

Abstract
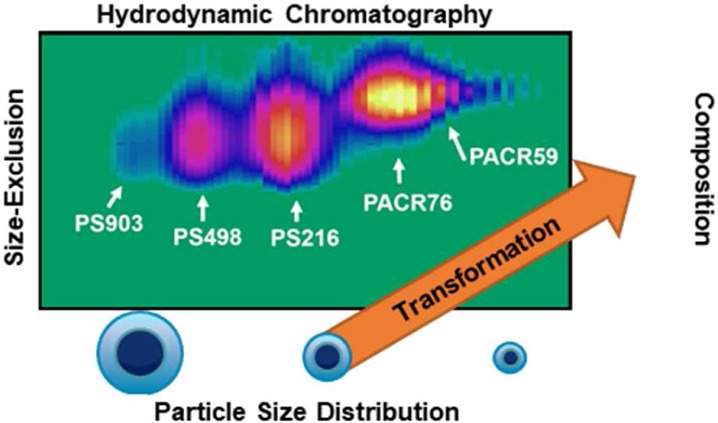
Polymeric nanoparticles have become indispensable in modern society with a wide array of applications ranging from waterborne coatings to drug-carrier-delivery systems. While a large range of techniques exist to determine a multitude of properties of these particles, relating physicochemical properties of the particle to the chemical structure of the intrinsic polymers is still challenging. A novel, highly orthogonal separation system based on comprehensive two-dimensional liquid chromatography (LC × LC) has been developed. The system combines hydrodynamic chromatography (HDC) in the first-dimension to separate the particles based on their size, with ultrahigh-performance size-exclusion chromatography (SEC) in the second dimension to separate the constituting polymer molecules according to their hydrodynamic radius for each of 80 to 100 separated fractions. A chip-based mixer is incorporated to transform the sample by dissolving the separated nanoparticles from the first-dimension online in tetrahydrofuran. The polymer bands are then focused using stationary-phase-assisted modulation to enhance sensitivity, and the water from the first-dimension eluent is largely eliminated to allow interaction-free SEC. Using the developed system, the combined two-dimensional distribution of the particle-size and the molecular-size of a mixture of various polystyrene (PS) and polyacrylate (PACR) nanoparticles has been obtained within 60 min.
Polymeric nanoparticles have become indispensable in modern society with applications ranging from electronics,1 pharmaceuticals,2 and templates for porous materials,3,4 to coatings5,6 and inks.7,8 There are various techniques for the preparation of these polymeric systems, including emulsion polymerization and nanoprecipitation. Emulsion polymerization involves the emulsification of relatively hydrophobic monomers in water by an emulsifier (e.g., a surfactant). Upon addition of a similarly hydrophobic initiator, the polymerization process yields stabilized, dispersed polymeric particles.9 In nanoprecipitation, preformed polymers are dissolved in a solvent (e.g., acetone). The solution is injected into a nonsolvent, typically an aqueous solution containing a stabilizing surfactant, to yield a suspension of particles.10,11 These and other preparation techniques provide a number of means to influence the various colloidal and physicochemical properties. They have been thoroughly reviewed by Rao and Geckeler.12
A wide array of analysis techniques exists to characterize nanoparticles based on their size. Notable examples are asymmetrical field field-flow fractionation (AF4), scanning electron microscopy (SEM), hydrodynamic chromatography (HDC), and dynamic light scattering (DLS). These techniques and their hyphenation with spectroscopic techniques have been compared and evaluated in a detailed review by Lespes and Gigault.13 Similarly, various analysis methods can be used to characterize the polymers and small molecules (e.g., for drug-delivery applications) which compose these nanoparticles. Typical examples are size-exclusion chromatography (SEC),14 field-flow fractionation (FFF),15 capillary electrophoresis (CE),16 and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS).17
All of these techniques have proven to provide highly useful data to study relevant properties of the sample of interest. It is no surprise that multiple detection techniques have been used in parallel or in series (i.e., online) to determine and relate different properties of nanoparticles simultaneously.18−21 However, contemporary nanoparticle analysis is typically limited to physicochemical properties at the particle level. In cases where compositional information is sought, spectroscopic methods yield exclusively average numbers on the elemental (e.g., by inductively coupled plasma–atomic-emission spectroscopy, ICP-AES) or structural (e.g., by FTIR) composition of the particle as a whole. Alternatively, dissolution of the nanoparticles and subsequent separation may yield compositional information yet forfeits information on the nanoparticle-size distribution.
Particle-size separation by HDC or FFF with subsequent fraction collection potentially provides both types of information but is labor- and time-intensive if multiple fractions are to be collected. Moreover, off-line coupling suffers from the common drawback that the yield (i.e., the mass of sample) per fraction is rather limited.
Another technique for the multidimensional characterization of complex mixtures is comprehensive two-dimensional liquid chromatography (LC × LC).22 In LC × LC, two substantially different (orthogonal) separation mechanisms are coupled online through a modulator. Typically, the modulator comprises a two-position switching valve equipped with two loops, which alternatingly connect to the first-dimension to fractionate the effluent or to the second dimension to inject the collected fraction. The rate at which the valve switches is determined by what is commonly referred to as the modulation time. For comprehensive 2D-LC, the premise is that the entire first-dimension effluent is subjected to a second-dimension separation, thus inducing the need for the second-dimension separation to be finished before the valve switches again (i.e., the modulation time equals the maximum second-dimension analysis time).
