

Abstract
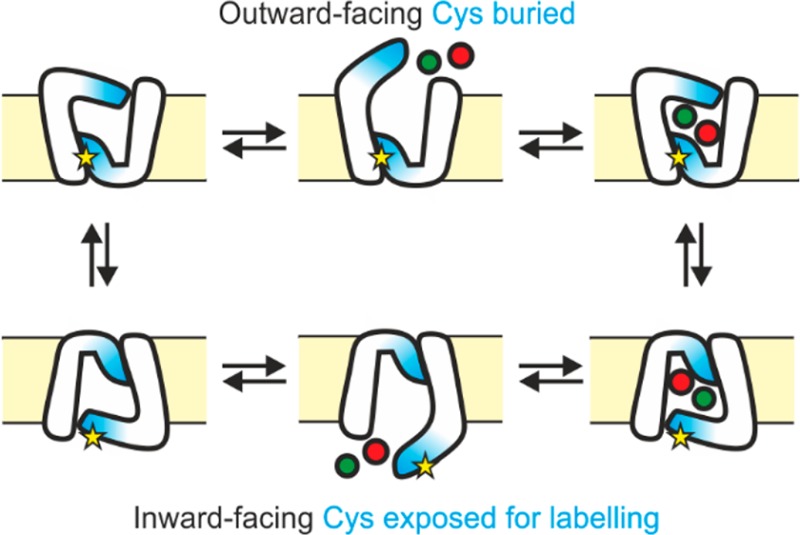
Cys accessibility and quantitative intact mass spectrometry (MS) analyses have been devised to study the topological transitions of Mhp1, the membrane protein for sodium-linked transport of hydantoins from Microbacterium liquefaciens. Mhp1 has been crystallized in three forms (outward-facing open, outward-facing occluded with substrate bound, and inward-facing open). We show that one natural cysteine residue, Cys327, out of three, has an enhanced solvent accessibility in the inward-facing (relative to the outward-facing) form. Reaction of the purified protein, in detergent, with the thiol-reactive N-ethylmalemide (NEM), results in modification of Cys327, suggesting that Mhp1 adopts predominantly inward-facing conformations. Addition of either sodium ions or the substrate 5-benzyl-l-hydantoin (L-BH) does not shift this conformational equilibrium, but systematic co-addition of the two results in an attenuation of labeling, indicating a shift toward outward-facing conformations that can be interpreted using conventional enzyme kinetic analyses. Such measurements can afford the Km for each ligand as well as the stoichiometry of ion–substrate-coupled conformational changes. Mutations that perturb the substrate binding site either result in the protein being unable to adopt outward-facing conformations or in a global destabilization of structure. The methodology combines covalent labeling, mass spectrometry, and kinetic analyses in a straightforward workflow applicable to a range of systems, enabling the interrogation of changes in a protein’s conformation required for function at varied concentrations of substrates, and the consequences of mutations on these conformational transitions.
Secondary active membrane transport proteins exploit the potential energy of ion gradients (e.g., proton or sodium) to drive the transport of solutes across membranes.1 The available structures of these proteins (and other biophysical analyses) suggest a common transport mechanism, termed alternating access, whereby substrate/ion binding sites on the two sides of the membrane are alternately exposed.2−6 Substrate binding on one side of the membrane leads to conformational changes that enable its release on the other side. Movements of transmembrane helices as well as external/internal helices and loops underlie this alternating access mechanism. The conformational state(s) of such proteins can be determined by means of X-ray crystallography, but elucidating the conformational state(s) and intermediates adopted in solution, and how the binding of ligands influences the conformational equilibrium of the protein, is of vital importance to enable full characterization of the transport cycle.7−10
Mass Spectrometry (MS) is being employed increasingly for the structural interrogation of proteins and protein assemblies,11−14 with recent methodological advances permitting the analysis of membrane proteins (MPs).15 Noncovalent MS can be employed to determine the stoichiometry of MP assemblies,16−18 observe protein binding to lipids and small molecules,19,20 and study conformational changes upon binding (when coupled with ion mobility spectrometry).21,22 Chemical cross-linking MS, where a bifunctional small molecule is used to chemically join spatially proximal residues, can be used to afford low residue distance restraints for the modeling of protein complex architecture and conformational changes.23−27 Mapping of solvent accessibility and dynamics can also be performed by implementing well-established covalent labeling workflows, including hydrogen–deuterium exchange,28,29 hydroxyl radical footprinting,30,31 carbene labeling,32,33 and amine reactivity.23,24 Additionally, reaction of Cys residues with maleimides, such as N-ethylmaleimide (NEM), which react with the thiol group of Cys residues by Michael addition, has been successfully implemented to interrogate Cys residue accessibility and to deduce conformational/topological information.9,34,35
The transport protein Mhp1 from Microbacterium liquefaciens (M. liquefaciens) mediates the uptake of 5-aryl-substituted hydantoins in a Na+-dependent fashion.36,37 Mhp1 is a member of the nucleobase–cation–symport-1 (NCS-1) family of secondary active transport proteins [part of the amino acid–polyamine–organocation (APC) superfamily], which is found widely in bacteria, archaea, fungi, and plants.2,7,38−41 NCS-1 family proteins are structurally related by a 5-helix/5-helix internal pseudosymmetry to proteins in different subfamilies of the APC superfamily, which is also called the 5-helix inverted repeat (5HIRT) or LeuT superfamily of ion-coupled transporters. In humans, membrane transport proteins of these families are involved in processes such as neurotransmitter, sugar, amino acid, and drug transport.42−44 The diversity of their biological functions has led to a burgeoning field of research pertaining to unravelling the structural basis by which this class of proteins transport their assorted substrates.2−4,6 Importantly, structures of proteins in the 5HIRT/LeuT superfamily differ completely from those in the major facilitator superfamily, a small number of which have been studied by MS previously.35,45
Mhp1 contains 12 transmembrane helices (TMHs), with 10 core TMHs, characteristic of the 5HIRT superfamily, and two additional C-terminal helices.48 Structures of Mhp1 in the outward-open, inward-open, and occluded states (Figure 1a) have been solved by X-ray crystallography.46−48 TMHs 1, 2, 6, and 7 form a four helix bundle (Figure 1a, red helices), and TMHs 3, 4, 8, and 9 form a motif that resembles a hash sign (#) (Figure 1a, yellow helices).46 The ligand and Na+ binding sites are located between the hash and bundle motifs and involve residues in TMHs 1 and 6 (where the helices break). Binding of the ligand to the outward-facing conformation causes TMH 10 to bend and occlude the substrate binding site; a subsequent transition to the inward-facing conformation occurs as a result of movement of the hashed domain relative to the bundle domain.46,48
Figure 1.

