

Abstract
G-quadruplexes are nucleic acids structures stabilized by physiological concentration of potassium ions. Because low stability G-quadruplexes are hardly detectable by mass spectrometry, we optimized solvent conditions: isopropanol in a triethylamine/hexafluoroisopropanol mixture highly increased G-quadruplex sensitivity with no modification of the physiological G-quadruplex conformation. G-quadruplexes/G-quadruplex-ligand complexes were also correctly detected at concentration as low as 40 nM. Detection of the physiological conformation of G4s and their complexes opens up the possibility to perform high-throughput screening of G-quadruplex ligands for the development of drug molecules effective against critical human diseases.
G-quadruplexes (G4s) are noncanonical nucleic acid structures, which form in guanine (G)-rich sequences by G-quartet stacking, and are stabilized by potassium (K+), the most relevant intracellular monovalent cation.1,2 G4s may adopt different conformations, i.e., parallel, antiparallel, or hybrid, depending on their strand orientation.3 G4s are involved in the modulation of important biological processes in eukaryotes, prokaryotes, and viruses.4−11 In eukaryotes and viruses, the use of G4-ligands has shown promising therapeutic activity.12−15 Electrospray ionization (ESI) mass spectrometry (MS) is a powerful tool to investigate both G4 structure and G4/small molecule binding.16 To this aim, G4s need to maintain their native conformation. The main problem is that G4s are folded in physiological concentration of K+ (100–150 mM), which strongly hampers G4 detection and data quality in ESI-MS. To circumvent this problem, different approaches have been developed: (i) substitution of K+ with the volatile NH4+ ion, which fits in the G4;17,18 however, G4s folded in NH4+ often adopt nonphysiological conformations.19 (ii) G4 folding in physiological concentration of K+ and removal of the noncoordinated ions by filtration or ethanol precipitation;20,21 this approach is suitable only for stable G4s with slow unfolding kinetics in low K+ concentration. (iii) G4 folding in MS-compatible amounts of K+ (<1 mM) in the presence of a volatile bulky buffer (e.g., triethylammonium acetate (TEAA) or trimethylammonium acetate (TMAA)22); the rationale is that the bulky buffer does not fit into the G4 cavity and thus it does not prevent K+ from coordinating the G4, but it provides physiological ionic strength. Using this approach we obtained very good results in MS detection of stable G4s, but we observed weak detection and resolution of low stability G4s. We thus deemed it worth searching for new solvent conditions to favorably compare with the TMAA-based method.
Experimental Section
Oligonucleotides
Oligonucleotides were purchased from Sigma-Aldrich, Milan, Italy, and used without further purification. Five G4s were tested: two form in the human immunodeficiency virus-1 (HIV-1) LTR promoter (LTR-III and LTR-IV),8 one in the herpes simplex virus-1 (HSV-1) genome (un2)12 and two in the human chromosome, at telomeres (hTel) and in the promoter of the c-myc oncogene (c-myc) (Table S1). Oligonucleotides (5 μM) in 1 mM KCl (Fluka, St. Louis, MO), 150 mM trimethylammonium acetate (TMAA) (Fluka) pH 7.4 (adjusted from pH ∼7 to 7.4 with triethylamine (TEA) (Fisher, Pittsburgh, PA)) were annealed by heating at 95 °C for 5 min, gradually cooled to room temperature, added of 20% of isopropanol (IPA) (Carlo Erba Reagenti S.p.A, Milan, Italy) or 20% of water and incubated overnight at 4 °C (final buffer composition, 0.8 mM KCl, 120 mM TMAA pH 7.4, (20% IPA)). Alternatively oligonucleotides (5 μM) in 1 mM KCl, 150 mM of hexafluoroisopropanol (Across, Pittsburgh, PA) neutralized to pH 7.4 with TEA (TEA/HFIP) were annealed by heating at 95 °C for 5 min, gradually cooled to room temperature, added of 20% of IPA or 20% of water and incubated overnight at 4 °C (final buffer composition: 0.8 mM KCl, 120 mM TEA/HFIP, pH 7.4, 20% IPA). BRACO-19 (Endotherm Life Science, Saarbrücken, Germany) was added to the annealed G4 at G4/compound molar ratio 1:2 and incubated one more day before analysis.