LC × LC has not been applied to nanoparticles for several good reasons. HDC and AF4 provide a very similar, size-based selectivity. No other LC mechanism is expected to provide any meaningful information on the intact particles, since these are typically too large to enter the pores fully (to allow sufficient enthalpic interaction) or partly (to allow size-exclusion chromatography). Thus, the particles need to be dissolved to allow characterization by LC. The only possible way of characterizing nanoparticles with LC × LC appears to perform HDC in the first dimension, and then dissolve the particles, and use one of several LC techniques (SEC, gradient-elution LC, etc.) in the second dimension to characterize the constituting polymers. Dissolution of the particles requires the addition of a strong organic solvent to the aqueous dispersion. However, it is likely that either the organic solvent or the remaining aqueous fraction pose detrimental effects on the second-dimension separation due to solvent incompatibility problems and excessive dilution. For example, the large amount of organic solvent required for dissolution increases the risk of breakthrough phenomena in RPLC,23 whereas the remaining water potentially engenders interaction in SEC.24 Fortunately, these effects can be minimized using efficient mixing and small injection volumes in the second dimension. The latter requires either very low flow rates in the first dimension or short modulation times in the second dimension, which in turn translates into very fast second-dimension separations. We have recently demonstrated ultrafast SEC separations using core–shell stationary phases25 which satisfies this premise.
One research interest of our group is to develop methods for the comprehensive characterization of nanoparticles to obtain complete information on mutually dependent particle-size and molecular distributions. Examples of the latter are molecular-weight distribution, chemical-composition distribution, functionality-type distribution, etc. To measure accurate molecular distributions we must address challenges, such as breakthrough or undesired interactions.
In this work, we demonstrate the comprehensive analysis of polystyrene (PS) and polyacrylate (PACR) nanoparticles by HDC × SEC, using online dissolution of the nanoparticles into the composite polymers and stationary-phase-assisted modulation. We will first address the development of the individual one-dimensional separation mechanisms before addressing the feasibility of coupling the two dimensions together with intermediate dissolution. Factors determining successful transfer and dissolution (i.e., mixing, solvent ratios, etc.) with minimal detrimental effects are addressed.
Experimental Section
Instrumentation
The experiments in this study were all carried out on an Agilent 1290 Infinity 2D-LC system (Agilent, Waldbronn, Germany). The system encompassed two binary pumps (model G4220A), one 1200 isocratic pump (model G1310A), an autosampler (model G4226A), two thermostated column compartments (model G1316C) each equipped with a 2D-LC 8-port 2-position modulation valve (model G4236A), and two diode-array detectors (model G4212A) outfitted with Agilent Max-Light cartridge cells (model G4212-6008, 10 mm, Vdet = 1.0 μL). The autosampler injector needle was set to draw and eject at a speed of 10 μL/min with two seconds equilibration time. All tubing connections were made from stainless steel. The entire system was controlled using Agilent OpenLAB CDS Chemstation Edition (Rev. C.01.04) software.
The first-dimension column was an Agilent PL–PSDA cartridge type-2 (800 × 7.5 mm i.d.). The second-dimension column combination comprised three Phenomenex (Torrance, CA) experimental core–shell (150 × 4.6 mm i.d., 3.6-μm particles, 500 Å pore size) columns coupled to each other with the exception of one experiment, in which the column at the terminus was packed with particles with an average pore size of 328 Å. In all cases, the first column contained unmodified silica particles, whereas the latter two were packed with XB-C18 modified silica particles.
For LC × LC experiments, in the valve used for modulation, 160 μL homemade loops or traps were installed, depending on the application. Phenomenex SecurityGuard ULTRA (P/N: AJ0–9000, V0 = 0.4 μL) guard-column holders were used in conjunction with UHPLC C18 2.1 mm i.d. SecurityGuard ULTRA cartridges (P/N: AJ0-8782). In the case of one trap per modulation loop, the trap was directly connected to the valve with tubing (Agilent, P/N: 5067-4650, 150 × 0.12 mm i.d.), connecting the outlet to the second valve connection. For experiments utilizing two traps per modulation loop, the two traps were directly coupled to each other. One end of the coupled traps was connected to the valve using a male-to-female tubing piece (Agilent, P/N: G1316-87313, 70 × 0.12 mm i.d., m/f), whereas standard tubing was used to couple the other end to the valve (Agilent, P/N: 5067-4649, 90 × 0.12 mm).
For experiments that required mixing, the flow eluting from the HDC column was combined with the flow from a pump delivering THF using a stainless-steel tee-connection (P/N: U-428, IDEX Corp, Illinois). The combined flow was then routed to the Agilent Jet Weaver V35 or V100 mixer (model G4220-60006) of the first-dimension binary pump using 500 × 0.12 mm stainless-steel tubing. The outlet of the mixer was connected with the valve using the same dimensions of tubing.
Configuration Chromatographic System
System A: Depending on the type of experiment, the equipment above was used in a specific setup. For one-dimensional HDC and SEC experiments (system A), one channel of the binary pump was connected to the autosampler, which was coupled to the column(s) and then linked to the DAD. LC × LC experiments were performed with the instrumental setup illustrated in Figure 1.
Figure 1.

Schematic overview of the setup used for HDC × SEC experiments.
