

Abstract
Adenylation is a crucial enzymatic process in the biosynthesis of nonribosomal peptide synthetase (NRPS) derived natural products. Adenylation domains are considered the gatekeepers of NRPSs since they select, activate, and load the carboxylic acid substrate onto a downstream peptidyl carrier protein (PCP) domain of the NRPS. We describe a coupled continuous kinetic assay for NRPS adenylation domains that substitutes the PCP domain with hydroxylamine as the acceptor molecule. The pyrophosphate released from the first-half reaction is then measured using a two-enzyme coupling system, which detects conversion of the chromogenic substrate 7-methylthioguanosine (MesG) to 7-methylthioguanine. From profiling substrate specificity of unknown or engineered adenylation domains to studying chemical inhibition of adenylating enzymes, this robust assay will be of widespread utility in the broad field NRPS enzymology
Keywords: Adenylation, Adenylate-forming, Hydroxamate, MesG, Enzyme assay
1 Introduction
Adenylation domains prime nonribosomal peptide synthetase (NRPS) biosynthetic pathways by catalyzing a two-step reaction (Fig. 1) [1, 2]. In the first step, the adenylation domain recognizes and binds ATP and its cognate carboxylic acid substrate 1, which is usually an amino acid but can also be an α-hydroxy acid, aryl acid, or fatty acid (Fig. 1). The enzyme then catalyzes the nucleophilic attack (step a) of the substrate carboxylic acid on the α-phosphate of ATP to form an acyl-adenylate intermediate 2 and pyrophosphate. In the second-half reaction (step b), the adenylation domain binds a peptidyl carrier protein (PCP) domain and transfers the activated acid onto the phosphopantetheinyl arm of the PCP domain to provide the thioester tethered carboxylic acid building block (3).
Fig. 1.
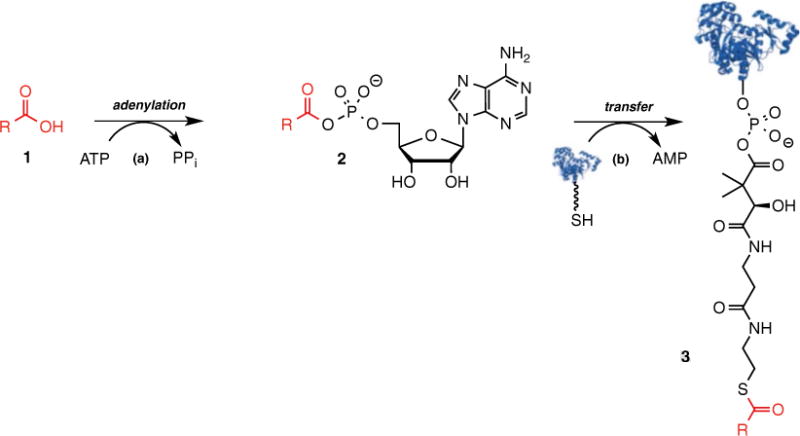
Two-step mechanism of adenylating enzymes
NRPS adenylation domains (A-domains) are usually located in cis to the PCP domain as part of a multifunctional NRPS protein; however, A-domains can also be located in trans as separate proteins [3]. In the former case, the A-domain cannot be analyzed by steady-state kinetic techniques since only a single turnover can be performed whereas in the latter case stoichiometric amounts of the cognate PCP domain are required. The most common method to measure A-domain activity that obviates the need for a PCP domain is the pyrophosphate exchange radioassay wherein one measures incorporation of [32P]PPi into ATP. Bachmann and coworkers have also reported an innovative mass spectrometric-based pyrophosphate exchange assay employing [18O]ATP that supplants the requirement for radioisotopes [4]. These exchange assays are useful for most A-domains, but not all A-domains undergo pyrophosphate exchange [5, 6]. Moreover, the pyrophosphate exchange assay only evaluates the adenylation partial reaction in the reverse direction and therefore may not provide physiologically meaningful kinetic parameters. In the absence of an acceptor molecule such as a PCP domain, the acyl-adenylate slowly leaks out of the active site, enabling slow turnover (the leak rates are typically 100-fold slower than the overall rates using the cognate PCP domain) [6, 7]. Garneau-Tsodikova and coworkers have exploited this phenomenon to develop a nonradioactive assay employing pyrophosphatase that cleaves the liberated pyrophosphate to inorganic phosphate, which is detected by malachite green [8]. All of these aforementioned assays are end-point assays. Ideally, it would be useful to develop a continuous assay, which measures the overall A-domain catalyzed reaction in the kinetically relevant forward direction and that employs a reactive surrogate for the PCP domain in order to provide fast enzyme turnover.