X-ray crystal structures of Mhp1. (a) X-ray crystal structures of Mhp1 in the inward-facing (Protein Data Bank (PDB) 2X79),46 outward-occluded (PDB 4D1B),47 and outward-facing (PDB 2JLN)48 conformations. Helices are represented as cylinders. The bundle46 helices (TMHs 1, 2, 6, and 7) are colored red, the hash46 motif helices (TMHs 3, 4, 8, and 9) are colored yellow, the flexible helices (TMHs 5 and 10) are colored blue, the C-terminal helices (TMHs 11 and 12) and the surface extracellular and cytoplasmic helices are colored gray. The Cys residues are shown in cyan. (b, c) Location of Cys327 (cyan) in the (b) inward-facing and (c) outward-facing conformations of Mhp1, showing that TMH9 protects Cys327 from solvent in the outward-facing conformation. (d) Average side-chain solvent-accessible surface area (SASA) values of the three Cys residues of Mhp1 in 20 ns MD simulations started from the inward-facing (IF), outward-occluded (OOc), and outward-facing (OF) crystal structures.
Here we combine Cys-accessibility determination by NEM reactivity, intact MS analysis, peptide mapping, and localization of NEM modification sites to gain insights into the topological states of Mhp1. The data suggest that detergent-solubilized Mhp1 adopts predominantly inward-facing conformations and that the presence of either Na+ or the substrate L-BH does not shift this conformational equilibrium. However, co-addition of both Na+ and L-BH results in the outward-facing conformation being populated significantly. By titrating in various combinations of L-BH/Na+, we demonstrate that the data obtained can be used to extract the stoichiometry of binding which induces the conformational change. We also demonstrate the suitability of the method to characterize variants of Mhp1 and provide a rationale for mutation-induced changes in substrate binding efficiency. Additionally, we show that the approach can be used to screen ligands and identify binders.
We envisage that the workflow developed will be widely transferrable and provide insights into membrane transport proteins and other protein systems, including (i) the conformational states adopted under different conditions; (ii) identifying and characterizing binding of (novel) substrates and inhibitors (by using the MS method as a library screening tool); and (iii) characterizing variants to determine whether they have the same conformational fingerprint as the wild-type protein.
Experimental Section
Expression and Purification of Mhp1 and Mhp1 Variants
Throughout the text, wild-type Mhp1 indicates the protein modified by the addition of a C-terminal His6 tag.37 Expression and purification of Mhp1 and Mhp1 variants was conducted as described previously.36,37,47−49
Labeling of Mhp1 with N-Ethylmaleimide
Solutions of purified Mhp1 (10 μM) in 10 mM Tris-HCl (pH 8), 2.5% (v/v) glycerol, 0.05% (w/v) DDM, and 2% (v/v) DMSO were supplemented with one or more of the following (see figure legends): 15-crown-5 (1.25 mM), NaCl (15, 140, or 1000 mM), choline chloride (125 mM, with 15 mM NaCl), and/or L-BH (2 mM). The protein was incubated with the appropriate additives for 10 min before labeling. NEM (1 mM final concentration) was added, and the solution was incubated for 1 h at 25 °C. The reaction was quenched by adding DTT (final concentration of 30 mM) and then incubating at 25 °C for 10 min.
Preparation of Samples for Mass Spectrometry
A sample of Mhp1 (unlabeled or labeled with NEM) (50 uL, 10 μM) was taken, and methanol (150 μL) and chloroform (50 μL) were added. The solution was mixed by vortexing, water (100 μL) was added, and the solution was mixed again before centrifuging (10000g, 2 min). The upper aqueous phase was carefully removed (leaving the white protein pellet and the lower organic phase), and methanol (150 μL) was then added. The solution was mixed by vortexing, centrifuged (10000g, 2 min), and the supernatant removed. The precipitated protein was air-dried in a laminar flow hood. The dried protein pellet was resuspended in formic acid (4 μL), and ddH2O was then added (46 μL) for subsequent MS analyses.
Measurement of the Intact Mass of Mhp1
Proteins were analyzed intact using online desalting LC-MS on a nanoAcquity LC system interfaced to a Xevo G2-S mass spectrometer (Waters Ltd., Wilmslow, Manchester, U.K.). Deconvolution was performed using UniDec.50 All expected and observed protein masses are shown in Table S1. See Supporting Information for details.
Proteolysis and Localization of the Reaction Sites for NEM in Individual Peptides
Mhp1 was digested by adding pepsin (at a 1:50 protease:substrate mass ratio) and incubating at 37 °C for 2 h with shaking. The protease was deactivated by boiling at 100 °C for 10 min. Peptides were then analyzed by LC-MS/MS on a nanoAcquity LC system interfaced to a Synapt G2-Si HDMS mass spectrometer (Waters, U.K.). See Supporting Information for details.
Mhp1 Ligand Binding Assays
The ability of Mhp1 to bind L-BH was determined by means of fluorescence emission spectroscopy on a QuantaMaster spectro-photofluorometer (Photon Technology International, Ford, West Sussex, U.K.), using previously published methods.37,47,48,51 See Supporting Information for details.
Calculation of the Solvent-Accessible Surface Area of Modified Residues
Trajectories of 20 ns all-atom molecular dynamics simulations in an explicit membrane and solvent environment of inward-facing (IF), outward-facing, and occluded with both the substrate and Na+ bound (OOc) and outward-facing with Na+ bound (OF) MhpI were obtained from a previous study.52 From the coordinates of frames saved in increments of 1 ps, the average SASA of Cys69, Cys234, and Cys327 was computed. The SASA of a residue (Ai) was calculated as the sum of the solvent accessibilities of the individual side-chain atoms (ai).
The atom-wise solvent-accessible surface area ai was calculated analytically as described previously.53
Results and Discussion
Cys Residues in Mhp1 Predicted To Have Different Solvent Accessibilities
The available structures of Mhp1 show that Cys327 (located in TMH 8, Figure 1) is located near the surface of the protein whereas Cys234 and Cys69 are buried within its structure (Figure 1a). Further comparisons of the position of Cys327 in the inward-facing open conformation (Protein Data Bank (PDB) 2X79)46 to its position in the outward-facing open form (PDB 2JLN)48 suggest that it becomes relatively protected from solvent in the outward-facing conformation via reorganization of TMH 9 (Figure 1b,c). To determine the accessibility of Cys residues in Mhp1, the residue-specific side-chain solvent-accessible surface area values were determined in silico from available molecular dynamics simulations in explicit membrane and solvent environments (Figure 1d).52 The analysis showed that the most accessible Cys residue in all three conformations of Mhp1 is Cys327, while the other two Cys residues (Cys69 and Cys234) are more protected from solvent (Figure 1d). In addition, Cys327 is more highly accessible in the inward-facing relative to the outward-facing conformation (Figure 1d). Combined, all these observations suggest that the accessibility of Cys327 could be a useful probe for discriminating between the inward-facing and outward-facing conformational states of Mhp1.
