHP may enhance the efficacy of camptothecin, 5-FU and hyperthermia (19). CDHP may be used in clinical practice with other therapeutic agents not investigated to date, as CDHP may possess mechanisms that increase the efficacy of other drugs, as it has in the results of the current study.
In the present study, CDHP (5–100 µg/ml) did not demonstrate cytostatic activity (Fig. 1A), and CDHP (2.0 mg/kg) did not exert any antitumor activity (Fig. 4B). In addition, it had a minor effect on the expression of PCNA (cell proliferation protein) in SAS cells (Fig. 3). However, CDHP is a potential therapeutic agent, as it enhanced the effect of CDDP in the current study and, in combination with CDDP, it effectively suppressed OSCC cell growth in vitro and OSCC tumor growth in vivo. Furthermore, as CDHP (2.0 mg/kg) was not associated with remarkable weight loss in mice (Fig. 4C), it may have less adverse effects clinically compared with chemotherapeutic agents typically used for cancer treatment. A mechanistic action of CDHP and CDDP combination treatment may be the suppression of DNA double strand break repair system in OSCC cells (Fig. 2) and tumors (Fig. 5). The combination of CDHP and CDDP may inhibit the DNA double strand break repair system by suppressing non-homologous end-joining-mediated DNA repair as well as HR-mediated DNA repair in OSCC cells. CDDP has the ability to cause crosslinking of the purine bases on the DNA, and this interferes with DNA repair mechanisms, which promotes DNA damage and subsequently induces apoptosis in cancer cells (20). Therefore, CDHP may also have suppressing effects on HR-mediated DNA repair pathways, and the combination of CDHP and CDDP may be able to inhibit the DNA double strand break repair system more effectively than CDDP alone. The authors of the present study have also demonstrated that CDHP suppresses the expression of excision repair cross-complementation group 1 in OSCC cells (data not shown).
In conclusion, the current study hypothesizes that CDHP may enhance the sensitivity of CDDP and other anticancer agents in clinical practice by promoting DNA damage in tumor cells.
Acknowledgements
The present study was supported in part by a Grant-in-Aid (JSPS; grant no. 26463045) from the Japanese Ministry of Education, Science and Culture.
References
- 1.Kish J, Drelichman A, Jacobs J, Hoschner J, Kinzie J, Loh J, Weaver A, Al-Sarraf M. Clinical trial of cisplatin and 5-FU infusion as initial treatment for advanced squamous cell carcinoma of the head and neck. Cancer Treat Rep. 1982;66:471–474. [PubMed] [Google Scholar]
- 2.Fujii M, Tomita K, Nishijima W, Tsukuda M, Hasegawa Y, Ishitoya J, Yamane H, Homma A, Tomita T. Phase I/II Study of S-1 plus Cisplatin Combination Chemotherapy in Patients with Advanced/Recurrent Head and Neck Cancer. Jpn J Clin Oncol. 2010;40:214–221. doi: 10.1093/jjco/hyp153. [DOI] [PubMed] [Google Scholar]
- 3.Fujii M. Combination therapy with S-1 and CDDP for head and neck cancer. Gan To Kagaku Ryoho. 2006;33:S150–S154. (Suppl 1) (In Japanese) [PubMed] [Google Scholar]
- 4.Shirasaka T, Shimamoto Y, Ohshimo H, Yamaguchi M, Kato T, Yonekura K, Fukushima M. Development of a novel form of 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anticancer Drugs. 1996;7:548–557. doi: 10.1097/00001813-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Shirasaka T, Nakano K, Takechi T, Satake H, Uchida J, Fujioka A, Saito H, Okabe H, Oyama K, Takeda S, et al. Antitumor effect of 1 M tegafur-0.4 M 5-chloro-2,4-dihydroxypyridine-1 M potassium oxonate (S-1) against human colon carcinoma orthotopically implanted into nude rats. Cancer Res. 1996;56:2602–2606. [PubMed] [Google Scholar]
- 6.Tatsumi K, Fukushima M, Shirasaka T, Fujii S. Inhibitory effects of pyrimidine, barbituric acid and pyridine derivatives on 5-fluorouracil degradation in rat liver extract. Jpn J Cancer Res. 1987;78:748–755. [PubMed] [Google Scholar]