System B: for studying the effects of various instrumental parameters including the dissolution process, modulation, traps and resulting SEC separation, the setup of Figure 1 was used without the HDC column (system B) as the experiments all comprised the analysis of a sample of 498 nm PS nanoparticles, for which no HDC separation was required. The first valve shown in Figure 1 was bypassed.
System C: all other LC × LC experiments utilized the system shown in Figure 1 without any modifications (system C). The first valve allowed for the redirection of the flow to waste during the flush program of the HDC column. The first detector functioned to monitor the premature elution of analytes from the traps.
In all setups, the diode-array detectors (DAD) recorded the full spectra from 190 to 640 nm, although the chromatograms shown in this paper reflect specific wavelengths. For 1D-HDC and 1D-SEC analyses, 254 nm was used as detection wavelength. Chromatograms recorded using setups B and C were obtained at 210 nm with 230 nm as reference (bandwidth 4 nm). For all experiments, the sampling rate of the detector was 160 Hz. The temperature for the stationary-phase-assisted modulation and SEC separation in all experiments was 60 °C, which lowered the operating pressures in comparison with room temperature operation. The modulation volumes were always emptied in the counter-current (i.e., backflush) mode, to flush out any nondissolved particles into the SEC dimension and protect the traps.
Chemicals
Nonstabilized tetrahydrofuran (THF, HPLC-S grade) was obtained from Biosolve (Valkenswaard, The Netherlands). Sodium dihydrogen phosphate (cat. no.: 106346, monohydrate) was obtained from Merck (Darmstadt, Germany). Sodium dodecyl sulfate (cat. no.: L4509-250G), Brij L23 nonionic surfactant (cat. no.: B4184-100 ML, specified as 30% w/v solution in water) and sodium azide (cat. no.: S2002-100G) were obtained from Sigma-Aldrich (Darmstadt, Germany). Polystyrene standards (PS) for determining MWD calibration curves were obtained from Polymer Laboratories (now Agilent Technologies, Church Stretton, Shropshire, UK). The 3000 series Nanosphere polystyrene nanoparticle standards were obtained from ThermoFisher Scientific (Bremen, Germany). The used particle diameters were 903 nm ± 12 nm (P/N: 3900A), 498 nm ± 9 nm (P/N: 3500A), 216 nm ± 4 nm (P/N: 3220A), 102 nm ± 3 nm (P/N: 3100A) and 46 nm ± 2 nm (P/N: 3050A). The polyacrylate nanoparticle samples of 76 and 59 nm (average particle sizes determined by 1D-HDC analysis) were provided by DSM Coating Resins (Waalwijk, The Netherlands).
Preparation Methods
For creating a calibration curve, all PS standards were dissolved in THF at concentrations of approximately 0.2 mg mL–1. A stock solution of HDC buffer was created by dissolving 6.2 g sodium dihydrogen orthophosphate, 10.0 g sodium lauryl sulfate, 134 mL Brij L23 nonionic surfactant (specified as a 30% (w/v) solution in water), and 4.0 g sodium azide in 866.7 mL Milli-Q purified water. The stock solution was diluted 20 times with Milli-Q purified water before use. Concentrations of the obtained PS and polyacrylate nanoparticles were reported as % (w/w). PS nanoparticles were diluted 10-fold to a concentration of approximately 0.1% (w/w). Polyacrylate nanoparticles were diluted to concentrations between 0.1 and 0.5% (w/w). For dilution, the buffer utilized for HDC was used.
Analytical Methods
For one-dimensional HDC analysis, system A (see Configuration Chromatographic System) was used with the flow rate set at 1 mL min–1 using the buffer as prepared according to Preparation Methods for the mobile phase. The analysis time was 15 min. The recorded pressure drop was on average 80 bar (8 MPa) at this flow rate. For investigating the effect of incorporating a flush program in the HDC separation on the width of the observed analyte bands, the flow rate was set as follows: 1 mL min–1 from 0.0 to 11.9 min and 0.04 mL min–1 from 11.9 to 60.0 min. The injection volume was 20 μL.
For one-dimensional SEC analysis, system A was used with the column combinations and one trap (for a more accurate comparison) as specified in the Instrumentation section and the flow rate set at 3.0 mL min–1. The mobile phase was 100% nonstabilized THF. The analysis time was 5 min. The injection volume was 5 μL. Typical operating pressures were 750–800 bar (75–80 MPa).
For experiments utilizing system B, the flow rates of the first-dimension pump A and dissolution pump B were varied, as is described in the section Coupling of SEC and HDC. The second-dimension flow rate was 3.3 mL min–1. The modulation time was 36 s. The injection volume was 20 μL.
For LC × LC experiments, system C was used. The first-dimension pump A operated at a flow rate of 40 μL min–1, unless stated otherwise, with an initial flush program analogous to that reported for system A. The dissolution pump B operated at a flow rate of 200 μL min–1, unless specified differently. The second-dimension pump C operated at 3.3 mL min–1, unless indicated otherwise. The analysis time was 60 min. The modulation time was 36 s. Observed backpressures ranged from 800 to 900 bar using one trap to 900–1000 bar using two traps, depending on the conditions of the traps. The injection volume was 20 μL.