Cys327, a Conformationally Sensitive Residue
Following incubation of Mhp1 with (or without) NEM (Figure 2a), electrospray ionization mass spectra of the proteins were obtained (Figure 2b and Figure S1) and the spectra deconvoluted (Figure 2c). In the absence of Na+ (ensured by adding the crown ether 15-crown-5 to sequester residual Na+), the predominant species observed corresponds to Mhp1 + 1NEM, with minimal unlabeled Mhp1 remaining and negligible Mhp1 + 2NEM and Mhp1 + 3NEM detected (Figure 2c, lower panel). The modified protein was digested with pepsin, and the modification site was localized by peptide mapping. This confirmed that Cys327 was the dominant residue modified (Figure S2), and that the other two Cys residues were only modified to a very low level (<1% relative abundance).
Figure 2.
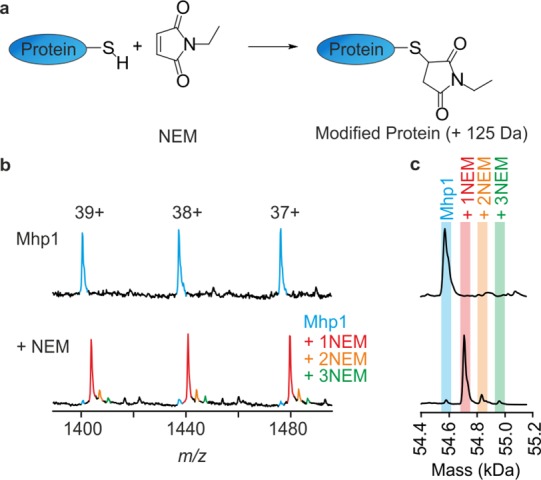
Mass spectrometry of wild-type Mhp1 and wild-type Mhp1 labeled with NEM. (a) Reaction of a Cys residue in Mhp1 with NEM, resulting in a mass addition of 125 Da. (b) Portion of a representative, unprocessed mass spectrum of Mhp1 (upper panel) and of NEM-labeled Mhp1 that was preincubated with 15-crown-5 to remove residual NaCl (lower panel). Spectra were obtained by online desalting-MS, and only three charge states are shown for clarity; the full unprocessed spectra are shown in Figure S1. (c) Deconvoluted mass distributions of labeled and unlabeled Mhp1 (spectra are shown next to their unprocessed counterpart in panel b). The shading indicates expected masses of unlabeled and labeled Mhp1.
Influence of Hydantoin and Na+ Binding on Cys Accessibility
To determine the effect (if any) of solution conditions on the labeling reaction, Mhp1 was preincubated with NaCl, the ligand L-BH, or a combination of both, before NEM was added. Intriguingly, the mass distributions obtained for Mhp1 that was labeled in the presence of either 140 mM NaCl or 2 mM L-BH were identical to those obtained in the absence of these species (Figure 3a(i).). Conversely, co-incubation with both 140 mM NaCl and L-BH resulted in a dramatically different distribution of masses, with both unlabeled Mhp1 and Mhp1 + NEM identified, now with comparable intensity (Figure 3a(i)). Increasing the NaCl concentration to 1 M and performing the NEM labeling reactions resulted in the same labeling pattern as in the presence of 140 mM NaCl (Figure 3b(i)). Again, both 1 M NaCl and 2 mM L-BH were required to observe a diminution in labeling, and the proportion of labeled Mhp1 was reduced further than with 140 mM NaCl (Figure 3b(i)). The modified Cys residue in all cases was identified as Cys327 using proteolysis and peptide mapping (e.g., see Figure S2).
Figure 3.
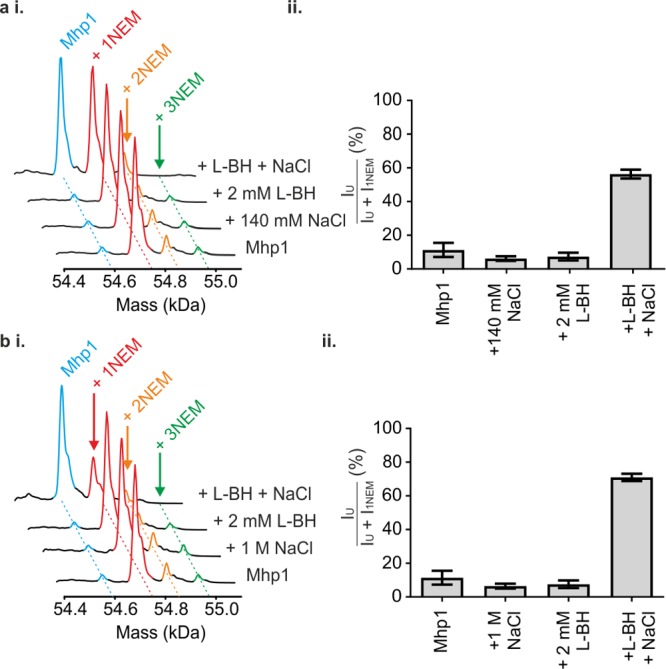
Mass spectrometry of wild-type Mhp1 after labeling with NEM under different conditions of substrate and/or Na+ inclusion. NaCl additions were performed at either (a) 140 mM NaCl or (b) 1 M NaCl. (i) Deconvoluted mass distributions of Mhp1 after NEM labeling under varied solution conditions. Note that at high concentrations of both ligands the relative abundance of the +2NEM peak decreases (although this is obscured in the figure). (ii) Relative abundances of unlabeled Mhp1 (IU) relative to Mhp1 with 1 NEM label (I1NEM) in the mass spectra. Data are shown as mean ± SEM of three independent experiments.
These data, combined with the structural and SASA data presented (Figure 1), suggest that addition of either Na+ or L-BH is insufficient to shift the conformational equilibrium of Mhp1. Thus, the inward-facing conformation is favored, as in their absence. However, the synergistic effect of both Na+ and L-BH together results in conformational conversion to a form of the protein in which Cys327 is protected from solvent, consistent with an alteration in the equilibrium so that the outward-facing conformation becomes favored.