Mass Spectrometry Analysis
Samples were analyzed by direct infusion electrospray ionization (ESI) on a Xevo G2-XS QTOF mass spectrometer (Waters, Manchester, U.K.). This is a high-resolution instrument that allowed us to visualize the isotopic pattern, identify the charge state, and therefore unambiguously calculate the neutral mass of the detected species. The injection was automatically performed by an Agilent 1290 Infinity HPLC (Agilent Technologies, Santa Clara, CA) equipped with an autosampler; the carrying buffer was TMAA 120 mM pH 7.4 with/without 20% IPA or TEA/HFIP 120 mM pH 7.4 with/without 20% IPA. A volume of 5 μL of each sample were typically injected for each analysis. In all experiments, ESI source settings were: electrospray capillary voltage 1.8 kV; source and desolvation temperatures 45 and 65 °C, respectively; sampling cone voltage 65 V. All these parameters ensured minimal fragmentation of the DNAs complexes. The instrument was calibrated using a 2 mg/mL solution of sodium iodide in 50% of IPA. Additionally, the use of the LockSpray during analysis provided a typical <5 ppm mass accuracy. The internal standard LockSpray consisted in a solution of leu-enkephalin 1 μg/mL in acetonitrile/water (50:50, v/v) containing 0.1% of formic acid. When comparing the same samples in different buffers, spectra were acquired the same day to avoid signal decrease due to source contamination.
Circular Dichroism Analysis
CD spectra were recorded on a Chirascan-Plus (Applied Photophysics, Leatherhead, U.K.) equipped with a Peltier temperature controller set at 20 °C, using a quartz cell of 5 mm optical path length, scanning at 1 nm bandwidth, 0.2 nm step size, and at 0.3 s per point over a wavelength range of 230–320 nm. Observed ellipticities were converted to molar ellipticity ([θ] = deg × cm2 × dmol–1). Oligonucleotides (5 μM) in 1–100 mM KCl, 150 mM TMAA (unless otherwise specified) or 20 mM potassium phosphate (Sigma-Aldrich, St. Louis, MO) buffer pH 7.4 were annealed by heating at 95 °C for 5 min, gradually cooled to room temperature, added 20% of the indicated solvent (Sigma or Carlo Erba) or 20% of water, and incubated overnight at 4 °C. Oligonucleotides in TEA/HFIP were annealed as previously discussed.
Results and Discussion
The conformation of the viral (LTR-III, LTR-IV, un2) and cellular (hTel and c-myc) G4s (Table S1) was initially tested. In 100 mM K+, LTR-IV folds in a bulged parallel G423 with Tm of 50 °C. LTR-III8 and hTel24 are hybrid G4 with Tm of 68 °C, while c-myc and un2 display parallel and antiparallel topology, respectively, and Tm > 90 °C.12,25 To assess the G4 conformation of the tested sequences in different buffer conditions, circular dichroism (CD) was used (Figure S1a). In 100 mM K+ and phosphate buffer, LTR-IV and c-myc showed a parallel G4 CD signature with one maximum at 260 nm and one minimum at 240 nm. LTR-III and hTel displayed a hybrid-like spectrum: LTR-III had a maximum at 260 nm and a shoulder at 290 nm, hTel a maximum at 290 nm and a shoulder at 250 nm; both structures displayed a minimum at 235–240 nm. Un2 displayed a typical antiparallel G4 structure with a minimum at 260 and two maxima at 240 and 290 nm.26 Substitution of the phosphate buffer with trimethylammonium acetate (TMAA) buffer, the cation of which is too bulky to fit the G4,22 did not lead to any CD-detectable structure modification, as long as the total ion concentration was maintained constant and K+ was kept above 10 mM (Figure S1a). When K+ concentration was reduced to 10 mM or below, decreased molar ellipticity was observed for LTR-IV, indicating less oligonucleotide folded into G4; yet the parallel conformation was maintained. LTR-III remained at the same fully folded conformation at both 100 and 10 mM K+, however at 0.8 mM K+ the molar ellipticity dramatically decreased and a structural change was monitored as inversion of relative peak intensity at 260 and 290 nm. HTel displayed a similar pattern, with identical CD spectra at 100 and 10 mM K+, turning into the CD signature characteristic of an antiparallel structure at 0.8 mM K+. In contrast, the structures with higher Tm (i.e., c-myc and un2) were poorly affected by K+ decrease (Figure S1a). These data indicate that low K+ concentration in TMAA is not optimal for several G4s, since they undergo conformational changes.