Data Treatment
Chromatograms obtained from LC × LC experiments were processed and analyzed by software written in-house in a MATLAB 2015a (Mathworks, Woodshole, MA) environment. For chromatograms recorded using system B, specific modulations are used and displayed in the Results and Discussion. To justly compare the effect of system parameters, such as the dissolution-solvent composition, mixing volumes, and trapping performance, on the SEC separation, the same modulation cycle was consistently selected (fourth modulation, from 1.8 to 2.4 min) and converted to the τ-scale (relative to τ = 1 at t0) and used for comparison. To allow calculation of the second derivatives without obstructing noise signals, a wide Savitsky-Golay smoothening filter was applied to all chromatograms used to construct Figure 4B (see Section S-5).
Figure 4.
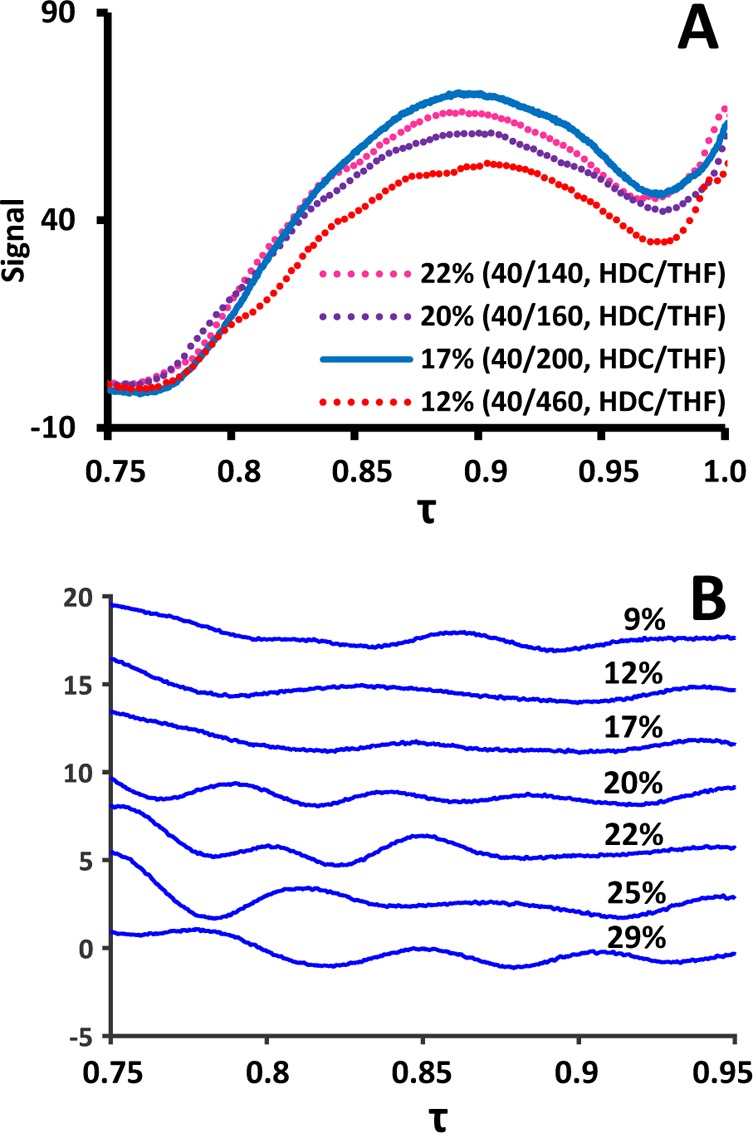
(A) SEC chromatograms of 498 nm PS nanoparticle obtained using different solvent ratios for in-line dissolution (see Figure S-3 for all chromatograms). (B) Second derivatives of chromatograms of all investigated solvent ratios for dissolution. Percentages represent fraction of aqueous first-dimension eluent. Each inflection point is visible as peak or valley and represents a distortion in the SEC curve.
Results and Discussion
Hydrodynamic Chromatography
Hydrodynamic chromatography (HDC) is a technique in which macromolecules and particles are separated according to size in a column packed with solid or porous beads. It is a rapid and, in principle, convenient method to obtain a fingerprint of the size distribution of particles. Large particles will migrate at average velocities corresponding to the faster streamlines, whereas the smaller particles can also occupy the much slower streamlines close to the walls of the channel (See Section S-1 for a more detailed explanation).
While HDC separations can be carried out on columns with porous particles, we chose to utilize nonporous particles as the stationary phase to rule out separation based on permeation. For such stationary phases, the HDC domain utilizes a rather narrow domain generally between 0.8 < τ < 1.0, where τ is the ratio between the analyte elution time (or volume) and the dead time (or volume) of the column (τ = te/t0 = Ve/V0). However, the used column featured a dead volume of approximately 14 mL. As a result, at a flow rate of 1 mL min–1, the first 12 mL (V0 ≈ 14 mL; τ ≈ 0.85) of the separation fell outside of the HDC domain and were not helpful (see Figure S-1A). In LC × LC, with a first-dimension flow rate at least 1 order of magnitude lower, the prospect of waiting for this volume to elute is certainly not attractive.
Fortunately, the band broadening does not heavily depend on the flow rate (see Figure S-1 for an expanded explanation).26−28 Consequently, we increased the flow rate to flush through the first 11.9 mL of eluent. This essentially reduced the analysis time for the 1D-HDC method to 3 min, and the analysis time for the 2D-LC method to 60 min, as will be shown later.