Herein we report a simple continuous coupled assay based on an amalgamation of several reported assays, which proceeds in the forward direction and employs hydroxylamine as an alternative and highly reactive acceptor molecule for the PCP domain [5, 9, 10]. The assay is rendered continuous by monitoring the pyrophosphate produced from the first-half reaction. The pyrophosphate is first cleaved to inorganic phosphate by inorganic pyrophosphatase (IP); the resulting phosphate is a substrate of purine nucleoside phosphorylase (PNP), which converts 7-methylthioguanosine (MesG, 5) to 7-methylthioguanine (6), whose formation can be continuously monitored at 360 nm. Our lab has validated this hydroxylamine-MesG coupled adenylation assay against several stand-alone A-domains as well as A-domains from multifunctional NRPS proteins. Moreover, the specificity constants (kcat/KM) obtained with this new assay are virtually identical to values obtained employing the standard, radioactive pyrophosphate exchange radioassay [6].
The following protocol details the hydroxylamine-MesG assay performed with the A-domain of the canonical NRPS known as GrsA that activates D-phenylalanine. GrsA is involved in the biosynthesis of the prototypical nonribosomal peptide gramicidin [11] and its adenylation domain (GrsA−APhe) has been successfully studied using the hydroxylamine-MesG continuous assay [6]. Below, we describe two assays that are run in separate wells (see Note 1); the first contains all assay components (GrsA−APhe enzyme, inorganic pyrophosphatase (IP), purine nucleoside phosphorylase (PNP), 7-methylthioguanosine (MesG), TCEP (tris(2-carboxyethyl)phosphine), hydroxylamine, ATP, and D-phenylalanine (D-Phe)), while the second reaction does not contain D-Phe and is used to measure background activity. Our lab uses a plate reader (Molecular Devices Spectramax M5e) that reads both absorbance in the kinetic mode and the pathlength of each individual well following the completion/end point of the assay. Additionally, this assay may be conveniently run on a UV-spectrophotometer using a micro-cuvette. As noted below, the slope of the absorbance progress curve (in mAU/ min) for each well is normalized to its own path length before being converted to concentration units (μM/min).
2 Materials
2.1 Expression of Adenylating Enzyme
Adenylating enzymes in our lab are routinely cloned into pET vectors and transformed into E. coli BL21 (DE3) for overexpression. His-tagged proteins are successively purified by Ni-NTA and gel filtration chromatography and aliquoted and stored at −80 °C in buffer containing 5 % glycerol [11].
2.2 Reagent Stock Solution Preparation
Water for this assay: Prepare ultrapure water for this assay by passing water through a Millipore-Q system or equivalent.
2× Adenylation Assay Buffer: 100 mM Tris-HCl, pH 8.0, 10 mM MgCl2. Weigh and transfer 12.1 g of Tris (Sigma) and 2 g of MgCl2∙6H2O (Sigma) to a 1 L graduated beaker. Add water to a volume of 900 mL, stir to dissolve, and then adjust the pH to 8.0 with 6 N HCl. Dilute up to 1 L with water and store at room temperature.
100 mM TCEP: Dissolve 29 mg of TCEP (HCl salt, Fisher Scientific) in 1 mL of water and store at −20 °C.
4 M hydroxylamine solution: Dissolve 2.78 g of hydroxyl-amine (HCl salt, Sigma) in 10 mL of water and store at 4 °C.
7 M NaOH solution: Dissolve 2.8 g of sodium hydroxide (Sigma) in 10 mL of water on ice and store at 4 °C.
40 U/mL inorganic pyrophosphatase (IP): add 2.5 mL of water to one vial of IP (Sigma I1643-100UN) and store at 4 °C.
100 U/mL purine nucleoside phosphorylase (PNP): add 1 mL of water to one vial of PNP (Sigma N8264-100UN) and store at 4 °C.