In order to perform a semiquantitative analysis, the relative intensities of peaks corresponding to unmodified Mhp1 and Mhp1 modified with a single NEM label were determined (Figure 3a(ii),b(ii)). These measurements reinforce the conclusion that adding either NaCl or L-BH alone does not significantly alter Mhp1 modification by NEM; however, co-addition of NaCl and L-BH leads to a significant shift in population toward unmodified Mhp1. Intriguingly, the amount of unmodified Mhp1 observed in the presence of 140 mM NaCl/2 mM L-BH (52.2 ± 5%) is significantly lower than that observed in the presence of 1 M NaCl/2 mM L-BH (68.2 ± 1%). The observation that significantly more protein remains unlabeled in the presence of higher concentrations of NaCl suggests that the conformational equilibrium of Mhp1 is shifted even further to the outward-facing form by higher NaCl concentrations. From these data we propose that the mass distribution after NEM labeling can be used as a conformational “fingerprint” to deduce the conformational state of the protein under varied conditions of Na+ and ligand concentrations.
Specificity for Substrates of the Protection against Labeling with NEM
We tested other known ligands of Mhp147 to determine whether they produced effects similar to L-BH. Indeed, 5-indolylmethyl-l-hydantoin (L-IMH), 5-(2-naphthylmethyl)-l-hydantoin (L-NMH), and 5-bromovinylhydantoin (BVH) in solution at a concentration of 2 mM in the presence of 140 mM Na+ resulted in a reduction in NEM labeling to approximately similar extents (Figure S3) as that of L-BH (Figure 3a), while addition of ligand alone had no effect (Figure S3). Importantly, we also tested the molecules hydantoin and allantoin, which do not bind to Mhp1.47 In the presence of these smaller molecules, the labeling of Mhp1 is not attenuated, consistent with a lack of binding to Mhp1, and no effect on conformation (Figure S3). Thus, in the presence of Na+, all ligands that are already known to bind to Mhp1 promote conversion of Mhp1 from the inward-facing to the outward-facing form. This NEM-MS method could be used therefore as a rapid screen for new ligands of transporters or for understanding allosteric switching in other proteins. Perhaps most appealing, the quantitative and direct readout of the population of different (NEM-accessible and -inaccessible) states, provides advantages over other methods used for such binding studies by avoiding the use of radioisotope-labeled candidates, reducing the amount of protein required (compared with e.g., ITC), and the need for protein immobilization (SPR) which can compromise function.
The failure of L-BH in the absence of Na+ to switch the conformation of Mhp1 from inward-facing to outward-facing is particularly significant, since measurements of tryptophan fluorescence of the protein show that L-BH binds to the protein in the absence of added Na+.37,47,48,51 This, in combination with the NEM-MS data presented here, suggests that, in the absence of Na+, L-BH binds with low affinity to the inward-facing binding site. Conversely, in the presence of added NaCl, the measured high-affinity binding reflects binding of L-BH to the outward-facing conformation (Figure 4a).
Figure 4.
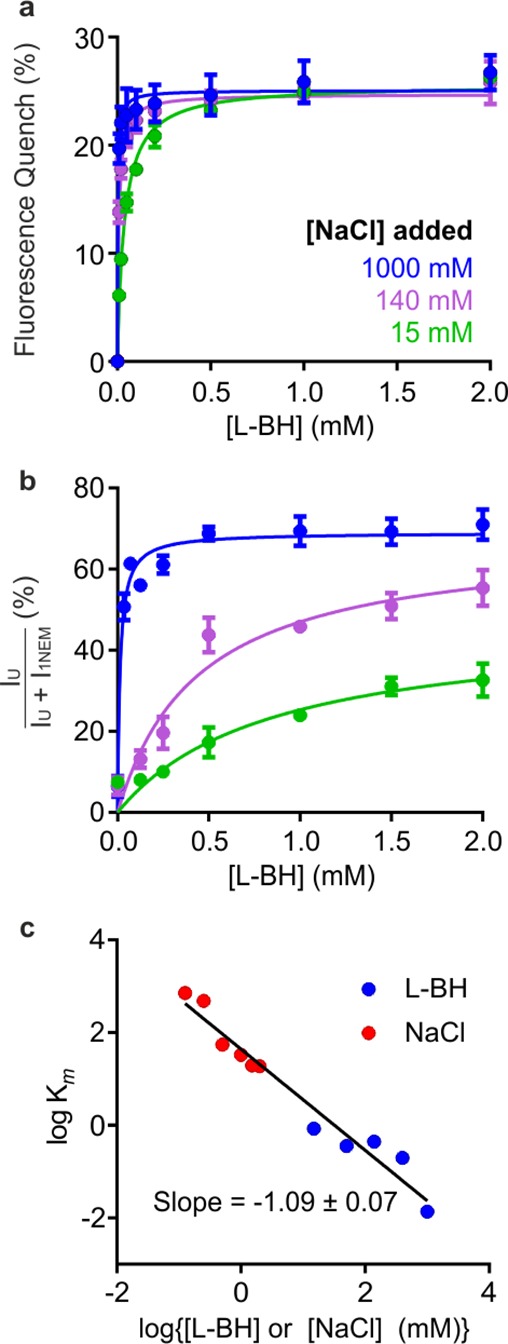
Titrations of Mhp1 with L-BH monitored by fluorescence and titratable effects of NEM labeling. (a) Tryptophan fluorescence quenching of Mhp1 upon titration with L-BH, in the presence of 15 mM (green), 140 mM (magenta), and 1000 mM (blue) added NaCl. (b) Quantification of unlabeled Mhp1 as a function of increasing [L-BH], at the same concentrations of NaCl as those in panel a. Data are shown as mean ± SEM of three independent measurements; additional titrations are shown in Figure S4. (c) Plot of log Km for either L-BH (blue) or Na+ (red) against either log[L-BH] (blue) or log [Na+] (red). The slope of the best fit line through all points was −1.09 ± 0.07, consistent with a 1:1 Na+:L-BH stoichiometry.56
Since the isolated Mhp1 in DDM appears to be largely in the inward-facing conformation, it is probable that Na+ alone does not affect the conformational distribution since the inward-facing species lacks the Na+-binding site present in the outward-facing form (as determined from X-ray crystallography).46,48 Indeed this conclusion is reinforced by molecular dynamics simulations, in which bound Na+ is seen to leave the inward-facing conformation rapidly.46
Next, we examined the concentration dependence of L-BH and NaCl on the extent of NEM labeling of Mhp1. Mhp1 was preincubated with various concentrations of NaCl (15, 50, 140, 500, or 1000 mM), L-BH was then titrated into the Mhp1-NaCl mixture, and labeling with NEM was subsequently performed (Figure 4b). From these experiments, a hyperbolic increase in unlabeled Mhp1 was observed as a function of L-BH concentration at each tested NaCl concentration (examples in Figure 4b).