With the use of the developed method, a calibration curve using the PS standards could be constructed (see Figure S-1D). To determine the reliability of the pump carrying out the flush program in terms of displacement volume (and thus the variation in the calibration curve), the experiments were repeated five times. A standard deviation of 0.11 min was found at 40 μL min–1, corresponding with 4.4 μL in terms of elution volume. It was concluded that the variation was not significant.
Size-Exclusion Chromatography
While SEC has been often applied as second-dimension separation,29,30 there is still room for improvement. SEC separations are typically carried out on relatively large columns, as the extent of separation (i.e., the difference ΔV in the elution volumes of different analytes) is proportional to the total pore volume.31 However, since the 2D analysis typically has to be performed under significant time-pressure, very fast separations are desired. Fast separations are achieved in |LC by using short columns packed with (very) small particles. We have recently investigated the feasibility of using core–shell particles as the stationary phase and found that, within specific domains, the gained efficiency in SEC compensates for the loss of resolution as a result of a loss in pore volume.25 To obtain as much resolution as possible under ultrahigh-pressure LC (UHPLC) conditions, three columns were coupled, the first of which was a nonmodified silica column to filter out any remaining water from the first-dimension fraction. After each experiment, all columns were regenerated to purge any remaining water.
One advantage of genuine SEC (in absence of adsorptive interactions) is that no compounds elute in the exclusion volume of the separation. This conveniently allowed the use of overlapping injections (Figure 2). With the three columns coupled together, the total column volume was approximately 5.1 mL. A calibration curve was recorded, which revealed that the total exclusion limit was around 3.6 mL. Consequently, the SEC separation could never comprise a range larger than approximately 1.5 mL or 30 s at a flow rate of 3.3 mL min–1. We therefore opted to use a modulation time of 36 s, which meant that at any point of the second-dimension separation there would be three modulations present simultaneously inside the column combination.
Figure 2.

Theoretical sketch illustrating the principle of overlapping injections. The modulation time is adapted such that the sample zones of subsequent injections do not overlap. Note: the three SEC columns coupled together for our study are shown here as a single column for simplicity (see also Figure S-2 for an illustration of how the corresponding chromatogram is established).
Coupling of SEC and HDC
The HDC and SEC separations were combined using the setup shown in Figure 1. Combination of the HDC and SEC separations required overcoming a set of challenges. In the HDC separation, PS particles were separated based on their particle sizes. In the second dimension, it was envisaged that the polymer molecules that make up the polymeric nanoparticles would be separated by SEC. For the latter purpose, it was required to transform the sample analytes from particles to a mixture of the composing polymer molecules. We thus combined the HDC-effluent with THF (pump B) through the use of a tee-piece. The resulting solvent blend was then mixed in an Agilent Jet Weaver mixer, belonging to one of the binary pumps. Such a mixer is usually used to mix two mobile phase components in gradient analysis. Due to the presence of THF, the nanoparticle dispersion destabilizes and the polymers dissolve in the organic solvent. In this section, we discuss the different challenges and effects using the separation of a standard mixture of PS nanoparticles with an average particle size of 498 nm as an example (see section Chemicals).
The presence of water in the polymer solution greatly complicated the SEC separation due to significant adsorption effects. Moreover, the solutions transferred to the SEC column were too dilute to yield clearly detectable signals, as is illustrated in Figure 3A. Relative to the dead volume signal (at τ = 1), the signal of the polymer fraction was diminished. To solve both the adsorption and dilution problems, the loops were replaced by traps with very short connections toward the valve to minimize extraneous volumes.
Figure 3.

Separation of dissolution products of 498 nm PS nanoparticles analyzed with setup B (see Configuration Chromatographic System) under various conditions. (A) No traps. (B) One trap (broken, green line) or two traps (solid, blue line). (C) 35 μL mixer (broken, purple line) or 100 μL mixer (solid, orange line). (D) Effect of the pore size in the last of three SEC columns; 328-Å (broken, blue line) vs 500 Å (solid orange line). Chromatograms were recorded using a 2D setup. They reflect one of many, highly repeatable modulations (see Data Treatment).
The effect of the use of a trap for each loop is shown in Figure 3B (blue, broken line). Indeed, the traps appeared to retain the dissolved polymers very well. Thanks to the small connection volumes to and from the traps, most of the water could be removed, while the polymers were retained on the traps. However, the molecular-weight distribution (MWD) was not consistent with reference data, especially in the low-molecular weight region (close to t0). Therefore, a second trap was added to each modulation connection. The addition of a second trap resulted in an improved MWD observed (Figure 3B, orange, solid line). This can be understood by realizing that the traps are in fact 3 mm long columns. While most polymer molecules are well-retained, low-molar-mass polymers may elute with the many column volumes of solvent that are flushed through the loops during each modulation (adsorption) cycle. The second trap catches most of the low-molar-mass polymers that are eluted from the first trap.
The Agilent Jet Weaver mixer features two modes of use, offering a mixing volume of 35 or 100 μL. To study the effect of the mixing volume, chromatograms were recorded using both mixing volumes. The results are shown in Figure 3C. The yield was found to be improved for the larger mixing volume (orange, solid line as compared to the purple, broken line), and the mixing volume of 100 μL was used for all the following experiments.
All experiments used three coupled SEC columns, each featuring core–shell stationary phase particles with pore sizes averaging 500 Å. To offer more resolution in the low-molar-mass region, the possibility of replacing the last column with one containing particles with smaller pore sizes (328 Å) was investigated. Unfortunately, the obtained traces (Figure 3D) suggested a pore-size mismatch when using the 328 Å terminus column combination (green, broken line), in comparison with the use of three 500-Å columns (blue, solid line).