To perform MS of G4s maintaining stability and conformation as close as possible to those in 100 mM K+, we initially explored the effect of organic solvents on G4-folding in TMAA. It has been reported that the presence of primary alcohols promotes G4-folding due to crowding effect27 and to water release during G4 assembly.28,29 Several organic solvents in 120 mM TMAA and 0.8 mM K+ were tested by CD for their effectiveness in promoting the physiological G4 conformation of LTR-IV. Isopropanol (IPA), acetonitrile, and methanol proved to be the most effective in stabilizing LTR-IV G4, whereas ethanol was slightly less efficient (Figure S1b). Molar ellipticity substantially increased after addition of 20% organic solvent to the TMAA solution with 0.8% K+, with the CD spectrum becoming practically identical to that measured in 100 mM K+, confirming the strong G4-folding-inducing properties of organic solvents (Figure S1b). IPA, being less toxic than methanol and acetonitrile, was chosen for further MS-buffer optimization. Molar ellipticity of LTR-IV G4 was proportional to the amount of solvent in the buffer, reaching a plateau at 30% of IPA; at 20% IPA the CD spectrum of LTR-IV G4 was similar to that measured in 100 mM K+ (Figure S1c), therefore 20% IPA was used in the following buffer optimization process. The G4-stabilizing effect of 20% IPA was maintained also on LTR-III and hTel, whereas un2 and c-myc were not modified (Figure 1a), in line with the previously observed stability of these G4s (Figure S1a).
Figure 1.

Analysis of the tested sequences in the different buffers optimized for MS detection of the G4 structure. (a) Dichroic spectra of G4 folding sequences in 100 mM K+, 20 mM K2HPO4, pH 7.4 (PB) (blue line), 0.8 mM K+, 120 mM TMAA (red line), 0.8 mM K+, 120 mM TMAA added of 20% IPA (green line), 0.8 mM K+, TEA/120 mM HFIP added of 20% IPA (violet line). (b) ESI-MS spectra of the tested sequences in the buffers indicated at the bottom. Spectra were zoomed to compare peaks with charge state that allows the highest relative intensity (RI) in TMAA buffer. (c) Base peak intensity of the tested sequences in the indicated buffers. (d) Representative ESI-MS spectra (LTR-IV) to show the entire peak charge distribution in the indicated buffers.
MS spectra in the presence/absence of IPA were obtained to confirm G4 folding by assessing the number of coordinated K+ ions, which are diagnostic for the number of G-quartets involved in the G4 structure (Figure 1b and Figure S2). The MS spectrum of LTR-IV in TMAA 0.8 mM K+ presented a major peak corresponding to LTR-IV and peaks of lower intensity that corresponded to LTR-IV-K+ adducts, which displayed a salt concentration-dependent statistical distribution, suggesting nonspecific K+ binding. The use of IPA drastically modified the spectrum: the intensity of the free DNA and 1K+-adduct became much lower compared to the 2K+-adduct. The 2K+-adduct is consistent with the reported structure of LTR-IV G4, comprising three G-quartets coordinating two K+23. The same rationale applies to LTR-III: in the absence of IPA the peak of the free LTR-III and small amounts of 1K+- and 2K+- adducts were present; upon addition of IPA, the peak corresponding to the 2K+-adduct became the main peak and those corresponding to the free LTR-III and 1K+-adducts disappeared. The behavior of hTel DNA was more complex: in the absence of IPA, hTel mainly showed 1K+- and 2K+-adducts, consistently with formation of two and three G-quartets. Addition of IPA increased the intensity of the 2K+-adduct compared to the 1K+-adduct, which however remained intense. These data are in line with the different G4 conformations adopted by the telomeric sequence, including “form 3” that displays two G-quartets coordinating one K+.30 MS spectra of un2 and c-myc showed the presence of a 3K+ and 2K+-adduct, respectively, which is consistent with their reported G4 structures12,25 and IPA addition did not affect K+-adduct distribution (Figure 1b and Figure S2). Relative peak intensity and K+-adduct distribution (Table 1) indicate that addition of 20% IPA positively influences MS detection of the least stable G4s (i.e., LTR-IV, LTR-III, and hTel), whereas it does not perturb the most stable G4s (i.e., un2 and c-myc).