These data show a [L-BH]- and [NaCl]-dependent increase in the amount of unlabeled Mhp1 when the other ligand is present at a fixed concentration. We fitted the data obtained to a Michaelis–Menten binding model (Figure 4b and Figure S4), as performed for the fluorescence titration experiments (Figure 4a), where the fitted Km value is the apparent dissociation constant, Kdapp. The binding model that was fitted to the MS data yields Km values that are uniformly higher than those using fluorescence (Figure S4), because the MS data reflect the ratio of all inward-facing to outward-facing Mhp1 at all the possible concentrations of [Na+]-[BH], rather than measuring only the percent of substrate bound, which is reflected by the change in tryptophan fluorescence. There is no reason to expect that these values would be similar, therefore, since different equilibria are being measured, as illustrated for a model alternating access transport mechanism54,55 in Figures S5 and S6. Thus, each technique provides complementary information that reveals insights into the transport mechanism. The MS data can be replotted to demonstrate the titratable effect of Na+ at fixed L-BH concentrations on the inward–outward equilibrium (Figure S4). Combined, these data demonstrate a synergistic effect of Na+ and L-BH in the transition of Mhp1 from inward-facing to outward-facing. Using the estimated Km values from all the titrations performed for the NEM-MS experiments, it is possible to extract the binding stoichiometry that is inducing the conformational change by plotting log Km as a function of log(ligand concentration) (here Na+ and L-BH; Figure 4c). These data are consistent with a 1:1 L-BH:Na+ binding stoichiometry, as the slope of the line of best fit is ∼1.56 For uncharacterized transporters, such an analysis may be used to unravel the stoichiometry of binding events that lead to conformational changes.
Susceptibility of Cys327 to Labeling by NEM in Mhp1 Altered by Single Residue Substitutions
In order to elucidate molecular mechanisms of transport, single point amino acid substitutions are often made in a protein to identify key residues in the transport process. However, it often remains unresolved whether such mutations alter the conformational distribution of species and/or the ability of the protein to interconvert between inward and outward-facing forms. A variety of mutations in Mhp1 have been made to interrogate the functional cycle of the protein.7,47 We thus subjected a number of these mutations to the same experimental probes used for the wild-type protein (Figure 5). These mutants all involved perturbing the substrate binding site of Mhp1 (Figure 5a), resulting in a reduction in uptake by Mhp1 and an increased Km (as determined by fluorescence).47 In most cases, mutation of residues in the L-BH binding site resulted in a NEM-MS labeling profile (in the presence of both L-BH and NaCl) that was comprised predominantly of singly-NEM-labeled Mhp1 (Figure 5b and Figure S7), indicating that the protein can no longer switch from inward-facing to outward-facing.
Figure 5.

Substitutions of individual amino acid in the ligand binding site, affecting the labeling of Mhp1 by NEM. (a) Representation of the L-BH binding site of Mhp1 in the inward-facing occluded state (PDB 4D1B).47 Key residues where substitutions have been made here are labeled. L-BH is shown in green. (b) Relative abundances of unlabeled protein (IU) relative to protein with 1 NEM label (I1NEM) in the mass spectra for Mhp1 and selected Mhp1 variants. “WT In” corresponds to Mhp1 in the presence of 1.25 mM 15-crown-5, and WT Out corresponds to labeling after preincubation with 2 mM L-BH and 140 mM NaCl. For all Mhp1 variants, labeling was performed after preincubation with 2 mM L-BH and 140 mM NaCl. Where the unmodified protein was not detected upon labeling, the relative abundance values are displayed as ±1%, reflecting the detection limit and signal-to-noise of the measurement (for G219S Mhp1). Values are displayed as mean ± SEM of three independent experiments. (c–g) Deconvoluted mass distributions of (c) Q42F Mhp1, (d) N318A Mhp1, (e) WT Mhp1 (f) WT Mhp1 heated to 80 °C and (g) Mhp1 in the presence of 0.5 % w/v SDS after NEM labeling in the presence of 15-crown-5 (1.25 mM).
In two instances, for the Q42F and N318A mutations, a strikingly different pattern was observed (Figure 5c,d), where all three Cys residues were labeled. These mutations presumably destabilize the whole protein, resulting in labeling of all three Cys residues with NEM. Consistent with this, denaturing conditions such as adding SDS or heating Mhp1 to 80 °C in the presence of Na+ and L-BH led to exposure of all three Cys residues in the protein and their consequent labeling with NEM (Figure 5e–g). It is therefore likely that the enhanced labeling of the protein seen with the N318A and Q42F mutants was due to (partial) unfolding of the three-dimensional structure, so that all the Cys residues became increasingly exposed to solvent and thus more amenable to labeling. Destabilization by such substitutions has also been reported in two fungal transporter homologues of Mhp1, FcyB (purine-adenine transporter), and FurD (uracil transporter).57,58
The NEM-MS approach could be applied to any transport protein containing cysteine residues by incubating it with NEM, determining the labeling fingerprint by MS, and then comparing the spectrum obtained with those of the modified variants to determine whether any changes in conformation are observed. Detection of unfolding does not necessarily rely on the Cys residues being in strategically placed positions, as would be required to monitor inward-to-outward (or the reverse) interconversion.
Conclusion
Covalent labeling, including Cys labeling using NEM, has been used previously to study membrane transport proteins.59 While radiolabeled NEM was used initially to monitor labeling,60−65 more recently MS strategies have been developed for such analyses.9,34,35 However, the application of such methods to study alternating access mechanisms and ion–substrate coupling has been limited.66 In several instances, NEM labeling of Cys residues has been shown to inhibit substrate binding by secondary active membrane transport proteins.64 The relatively narrow range of side-chain reactivity of NEM has made this reagent a valuable tool for the study of membrane protein topology, for example by scanning mutagenesis approaches.60,61 Alternative covalent labeling strategies to probe membrane protein topology are also available, such as hydroxyl radical footprinting67−70 or hydrogen–deuterium exchange,71−73 but such applications are limited by the need for specialized equipment, intricate workflows, time scales available (including issues with back-exchange in hydrogen–deuterium exchange and limited reactivity of some side chains in oxidative labeling29,67,68), and the complexity of the data analysis. Such structural analyses, therefore, are far from routine, especially for those interested in membrane proteins. The biochemical and analytical methodologies employed here to study NEM-labeled membrane proteins by MS, by contrast, are relatively simple to perform and enable quantitation at the intact protein level, opening up the method for application of the workflow described by a broad spectrum of biochemists interested in studying alternating access mechanisms, substrate binding, and ion–substrate coupling. Additionally, quantitation by MS at the intact protein level obviates the need for protease digestion to which many membrane proteins are refractory.74
An important, unique, feature of the NEM-MS experiments is that chemical modification took place after the incubation with the ligands that were expected to affect the conformational equilibrium of the protein. This sets the approach described apart from other methods, e.g., FRET or EPR,7,10,75,76 which require amino acid substitution and/or derivatization with bulky probes. Membrane proteins can be refractory to conventional analytical methods, and the added benefit that the analyses do not require the protein to retain its three-dimensional structure during the MS measurements also constitutes a significant advantage of this method.