Effect of Dissolution-Solvent Composition and Flow Rate
Not surprisingly, the dissolution ratio of analyte-containing-HDC eluent with THF proved to greatly affect several factors related to the SEC performance, including the consistency of the MWD and the performance of the traps. To assess this performance, we used the MWD obtained from 1D-UHPLC SEC analysis of the 498 nm PS nanoparticle standard recorded using exclusively THF in the mobile phase as a benchmark (see Figure S-4). The flow rate for the HDC separation was fixed to 40 μL min–1 to yield a reasonable analysis time. The flow rate for the THF pump was varied to allow investigation of the effect of the mixing ratio and the total flow rate on the SEC performance (Figure 4).
The resulting chromatograms confirmed the effect of the mixing ratio (Figure 4A, see Figure S-3 for all chromatograms). One of the observed effects was significant distortion in the SEC separation resulting in irregular peaks. To assess the extent of these inconsistencies, the second derivative was calculated for the SEC domains of each chromatogram Figure 4B. Each inflection point corresponds to an irregularity in the SEC behavior. The extent of distortions was found to be minimal for a ratio of 17% (HDC buffer/THF [17:83, v/v]). This was in accordance with the resemblance to the benchmark when using this composition (see Figure S-4).
However, it is good to realize that an increase in THF content under the present conditions results in an increase in the total flow rate and which increases the risk of (low-molar-mass) polymers eluting from the traps (see also Figure 3B). To assess this issue, the SEC elution curve was integrated relative to the baseline within the domain of the calibration curve (thus not including the rise of the signal toward the dead volume marker).
The integration area was defined as the trap yield and is shown in Table 1. Generally, two trends can be seen. First, high amounts of THF allow good dissolution of the particles, which is reflected in the relatively high trap yield compared to lower amounts of THF. However, higher amounts of THF also appear to hinder reliable trapping of the analytes with the yield decreasing slightly above 83%, possibly due to retention problems. Second, relatively high amounts of aqueous buffer (and thus analytes) of course lead to lower dilution factors and thus increasing yields, until the fraction of THF becomes too small to timely dilute the entire particle. Compositions with more than 22% aqueous buffer resulted in occasional trap clogging. Above 30% aqueous buffer no signal was observed in the SEC dimension and almost every experiment resulted in clogging of the traps. The above observations point to an optimum at around 17% aqueous buffer and 83% THF. Such a composition also showed favorable results in Figure 4B and thus was chosen for the remainder of the study.
Table 1. Performance of Traps at Different Dissolution-Solvent Compositions and Total Flow Rates.
| buffer | THF | total | ϕ | trap yield |
|---|---|---|---|---|
| μL min–1 | μL min–1 | μL min–1 | buffer | area × min |
| 40 | 100 | 140 | 29% | 11.849 |
| 40 | 120 | 160 | 25% | 12.92 |
| 40 | 140 | 180 | 22% | 13.495 |
| 40 | 160 | 200 | 20% | 12.728 |
| 40 | 200 | 240 | 17% | 13.949 |
| 40 | 300 | 340 | 12% | 13.26 |
| 40 | 400 | 440 | 9% | 13.346 |
| 60 | 460 | 520 | 12% | 10.572 |
Analysis of PS and Polyacrylate Nanoparticles
A mixture of 903, 498, and 216 nm PS and 76 and 59 nm polyacrylate nanoparticles, all at concentrations of 0.1–0.5% (w/w) (see Preparation Methods), was injected and analyzed using the developed separation system. The resulting LC × LC (HDC × SEC) chromatogram is shown in Figure 5. The corresponding information related to the particle-size distribution and the molecular weight distribution was tentatively added on the top and right axes, respectively. As can be seen, the three different kinds of PS particles are clearly separated from each other and from the polyacrylate particles. This is not entirely the case for the two sizes of polyacrylate particles, where clearly the resolution is insufficient. One solution here could be to use an HDC column covering a range of smaller particle sizes.
Figure 5.

HDC × SEC-DAD chromatogram of a mixture of nanoparticles containing PS 903 nm, PS 498 nm, PS 216 nm, polyacrylate 76 nm, and polyacrylate 59 nm. The corresponding information related to the particle size distribution and molecular weight distribution was tentatively added to the top and right axes, respectively.
The obtained separation also gives good insight in the differences in molecular weight distributions of the polymers, with larger particles generally comprising larger polymers. For some particles, the MWD was found to be narrower than for other particles.
The chromatogram in Figure 5 displays some slight perpetual distortions on one of the modulation traps. The extent of the distortion increases with analysis time and can be explained from deteriorating performance of one of the modulation traps. This exposes a weakness of the present HDC × SEC separation system. We observed that minor obstruction points form over time (e.g., by noneluting analytes or poorly processed nanoparticles), causing partial blockage of the traps and resulting in increased pressure drops. These obstructions may affect the loop filling, trapping efficiency, and the subsequent SEC separation because the extent of blockage is unlikely to be the same for both loops, causing the performance of the traps to become unbalanced. Indeed, for reliable performance it is imperative that both traps are as identical as possible. Fortunately, the traps could be regenerated in most cases by ultrasonication.