1 mM MesG: dissolve 3.0 mg of MesG (Berry & Associates) in 10 mL of water, aliquot into 1 mL aliquots, and store at −80 °C for up to 1 month.
100 mM ATP: dissolve 55 mg of ATP (disodium salt hydrate, Sigma) in 1 mL of 1 M Tris-HCl pH 8 and stored at −80 °C (see Note 2).
100 mM acid substrate in 100 mM Tris-HCl, pH 8.0: (see Note 3).
Concentrated adenylating enzyme: Make intermediate enzyme stock in 2× adenylation buffer (see Subheading 2.1).
Preparation of working solution of hydroxylamine: On the day that the assay is to be run, prepare a 2 M solution of hydroxylamine, pH 7.0. To a 1.5 mL centrifuge tube on ice, add 400 μL of the 4 M hydroxylamine stock solution. To this, add 175 μL of water and 225 μL of 7 M NaOH stock solution dropwise. Confirm that the pH of this solution is 7–8 using pH paper.
-
Prepare Master Mix: The assay will be initiated by adding 95 μL of master mix (GrsA−APhe enzyme, IP, PNP, MesG, TCEP, hydroxylamine, and ATP in Tris-HCl buffer pH 8.0) to 5 μL D-Phe (or buffer alone) in a well of a UV clear half-area 96-well plate. Therefore, all of the components in the master mix are made at 1.05× the concentration, so that they are at a final concentration of 1× when diluted to a final volume of 100 μL in the assay plate.
Make stock concentrations of assay components (see Subheading 2.1 and Column A in Table 1).
Determine final concentrations of enzyme and substrates (Column B, Table 1) that will provide initial velocity conditions (see Note 5).
Multiply values in Column B by a factor of 1.05 to obtain values in Column C.
Calculate the volume of each assay component for one assay well (Column D).
Multiply the volumes in Column D by 2.5 to obtain the total volume needed for all of the assay wells (Column E). In the assay example described here, two assays are run in separate wells (with and without D-Phe). The multiplication factor is increased from 2 to 2.5 in order to account for small volume losses due to pipetting.
Table 1.
Master mix preparation
| Column A | Column B | Column C | Column D | Column E | |
|---|---|---|---|---|---|
| [Stock] | [Final] in 100 μL well | [Final] in 95 μL | Volume needed for 1 well (μL) | Total volume needed in master mix (μL) | |
| 2× Adenylation assay buffer | – | 1× | 1× | 50 | 125 |
| Water | – | – | − | 22.9 | 70.3 |
| GrsA-A | 1 μM | 5 nM | 5.25 nM | 0.53 | 1.3 |
| IP | 40 U/mL | 0.04 U | 0.042 U | 1.05 | 2.6 |
| PNP | 100 U/mL | 0.1 U | 0.105 U | 1.05 | 2.6 |
| MesG | 1 mM | 100 μM | 105 μM | 5.25 | 13.2 |
| TCEP | 100 mM | 1 mM | 1.05 mM | 1.05 | 2.6 |
| Hydroxylamine | 2 M | 150 mM | 157.5 mM | 7.7 | 19.3 |
| ATP | 100 mM | 5 mM | 5.25 mM | 5.3 | 13.1 |
2.3 Additional Supplies and Equipment
96-well UV clear half-area plates (Corning #3679).
Plate reader capable of reading UV absorbance at 360 nm and calculating the path length of the wells (see Note 4).
3 Methods
3.1 Initiate Assay
To one well of a 96-well UV clear half-area plate add 5 μL of a 20 mM solution of D-Phe (made in 100 mM Tris, pH 8.0) and to another well add 5 μL of 100 mM Tris–HCl, pH 8.0. Add 95 μL of the Master Mix to both wells and ensure that no bubbles form.
3.2 Read Plate and Path Length
Read the absorbance at 360 nm for 5 min in the kinetic read mode (Fig. 2).