In the case of wild-type Mhp1, one Cys residue, Cys327, is fortuitously in a position where conformational changes were likely to affect access of the thiol-reactive reagent NEM. However, it is simple to introduce Cys residues into positions known to be conformationally sensitive and then implement the strategy described here to provide conformational insights. General application of such an approach to other proteins, including eukaryotic homologues of transporters that often contain an abundance of Cys residues, may also require judicious deletion of naturally occurring Cys residues or introduction of a Cys uniquely sensitive to labeling in different conformational forms. Importantly, such deletions/insertions must not alter the structure–activity relationship of the individual protein under investigation.
From the experiments described here we show that purified wild-type Mhp1 in DDM is in an inward-facing conformation and remains so when either a hydantoin substrate or Na+ is added, but changes to outward-facing when the two are added together in a concentration-dependent manner. This alone is an important constraint when attempting to understand the individual steps of the transport cycle. Equally as important is that our experiments have illuminated the effects of single residue substitutions on the conformational state of the protein. While only a small number of examples are given here, a wide range of single residue mutations of Mhp1 have been generated for study.47 Previously, information on the conformation of the protein required crystallographic structure determination, often impossible due to the perturbation(s) imposed by the mutation itself, highlighting the power of NEM-MS for a simple readout and quantification of the different conformational states of the protein ensemble.
For Mhp1, three conformationally different structures were identified by crystallography,46−48 but in a more likely scenario, for other proteins for which only a single structure is available, the MS-based strategy described here could be implemented to discriminate hypotheses about conformational changes, to identify residues important for structural maintenance, and to screen a number of variants quickly. Even when no structure is available, a model of the structure of any protein and its conformational flexibility could be tested using the strategy described, which requires only microgram quantities of protein and provides rapid insights about the relative population of different conformational states that is not possible using crystallography.
In the case of Mhp1 it was not necessarily anticipated that the protein would remain in the inward-facing state in DDM micelles when either L-BH or Na+ were present, nor that very high concentrations of Na+ would be necessary to drive the change from inward-facing to outward-facing. Moreover, it was previously unknown which mutations would affect the balance of conformations, nor which ones would promote unfolding/denaturing of its structure. These are all important observations that, alongside others,7,36,39,46,47,77 illuminate our understanding of the complete reaction cycle of this transport protein. For many other transporters, and indeed for any protein, the NEM-MS strategy described can be readily implemented to interrogate conformational transitions, elucidate ion–substrate coupling stoichiometry, screen widely for ligand specificity and illuminate mechanistic features that have eluded characterization to date.
Acknowledgments
This research was funded by the BBSRC (Grant No. BB/K000659/1) to A.E.A., S.E.R., and P.J.F.H. including support for D.S. A.N.C. was supported by the Wellcome Trust Institutional Strategic Support Fund (ISSF; Grant No. 015615/Z/14/Z). A.E.A. and S.E.R. are also supported by the ERC under the EU’s Seventh Framework Programme (FP7.2007-2013/Grant No. 322408) and P.J.F.H. by an Emeritus Research Fellowship from the Leverhulme Trust (Grant No. EM-2014-045). S.M.J. and L.N.J. were supported by Research Studentships from the MRC and BBSRC, respectively. M.K. is supported by a Ph.D. studentship from the Joachim Herz Foundation. M.S. and A.R.P. are supported by the German Federal Excellence Cluster “The Hamburg Centre for Ultrafast Imaging”. J.G. and F.H. acknowledge funding from NSERC (Natural Sciences and Engineering Research Council of Canada). O.B. was supported by the National Institute of General Medical Sciences of the National Institutes of Health (Award No. R01GM118772). The Waters Synapt G2-Si and Xevo mass spectrometers were purchased with funding from the BBSRC (Grant No. BB/M012573/1), and fermenter and allied equipment for protein production were funded by the BBSRC (Grant No. MPSI BBS/B/14418), the Wellcome Trust (Grant No. JIF 062164/Z/00/Z), and the University of Leeds. The MD trajectories of Mhp1 were kindly provided by Sergei Noskov (University of Calgary). We thank members of the Henderson, Ashcroft, and Radford research groups for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01310.
Supplementary methods for measurements of mass and tryptophan fluorescence, mass spectra, table of expected and observed masses, effects of some alternative substrates and of some additional mutations on mass spectra, and expected equilibria in a model alternating access mechanism (PDF)
Author Present Address
∇ Department of Biology, Institute of Molecular Biology and Biophysics, ETH Zurich, 8093 Zurich, Switzerland.
The authors declare no competing financial interest.