Another discussion point is the width of the peaks in HDC, which limits the extent of separation. One solution could be to mathematically reduce the peak width as is done in analytical (one-dimensional) HDC practice.32 Furthermore, the large separation volumes of the HDC method used in this study are subject to discussion, and this is something that our group aims to investigate.
The extraneous volumes prior, during, and after the mixing process are also a source of concern. In the current setup, the first-dimension column is connected by a long piece of capillary tubing to the 100 μL Jet Weaver V100 mixer and the exit of the mixer is again connected by a long piece of tubing back to the modulation valve. Ideally, the first-dimension column would be directly connected to an extremely low-volume mixer inside the column oven. In the present setup, we observed significant peak tailing in the SEC dimension under UHPLC conditions.
Ultimately, we aim to combine the HDC separation with other retention mechanisms, such as gradient-elution reversed-phase or normal-phase LC or ion-exchange chromatography, to characterize nanoparticles consisting of copolymers or charged polymers. We envisage that, depending on the type of application, different stationary phases can be used to retain the analytes.
Conclusion
An online separation system for the comprehensive analysis of nanoparticle formulations based on LC × LC technology was successfully developed. The method incorporates in-line transformation of the particulate sample to a molecular solution. It allows relating the particle-size distribution of the particles to the molecular-weight distribution of the constituting polymers. In the first dimension, hydrodynamic chromatography (HDC) was used to obtain information on the particle-size distribution. The first 12 mL of the 14 mL dead volume of the column were flushed through rapidly at the start of each analysis to reduce the analysis time without sacrificing chromatographic efficiency. The remaining 2 mL contained the effective HDC separation and was fractioned after blending with THF using a mixer. A ratio of 17% first-dimension aqueous buffer eluent/83% THF (v/v) was found to provide good yields of polymers with respect to dissolution and trapping efficiency. A mixing volume of 100 μL was found to improve the consistency of the SEC separation in comparison with a 35 μL mixing volume. To prevent adsorption effects in the SEC separation, the water was eliminated from the dissolution mixture using traps in the modulation loops. Two traps connected in series in each loop were found to yield a SEC separation that resembled the results obtained in conventional off-line SEC. Core–shell particles were used at UHPLC conditions in the second dimension to provide the necessary resolution. To efficiently utilize the separation space and reduce the modulation times, overlapping injections were applied.
Parameters such as the particle-size distribution, the MMD of the intrinsic polymers, and other properties greatly affect the performance of coatings. For example, smaller particles provide better chemical stability and, more importantly, optical properties of the coating, whereas large polymers improve the mechanical stability of the coating. Often, bi- or multimodal (co)polymer distributions are employed to obtain specific properties, including surface reflectance and tactual perception. The developed separation platform visualizes the information needed to reach the best compromise.
Acknowledgments
The MANIAC project is funded by The Netherlands Organisation for Scientific Research (NWO) in the framework of the Programmatic Technology Area PTA-COAST3 of the Fund New Chemical Innovations (Project 053.21.113). Phenomenex is acknowledged for supporting this work by providing the columns for SEC experiments. DSM Coating Resins, Waalwijk, is kindly acknowledged for providing the polyacrylate nanoparticle samples. Sanne Berbers, Denice van Herwerden, Chris Lukken, Sanne Brekelmans, Eline van der Galie, Mari-Anne Asseler, and Esmee Wierenga are acknowledged for assisting with the experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01906.
Principles of hydrodynamic chromatography and effect of flow rate; subsequent injections; effect of dissolution composition on trapping yield and curve stability, and second derivative of SEC chromatogram through a Savitsky-Golay filter (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Coll A.; Bermejo S.; Hernández D.; Castañer L. Nanoscale Res. Lett. 2013, 8 (1), 26. 10.1186/1556-276X-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treleaven J. G.; Ugelstad J.; Philip T.; Gibson F. M.; Rembaum A.; Caine G. D.; Kemshead J. T. Lancet 1984, 323 (8368), 70–73. 10.1016/S0140-6736(84)90004-7. [DOI] [PubMed] [Google Scholar]