Table 1. Relative Amounts of Free DNA (No K+) and K+ Adducts Detected by MS in TMAA and TEA/HFIP Buffers in the Presence/Absence of 20% IPAa.
| intensity of K+-coordinated adducts |
||||||||
|---|---|---|---|---|---|---|---|---|
| G4 | buffer | no K+ | 1K+ | 2K+ | 3K+ | 4K+ | charge state | base peak |
| LTR-IV | TMAA | 53.5 ± 0.8 | 23.6 ± 1.0 | 12.2 ± 0.6 | 6.6 ± 0.4 | 4.1 ± 0.6 | 4–, 5– | [G4]4– |
| TMAA/IPA | 2.6 ± 0.8 | 4.4 ± 0.3 | 54.3 ± 2.1 | 26.3 ± 1.2 | 12.4 ± 0.6 | 4–, 5– | [G4 + 2K+]4– | |
| TEA/HFIP | 50.6 ± 4.3 | 20.5 ± 2.5 | 20.3 ± 0.8 | 6.8 ± 0.9 | 1.8 ± 0.0 | 3– to 10– | [G4]9– | |
| TEA/HFIP/IPA | 4.1 ± 0.5 | 0.5 ± 0.0 | 92.0 ± 0.1 | 3.0 ± 0.1 | 0.3 ± 0.0 | 3– to 9– | [G4 + 2K+]7– | |
| LTR-III | TMAA | 34.4 ± 2.6 | 15.2 ± 0.8 | 23.0 ± 2.0 | 17.2 ± 2.3 | 10.3 ± 0.70 | 5– to 7– | [G4 + 2K+]5– |
| TMAA/IPA | 1.3 ± 0.12 | 1.9 ± 0.19 | 54.0 ± 6.0 | 27.4 ± 3.5 | 15.3 ± 2.8 | 5–, 6– | [G4 + 2K+]5– | |
| TEA/HFIP | 42.2 ± 1.3 | 7.9 ± 1.1 | 41.1 ± 5.3 | 7.6 ± 1.3 | 0.0 ± 0.0 | 6– to 13– | [G4]11– | |
| TEA/HFIP/IPA | 5.5 ± 0.9 | 1.4 ± 0.4 | 81.4 ± 1.6 | 10.6 ± 1.2 | 1.0 ± 0.6 | 5– to 12– | [G4 + 2K+]8– | |
| un2 | TMAA | 0.0 ± 0.0 | 0.0 ± 0.0 | 3.1 ± 0.15 | 72.7 ± 1.1 | 24.2 ± 1.3 | 5–, 6– | [G4 + 3K+]5– |
| TMAA/IPA | 0.0 ± 0.0 | 0.0 ± 0.0 | 4.8 ± 0.9 | 67.3 ± 0.8 | 27.8 ± 1.0 | 5–, 6– | [G4 + 3K+]5– | |
| TEA/HFIP | 0.0 ± 0.0 | 0.0 ± 0.0 | 4.9 ± 0.9 | 80.4 ± 3.3 | 14.1 ± 2.6 | 6– to 9– | [G4 + 3K+]8– | |
| TEA/HFIP/IPA | 1.7 ± 0.1 | 1.5 ± 0.0 | 12.5 ± 0.3 | 73.2 ± 0.3 | 11.1 ± 0.1 | 6– to 9– | [G4 + 3K+]8– | |
| hTel | TMAA | 7.3 ± 1.2 | 39.8 ± 2.7 | 31.4 ± 1.6 | 13.9 ± 1.4 | 7.5 ± 1.5 | 4– to 6– | [G4 + 2K+]4– |
| TMAA/IPA | 0.0 ± 0.0 | 26.7 ± 1.6 | 44.5 ± 0.6 | 19.7 ± 1.0 | 9.1 ± 0.7 | 3– to 5– | [G4 + 2K+]4– | |
| TEA/HFIP | 57.2 ± 6.7 | 8.9 ± 2.8 | 20.7 ± 2.1 | 12.6 ± 1.0 | 0.0 ± 0.0 | 4– to 10– | [G4]9– | |
| TEA/HFIP/IPA | 27.3 ± 1.0 | 8.0 ± 0.0 | 62.4 ± 0.5 | 2.2 ± 0.6 | 0.0 ± 0.0 | 3– to 10– | [G4 + 2K+]7– | |
| c-myc | TMAA | 0.0 ± 0.0 | 0.0 ± 0.0 | 61.3 ± 5.3 | 25.8 ± 1.6 | 12.8 ± 3.7 | 4– to 6– | [G4 + 2K+]5– |
| TMAA/IPA | 0.0 ± 0.0 | 0.0 ± 0.0 | 59.5 ± 2.6 | 26.4 ± 1.5 | 14.0 ± 1.3 | 4– to 6– | [G4 + 2K+]5– | |
| TEA/HFIP | 0.0 ± 0.0 | 0.0 ± 0.0 | 52.0 ± 5.2 | 33.1 ± 2.5 | 14.0 ± 2.5 | 6– to 10– | [G4 + 2K+]8– | |
| TEA/HFIP/IPA | 3.9 ± 0.4 | 1.2 ± 0.2 | 75.4 ± 0.2 | 19.4 ± 0.4 | 0.0 ± 0.0 | 4– to 11– | [G4 + 2K+]8– | |