Supplementary Material
References
- Mitchell P. Nature 1957, 180, 134–136. 10.1038/180134a0. [DOI] [PubMed] [Google Scholar]
- Shi Y. Annu. Rev. Biophys. 2013, 42, 51–72. 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- Forrest L. R.; Kramer R.; Ziegler C. Biochim. Biophys. Acta, Bioenerg. 2011, 1807, 167–188. 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy H.; Piscitelli C. L.; Gouaux E. Nature 2009, 459, 347–355. 10.1038/nature08143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardetzky O. Nature 1966, 211, 969–970. 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Abramson J.; Wright E. M. Curr. Opin. Struct. Biol. 2009, 19, 425–432. 10.1016/j.sbi.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmier K.; Sharma S.; Islam S. M.; Roux B.; McHaourab H. S. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 14752–14757. 10.1073/pnas.1410431111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton D. P.; Kazmier K.; Mishra S.; McHaourab H. S. Methods Enzymol. 2015, 564, 349–387. 10.1016/bs.mie.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai A. W.; Rajagopal S.; Sun J.; Xiao K. Nat. Protoc. 2014, 9, 1301–1319. 10.1038/nprot.2014.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmier K.; Sharma S.; Quick M.; Islam S. M.; Roux B.; Weinstein H.; Javitch J. A.; McHaourab H. S. Nat. Struct. Mol. Biol. 2014, 21, 472–479. 10.1038/nsmb.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Nature 2016, 537, 347–355. 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Rajabi K.; Ashcroft A. E.; Radford S. E. Methods 2015, 89, 13–21. 10.1016/j.ymeth.2015.03.004. [DOI] [PubMed] [Google Scholar]
- Lanucara F.; Holman S. W.; Gray C. J.; Eyers C. E. Nat. Chem. 2014, 6, 281–294. 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- Konijnenberg A.; Butterer A.; Sobott F. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 1239–1256. 10.1016/j.bbapap.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Robinson C. V. Curr. Opin. Struct. Biol. 2014, 28, 122–130. 10.1016/j.sbi.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Hopper J. T.; Robinson C. V. Nat. Protoc. 2013, 8, 639–651. 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper J. T.; Yu Y. T.; Li D.; Raymond A.; Bostock M.; Liko I.; Mikhailov V.; Laganowsky A.; Benesch J. L.; Caffrey M.; Nietlispach D.; Robinson C. V. Nat. Methods 2013, 10, 1206–1208. 10.1038/nmeth.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffrin B.; Calabrese A. N.; Devine P. W. A.; Harris S. A.; Ashcroft A. E.; Brockwell D. J.; Radford S. E. Nat. Struct. Mol. Biol. 2016, 23, 786–793. 10.1038/nsmb.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood S.; Marcoux J.; Gault J.; Quigley A.; Michaelis S.; Young S. G.; Carpenter E. P.; Robinson C. V. Nat. Chem. 2016, 8, 1152–1158. 10.1038/nchem.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Allison T. M.; Ulmschneider M. B.; Degiacomi M. T.; Baldwin A. J.; Robinson C. V. Nature 2014, 510, 172–175. 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Politis A.; Davies R. B.; Liko I.; Wu K. J.; Stewart A. G.; Stock D.; Robinson C. V. Nat. Chem. 2014, 6, 208–215. 10.1038/nchem.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Morgner N.; Barrera N. P.; Politis A.; Isaacson S. C.; Matak-Vinkovic D.; Murata T.; Bernal R. A.; Stock D.; Robinson C. V. Science 2011, 334, 380–385. 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C.; Zhou M.; Marriott H.; Morgner N.; Politis A.; Robinson C. V. Nat. Commun. 2013, 4, 1985. 10.1038/ncomms2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C.; Robinson C. V. Nat. Protoc. 2014, 9, 2224–2236. 10.1038/nprot.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahraman A.; Herzog F.; Leitner A.; Rosenberger G.; Aebersold R.; Malmstrom L. PLoS One 2013, 8, e73411. 10.1371/journal.pone.0073411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese A. N.; Pukala T. L. Aust. J. Chem. 2013, 66, 749–759. 10.1071/CH13164. [DOI] [Google Scholar]
- Sinz A. Expert Rev. Proteomics 2014, 11, 733–743. 10.1586/14789450.2014.960852. [DOI] [PubMed] [Google Scholar]
- Engen J. R.; Wales T. E. Annu. Rev. Anal. Chem. 2015, 8, 127–148. 10.1146/annurev-anchem-062011-143113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L.; Pan J.; Liu Y. H. Chem. Soc. Rev. 2011, 40, 1224–1234. 10.1039/C0CS00113A. [DOI] [PubMed] [Google Scholar]
- Calabrese A. N.; Ault J. R.; Radford S. E.; Ashcroft A. E. Methods 2015, 89, 38–44. 10.1016/j.ymeth.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L.; Pan Y. Expert Rev. Proteomics 2012, 9, 497–504. 10.1586/epr.12.42. [DOI] [PubMed] [Google Scholar]
- Jumper C. C.; Schriemer D. C. Anal. Chem. 2011, 83, 2913–2920. 10.1021/ac102655f. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Rempel D. L.; Gross M. L. J. Am. Soc. Mass Spectrom. 2016, 27, 552–555. 10.1007/s13361-015-1313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Casey J. R. Methods 2007, 41, 439–450. 10.1016/j.ymeth.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Jones L. N.; Baldwin S. A.; Henderson P. J.; Ashcroft A. E. Rapid Commun. Mass Spectrom. 2010, 24, 276–284. 10.1002/rcm.4387. [DOI] [PubMed] [Google Scholar]
- Jackson S. M.; Patching S. G.; Ivanova E.; Simmons K.; Weyand S.; Shimamura T.; Brueckner F.; Suzuki S.; Iwata S.; Sharples D. J.; Baldwin S. A.; Sansom M. P. S.; Beckstein O.; Cameron A. D.; Henderson P. J. F. In Encyclopedia of Biophysics; Roberts G. C. K., Ed.; Springer: Berlin, Heidelberg, 2013; pp 1514–1521. [Google Scholar]
- Suzuki S.; Henderson P. J. J. Bacteriol. 2006, 188, 3329–3336. 10.1128/JB.188.9.3329-3336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier M. H. Jr.; Tran C. V.; Barabote R. D. Nucleic Acids Res. 2006, 34, D181–D186. 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman J. L.; Dale A. L.; Zwier M. C.; Bhatt D.; Chong L. T.; Zuckerman D. M.; Grabe M. Biophys. J. 2011, 101, 2399–2407. 10.1016/j.bpj.2011.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diallinas G. Front. Pharmacol. 2014, 5, 207. 10.3389/fphar.2014.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala-Rabanal M.; Hirayama B. A.; Loo D. D.; Chaptal V.; Abramson J.; Wright E. M. Am. J. Physiol Cell Physiol 2012, 302, C1293–C1305. 10.1152/ajpcell.00397.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright E. M. Mol. Aspects Med. 2013, 34, 183–196. 10.1016/j.mam.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Bröer S.; Palacin M. Biochem. J. 2011, 436, 193–211. 10.1042/BJ20101912. [DOI] [PubMed] [Google Scholar]