Obtain slope (in mAU/min) using the plate reader software.
Read the pathlength of each well.
Fig. 2.
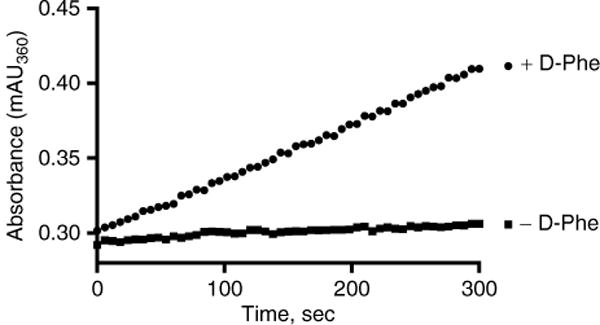
Time course of 7-methylguanine production. Each reaction contained 50 mM Tris–HCl, pH 8.0, 5 mM MgCl2, 5 nM GrsA−APhe, 0.04 U IP, 0.1 U PNP, 200 μM MesG, 1 mM TCEP, 150 mM hydroxylamine, 5 mM ATP. The control reaction contained no D-Phe while the positive reaction contained 1 mM D-Phe
3.3 Calculate Initial Velocity
- The slopes measured by the plate reader for the two assays in Fig. 2 are:
- +D-Phe = 21.972 mAU/min
- −D-Phe = 2.195 mAU/min
- The path lengths at the end point of the assays are:
- +D-Phe = 0.512 cm
- −D-Phe = 0.505 cm
- Calculate the path length normalized slope by dividing the slopes by the pathlength of each well:
- +D-Phe: 21.972 mAU/min ÷ 0.512 cm = 42.914 mAU/min/cm
- −D-Phe: 2.195 mAU/min ÷ 0.505 cm = 4.347 mAU/min/cm
- Convert units from mAU to AU (see Note 7):
- +D-Phe: 42.914 mAU/min/cm ÷ 1000 = 0.0429 min−1 cm−1
- −D-Phe: 4.347 mAU/min/cm ÷ 1000 = 0.0043 min−1 cm−1
- Divide by the extinction coefficient of 7-methylthioguanine (7, Fig. 3):
- +D-Phe: 0.0429 min−1 cm−1 ÷ 11,000 M−1 cm−1 = 3.9 × 10−6 M/min
- −D-Phe: 0.0043 min−1 cm−1 ÷ 11,000 M−1 cm−1 = 3.9 × 10−7 M/min
- Convert to μM:
- +D-Phe: 3.9 × 106 M/min × 106 μM/M = 3.9 μM/min
- +D-Phe: 3.9 × 10−7 M/min × 106 μM/M = 0.39 μM/min
- Divide by 2 since there are two molecules of 7-methylguanine formed per turnover:
- +D-Phe: 3.9 μM/min ÷ 2 = 1.95 μM/min
- +D-Phe: 0.39 μM/min ÷ 2 = 0.195 μM/min
- Subtract the background rate (−D-Phe) from the full enzymatic rate (+D-Phe):
- Rate: 1.95 μM/min − 0.195 μM/min = 1.76 μM/min
The rate value obtained in step 8 can now be used to obtain Michaelis-Menten kinetic parameters of all substrates (by varying substrate concentrations) or study enzyme inhibition (by varying inhibitor concentrations) (see Note 6).
Fig. 3.
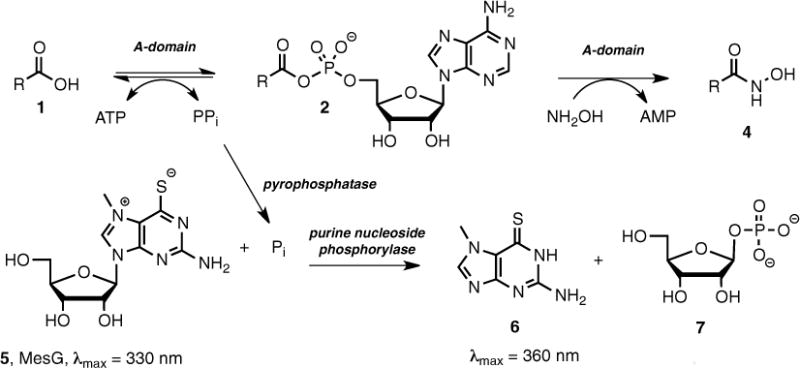
Continuous hydroxamate-MesG assay for measuring adenylation activity
Acknowledgments
CCA. acknowledges membership in and support from the Region V “Great Lakes” Regional Center of Excellence in Biodefense and Emerging Infectious Diseases Consortium (National Institutes of Health award 1-U54-AI-057153) as support from R01-AI-070219 for this work.