- Iskandar F.; Mikrajuddin; Okuyama K. Nano Lett. 2001, 1 (5), 231–234. 10.1021/nl0155227. [DOI] [Google Scholar]
- Velev O. D.; Jede T. A.; Lobo R. F.; Lenhoff A. M. Nature 1997, 389 (6650), 447–448. 10.1038/38921. [DOI] [Google Scholar]
- Lee D. I. In Polymeric Dispersions: Principles and Applications; Asua J. M., Ed.; Springer: Dordrecht, Netherlands, 1997; pp 497–513. [Google Scholar]
- Defusco A. J.; Sehgal K. C.; Bassett D. R. In Polymeric Dispersions: Principles and Applications; Asua J. M., Ed.; Springer: Dordrecht, Netherlands, 1997; pp 379–419. [Google Scholar]
- Hirose M.; Kadowaki F.; Zhou J. Prog. Org. Coat. 1997, 31 (1–2), 157–169. 10.1016/S0300-9440(97)00032-5. [DOI] [Google Scholar]
- Zhang J.; Li X.; Shi X.; Hua M.; Zhou X.; Wang X. Prog. Nat. Sci. 2012, 22 (1), 71–78. 10.1016/j.pnsc.2011.12.012. [DOI] [Google Scholar]
- Chern C. S. Prog. Polym. Sci. 2006, 31 (5), 443–486. 10.1016/j.progpolymsci.2006.02.001. [DOI] [Google Scholar]
- Niwa T.; Takeuchi H.; Hino T.; Kunou N.; Kawashima Y. J. Controlled Release 1993, 25 (1–2), 89–98. 10.1016/0168-3659(93)90097-O. [DOI] [Google Scholar]
- Lebouille J. G. J. L.; Stepanyan R.; Slot J. J. M.; Cohen Stuart M. A.; Tuinier R. Colloids Surf., A 2014, 460, 225–235. 10.1016/j.colsurfa.2013.11.045. [DOI] [Google Scholar]
- Rao J. P.; Geckeler K. E. Prog. Polym. Sci. 2011, 36 (7), 887–913. 10.1016/j.progpolymsci.2011.01.001. [DOI] [Google Scholar]
- Lespes G.; Gigault J. Anal. Chim. Acta 2011, 692 (1–2), 26–41. 10.1016/j.aca.2011.02.052. [DOI] [PubMed] [Google Scholar]
- Uliyanchenko E.; Schoenmakers P. J.; van der Wal S. J. Chromatogr. A 2011, 1218 (11), 1509–1518. 10.1016/j.chroma.2011.01.053. [DOI] [PubMed] [Google Scholar]
- Malik M. I.; Pasch H. Prog. Polym. Sci. 2016, 63, 42–85. 10.1016/j.progpolymsci.2016.03.004. [DOI] [Google Scholar]
- Cottet H.; Simó C.; Vayaboury W.; Cifuentes A. J. Chromatogr. A 2005, 1068 (1), 59–73. 10.1016/j.chroma.2004.09.074. [DOI] [PubMed] [Google Scholar]
- Montaudo G.; Samperi F.; Montaudo M. S. Prog. Polym. Sci. 2006, 31 (3), 277–357. 10.1016/j.progpolymsci.2005.12.001. [DOI] [Google Scholar]
- Liu F. K.; Lin Y. Y.; Wu C. H. Anal. Chim. Acta 2005, 528 (2), 249–254. 10.1016/j.aca.2004.08.052. [DOI] [Google Scholar]
- Augsten C.; Kiselev M. A.; Gehrke R.; Hause G.; Mäder K. J. Pharm. Biomed. Anal. 2008, 47 (1), 95–102. 10.1016/j.jpba.2007.12.031. [DOI] [PubMed] [Google Scholar]
- Lyvén B.; Hassellöv M.; Turner D. R.; Haraldsson C.; Andersson K. Geochim. Cosmochim. Acta 2003, 67 (20), 3791–3802. 10.1016/S0016-7037(03)00087-5. [DOI] [Google Scholar]
- Kammer F. V. D.; Baborowski M.; Friese K. Anal. Chim. Acta 2005, 552 (1–2), 166–174. 10.1016/j.aca.2005.07.049. [DOI] [Google Scholar]
- Stoll D. R.; Carr P. W. Two-Dimensional Liquid Chromatography: A State of the Art Tutorial. Anal. Chem. 2016, 100, 519–531. 10.1021/acs.analchem.6b03506. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Van Der Horst A.; Schoenmakers P. J. J. Chromatogr. A 2002, 982 (1), 55–68. 10.1016/S0021-9673(02)01483-8. [DOI] [PubMed] [Google Scholar]
- Reingruber E.; Jansen J. J.; Buchberger W.; Schoenmakers P. J. Chromatogr. A 2011, 1218 (8), 1147–1152. 10.1016/j.chroma.2010.12.080. [DOI] [PubMed] [Google Scholar]
- Pirok B. W. J.; Breuer P.; Hoppe S. J. M.; Chitty M.; Welch E.; Farkas T.; van der Wal S.; Peters R.; Schoenmakers P. J. J. Chromatogr. A 2017, 1486, 96–102. 10.1016/j.chroma.2016.12.015. [DOI] [PubMed] [Google Scholar]
- Striegel A. M.; Brewer A. K. Annu. Rev. Anal. Chem. 2012, 5 (1), 15–34. 10.1146/annurev-anchem-062011-143107. [DOI] [PubMed] [Google Scholar]
- Vander Heyden Y.; Popovici S. T.; Schoenmakers P. J. J. Chromatogr. A 2002, 957 (2), 127–137. 10.1016/S0021-9673(02)00311-4. [DOI] [PubMed] [Google Scholar]
- Popovici S. T.; Schoenmakers P. J. J. Chromatogr. A 2005, 1099 (1–2), 92–102. 10.1016/j.chroma.2005.08.071. [DOI] [PubMed] [Google Scholar]
- Uliyanchenko E.; Cools P. J. C. H.; van der Wal S.; Schoenmakers P. J. Anal. Chem. 2012, 84 (18), 7802–7809. 10.1021/ac3011582. [DOI] [PubMed] [Google Scholar]
- Murphy R. E.; Schure M. R.; Foley J. P. Anal. Chem. 1998, 70 (8), 1585–1594. 10.1021/ac971184b. [DOI] [Google Scholar]
- Striegel A. M.; Yau W. W.; Kirkland J. J.; Bly D. D.. Modern Size-Exclusion Liquid Chromatography, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2009. [Google Scholar]
- McGowan G. R.; Langhorst M. A. J. Colloid Interface Sci. 1982, 89 (1), 94–106. 10.1016/0021-9797(82)90124-2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.