- Gether U.; Andersen P. H.; Larsson O. M.; Schousboe A. Trends Pharmacol. Sci. 2006, 27, 375–383. 10.1016/j.tips.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Weinglass A. B.; Whitelegge J. P.; Hu Y.; Verner G. E.; Faull K. F.; Kaback H. R. EMBO J. 2003, 22, 1467–1477. 10.1093/emboj/cdg145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T.; Weyand S.; Beckstein O.; Rutherford N. G.; Hadden J. M.; Sharples D.; Sansom M. S.; Iwata S.; Henderson P. J.; Cameron A. D. Science 2010, 328, 470–473. 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons K. J.; Jackson S. M.; Brueckner F.; Patching S. G.; Beckstein O.; Ivanova E.; Geng T.; Weyand S.; Drew D.; Lanigan J.; Sharples D. J.; Sansom M. S.; Iwata S.; Fishwick C. W.; Johnson A. P.; Cameron A. D.; Henderson P. J. EMBO J. 2014, 33, 1831–1844. 10.15252/embj.201387557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand S.; Shimamura T.; Yajima S.; Suzuki S.; Mirza O.; Krusong K.; Carpenter E. P.; Rutherford N. G.; Hadden J. M.; O’Reilly J.; Ma P.; Saidijam M.; Patching S. G.; Hope R. J.; Norbertczak H. T.; Roach P. C.; Iwata S.; Henderson P. J.; Cameron A. D. Science 2008, 322, 709–713. 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach P. C. J.; O’Reilly J.; Norbertczak H. T.; Hope R. J.; Venter H.; Patching S. G.; Jamshad M.; Stockley P. G.; Baldwin S. A.; Herbert R. B.; Rutherford N. G.; Bill R. M.; Henderson P. J. F. In Practical Fermentation Technology; McNeil B., Harvey L., Eds.; John Wiley & Sons: Chichester, U.K., 2008; pp 37–67. [Google Scholar]
- Marty M. T.; Baldwin A. J.; Marklund E. G.; Hochberg G. K.; Benesch J. L.; Robinson C. V. Anal. Chem. 2015, 87, 4370–4376. 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese A. N.; Watkinson T. G.; Henderson P. J.; Radford S. E.; Ashcroft A. E. Anal. Chem. 2015, 87, 1118–1126. 10.1021/ac5037022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Noskov S. Y. PLoS Comput. Biol. 2013, 9, e1003296. 10.1371/journal.pcbi.1003296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasel W.; Hendrickson T. F.; Still W. C. Tetrahedron Comput. Methodol. 1988, 1, 103–116. 10.1016/0898-5529(88)90015-2. [DOI] [Google Scholar]
- Henderson P. J. F. Comprehensive Biophysics, Vol 8: Bioenergetics 2012, 8, 265–288. 10.1016/B978-0-12-374920-8.00822-5. [DOI] [Google Scholar]
- Boudker O.; Verdon G. Trends Pharmacol. Sci. 2010, 31, 418–426. 10.1016/j.tips.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudker O.; Ryan R. M.; Yernool D.; Shimamoto K.; Gouaux E. Nature 2007, 445, 387–393. 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- Krypotou E.; Kosti V.; Amillis S.; Myrianthopoulos V.; Mikros E.; Diallinas G. J. Biol. Chem. 2012, 287, 36792–36803. 10.1074/jbc.M112.400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krypotou E.; Evangelidis T.; Bobonis J.; Pittis A. A.; Gabaldon T.; Scazzocchio C.; Mikros E.; Diallinas G. Mol. Microbiol. 2015, 96, 927–950. 10.1111/mmi.12982. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Konermann L. Analyst 2010, 135, 1191–1200. 10.1039/b924805f. [DOI] [PubMed] [Google Scholar]
- Guan L.; Kaback H. R. Nat. Protoc. 2007, 2, 2012–2017. 10.1038/nprot.2007.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaback H. R.; Dunten R.; Frillingos S.; Venkatesan P.; Kwaw I.; Zhang W.; Ermolova N. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 491–494. 10.1073/pnas.0609968104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T. H. D.; Kennedy E. P. J. Biol. Chem. 1969, 244, 5981–5987. [PubMed] [Google Scholar]
- Macpherson A. J.; Jones-Mortimer M. C.; Horne P.; Henderson P. J. J. Biol. Chem. 1983, 258, 4390–4396. [PubMed] [Google Scholar]
- Macpherson A. J. S.; Jonesmortimer M. C.; Henderson P. J. F. Biochem. J. 1981, 196, 269–283. 10.1042/bj1960269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald T. P.; Henderson P. J. F. Biochem. J. 2001, 353, 709–717. 10.1042/0264-6021:3530709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B. L.; Gross A. Nat. Struct. Biol. 2003, 10, 280–284. 10.1038/nsb908. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Stocks B. B.; Brown L.; Konermann L. Anal. Chem. 2009, 81, 28–35. 10.1021/ac8020449. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Ruan X.; Valvano M. A.; Konermann L. J. Am. Soc. Mass Spectrom. 2012, 23, 889–898. 10.1007/s13361-012-0342-x. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Zhang H.; Niedzwiedzki D. M.; Jiang J.; Blankenship R. E.; Gross M. L. Anal. Chem. 2016, 88, 8827–8834. 10.1021/acs.analchem.6b01945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Bavro V. N.; D’Mello R.; Tucker S. J.; Venien-Bryan C.; Chance M. R. Structure 2010, 18, 839–846. 10.1016/j.str.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebling C. M.; Morgan C. R.; Stafford D. W.; Jorgenson J. W.; Rand K. D.; Engen J. R. Anal. Chem. 2010, 82, 5415–5419. 10.1021/ac100962c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker C. H.; Morgan C. R.; Rand K. D.; Engen J. R.; Jorgenson J. W.; Stafford D. W. Biochemistry 2014, 53, 1511–1520. 10.1021/bi401536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Chien E. Y.; Chalmers M. J.; Pascal B. D.; Gatchalian J.; Stevens R. C.; Griffin P. R. Anal. Chem. 2010, 82, 1100–1108. 10.1021/ac902484p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter H.; Ashcroft A. E.; Keen J. N.; Henderson P. J. F.; Herbert R. B. Biochem. J. 2002, 363, 243–252. 10.1042/bj3630243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen A.; Hall J.; Diegel J.; Hassan I.; Fey A.; MacMillan F. Biochem. Soc. Trans. 2016, 44, 905–915. 10.1042/BST20160024. [DOI] [PubMed] [Google Scholar]
- Majumdar D. S.; Smirnova I.; Kasho V.; Nir E.; Kong X.; Weiss S.; Kaback H. R. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12640–12645. 10.1073/pnas.0700969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand S.; Shimamura T.; Beckstein O.; Sansom M. S.; Iwata S.; Henderson P. J.; Cameron A. D. J. Synchrotron Radiat. 2011, 18, 20–23. 10.1107/S0909049510032449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.