- 7.Inuyama Y, Kida A, Tsukuda M, Kohno N, Satake B. S-1 Cooperative Study Group (Head and Neck Cancer Working Group): Late phase II study of S-1 in patients with advanced head and neck cancer. Gan To Kagaku Ryoho. 2001;28:1381–1390. (In Japanese) [PubMed] [Google Scholar]
- 8.Harada K, Sato M, Ueyama Y, Nagayama M, Hamakawa H, Nagahata S, Yoshimura Y, Osaki T, Ryoke K. Oral Cancer Study Group of Chugoku-Shikoku: Multi-institutional phase II trial of S-1 in patients with oral squamous cell carcinoma. Anticancer Drugs. 2008;19:85–90. doi: 10.1097/CAD.0b013e3282f0ce4d. [DOI] [PubMed] [Google Scholar]
- 9.Asaumi Y, Miyanaga T, Hosokawa O, Nishida Y, Matsunaga T, Shimizu S, Tanaka N, Ito T, Sato Y, Hirano Y, et al. Effectiveness of postoperative adjuvant chemotherapy using S-1 plus CDDP for type 4 gastric cancer. Gan To Kagaku Ryoho. 2013;40:323–325. (In Japanese) [PubMed] [Google Scholar]
- 10.Rino Y, Murakami H, Yukawa N, Wada N, Oshima T, Matsuura H, Sugano N, Arai H, Masuda M, Imada T. Feasibility of S-1/CDDP therapy for outpatients with advanced gastric cancer. Gan To Kagaku Ryoho. 2009;36:1829–1831. (In Japanese) [PubMed] [Google Scholar]
- 11.Fujii M. Combination therapy with S-1 and CDDP for head and neck cancer. Gan To Kagaku Ryoho. 2006;33:S150–S154. (Suppl 1) (In Japanese) [PubMed] [Google Scholar]
- 12.Uchida K, Hayashi K, Kuramochi H, Nakajima G, Inoue Y, Yamamoto M. Combination therapy of S-1 and CDDP for patients with colorectal cancer. J Cancer Res Clin Oncol. 2007;133:841–846. doi: 10.1007/s00432-007-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujii M, Sato H, Ogasawara T, Ando T, Tsujii S, Nagahori J, Komatsu Y, Matsuoka A. A case of liver metastasis of pancreatic acinar cell carcinoma treated with S-1 and intra-arterial CDDP combination therapy. Gan To Kagaku Ryoho. 2010;37:1987–1990. (In Japanese) [PubMed] [Google Scholar]
- 14.Nishiyama M, Yamamoto W, Park JS, Okamoto R, Hanaoka H, Takano H, Saito N, Matsukawa M, Shirasaka T, Kurihara M. Low-dose cisplatin and 5-fluorouracil in combination can repress increased gene expression of cellular resistance determinants to themselves. Clin Cancer Res. 1999;5:2620–2628. [PubMed] [Google Scholar]
- 15.Scanlon KJ, Newman EM, Lu Y, Priest DG. Biochemical basis for cisplatin and 5-fluorouracil synergism in human ovarian carcinoma cells. Proc Natl Acad Sci USA. 1986;83:8923–8925. doi: 10.1073/pnas.83.23.8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takagi M, Sakata K, Someya M, Tauchi H, Iijima K, Matsumoto Y, Torigoe T, Takahashi A, Hareyama M, Fukushima M. Gimeracil sensitizes cells to radiation via inhibition of homologous recombination. Radiother Oncol. 2010;96:259–266. doi: 10.1016/j.radonc.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 17.Sakata K, Someya M, Matsumoto Y, Tauchi H, Kai M, Toyota M, Takagi M, Hareyama M, Fukushima M. Gimeracil, an inhibitor of dihydropyrimidine dehydrogenase, inhibits the early step in homologous recombination. Cancer Sci. 2011;102:1712–1716. doi: 10.1111/j.1349-7006.2011.02004.x. [DOI] [PubMed] [Google Scholar]
- 18.Harada K, Ferdous T, Ueyama Y. Gimeracil exerts radiosensitizing effects on oral squamous cell carcinoma cells in vitro and in vivo. Anticancer Res. 2016;36:5923–5930. doi: 10.21873/anticanres.11179. [DOI] [PubMed] [Google Scholar]
- 19.Takagi M, Sakata K, Someya M, Matsumoto Y, Tauchi H, Hareyama M, Fukushima M. The combination of hyperthermia or chemotherapy with gimeracil for effective radiosensitization. Strahlenther Onkol. 2012;188:255–261. doi: 10.1007/s00066-011-0043-6. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg B. Possible mechanisms for the antitumor activity of platinum coordination complexes. Cancer Chemother Rep. 1975;59:589–598. [PubMed] [Google Scholar]