Footnotes
Assays should be run in triplicate. The examples shown here are run in singlicate for clarity of presentation.
To dissolve ATP, a high concentration of Tris–HCl Buffer (1 M, pH 8.0) must be used in order to neutralize ATP, which is acidic. ATP should not be stored for more than 1 week at −80 °C.
Our lab routinely makes 10–100 mM solutions of the acid substrate in 100 mM Tris-HCl, pH 8.0. The concentration of the acid stock will depend on the acid’s aqueous solubility.
The amount of adenylating enzyme (in units) should always be much lower (<50×) than the amount of coupling enzymes. This will ensure that the coupling enzymes are not the rate limiting enzymes.
Our lab uses a Spectramax M5e plate reader, which reads and calculates path length using the PathCheck® function.
If inhibition of adenylating enzymes is to be studied, add the Master Mix to the inhibitor (final DMSO ≤1 %) and incubate for 10 min prior to initiating the reaction by adding to the well containing the substrate.
Absorbance is unitless. Therefore, the unit AU is not shown after converting from mAU to AU.
References
- 1.Schmelz S, Naismith JH. Adenylate-forming enzymes. Curr Opin Struct Biol. 2009;19:666–671. doi: 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gulick AM. Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hur GH, Vickery CR, Burkart MD. Explorations of catalytic domains in nonribosomal peptide synthetase enzymology. Nat Prod Rep. 2012;29:1074–1098. doi: 10.1039/c2np20025b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phelan VV, Du Y, McLean JA, Bachmann BO. Adenylation enzyme characterization using γ-18O4-ATP pyrophosphate exchange. Chem Biol. 2009;16:473–478. doi: 10.1016/j.chembiol.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kadi N, Challis GL. Chapter 17 siderophore biosynthesis: a substrate specificity assay for nonribosomal peptide synthetase-independent siderophore synthetases involving trapping of acyl-adenylate intermediates with hydroxylamine. Methods Enzymol. 2009;458:431–457. doi: 10.1016/S0076-6879(09)04817-4. [DOI] [PubMed] [Google Scholar]
- 6.Wilson DJ, Aldrich CC. A continuous kinetic assay for adenylation enzyme activity and inhibition. Anal Biochem. 2010;404:56–63. doi: 10.1016/j.ab.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehmann DE, Shaw-Reid CA, Losey HC, et al. The EntF and EntE adenylation domains of Escherichia coli enterobactin synthetase: sequestration and selectivity in acyl-AMP transfers to thiolation domain cosubstrates. Proc Natl Acad Sci U S A. 2000;97:2509–2514. doi: 10.1073/pnas.040572897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McQuade TJ, Shallop AD, Sheoran A, et al. A nonradioactive high-throughput assay for screening and characterization of adenylation domains for nonribosomal peptide combinatorial biosynthesis. Anal Biochem. 2009;386:244–250. doi: 10.1016/j.ab.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc Natl Acad Sci U S A. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Upson RH, Huagland RP, Malekzadeh MN, et al. A spectrophotometric method to measure enzymatic activity in reactions that generate inorganic pyrophosphate. Anal Biochem. 1996;243:41–45. doi: 10.1006/abio.1996.0479. [DOI] [PubMed] [Google Scholar]
- 11.Stachelhaus T, Marahiel M. Modular structure of peptide synthetases revealed by dissection of the multifunctional enzyme GrsA. J Biol Chem. 1995;270:6163–6169. doi: 10.1074/jbc.270.11.6163. [DOI] [PubMed] [Google Scholar]