In bold is the intensity of the prevalent adduct in the corresponding sample. The numbers represent the intensity of the free DNA (no K) or K+ adducts (1K+, 2K+, 3K+, 4K+). The intensity is normalized to the sum of the intensities of all species (no K, 1K+, 2K+, 3K+, 4K+) and expressed in %. All charge states present in the spectra were included in the calculation; charge states per each sample are indicated. Each sample was assayed at least three times and standard deviation is reported. The base peak, i.e., the peak with the highest intensity within each spectrum, is shown.
The ability of triethylamine/hexafluoroisopropanol (TEA/HFIP) to improve MS detection of nucleic acids has been described.31,32 We initially compared by CD the effect of TEA/HFIP/IPA vs TMAA/IPA on G4-folding. All oligonucleotides essentially maintained the same conformation in TEA/HFIP/IPA and TMAA/IPA (Figure 1a), with the exception of hTel, which in TEA/HFIP/IPA increased at 265 nm and decreased at 250 nm. K+-adduct distribution in the two buffers was next tested by ESI/MS (Figure 1b and Figure S2): all G4s displayed similar MS spectra in the two buffers, with hTel representing the only exception: in TEA/HFIP ± IPA, the peaks corresponding to the 1K+-adduct almost disappeared. The hTel 1K+-adduct likely represents hTel G4 form 3: in the same buffer conditions, the CD spectrum exhibited a decrease of the shoulder at 250 nm, reported as characteristic of hTel G4 form 3.30 K+-adduct distribution of the tested G4s in TEA/HFIP buffer in the presence/absence of 20% IPA are summarized in Table 1: as in the case of TMAA, the presence of IPA highly increased detection of MS peaks corresponding to the fully folded G4s, especially with the least stable G4s, i.e., LTR-IV, LTR-III, and hTel. In TEA/HFIP/IPA, however, and in contrast to what was observed in TMAA/IPA, peaks corresponding to the folded G4 increased also for the most stable G4s, i.e., un2 and c-myc. In general, peak intensity was highly augmented in TEA/HFIP ± IPA: the most intense peak (base peak) in each MS spectrum increased by 1 order of magnitude in TEA/HFIP/IPA vs TMAA/IPA (Figure 1c). In addition, charge distribution in TEA/HFIP and TMAA was different: for instance, LTR-IV in TEA/HFIP/IPA presented peaks with charge states from 3– to 9–, whereas in TMAA/IPA 4– and 5– charge states only were detected (Figure 1d). K+ binding was not altered in TEA/HFIP/IPA buffer: more than 95% of peaks with charge state 7– corresponded to LTR-IV coordinated to two K+; when considering all charge states, the intensity of 2K+-adducts, indicating full G4-folding, represented more than 90% of the intensity of all peaks (Table 1). Around 4% peak intensity corresponded to free DNA, diagnostic of the unfolded oligonucleotide, and similar percentage to multi-K+ or TEA adducts, mostly at lower charge states (4–, 3−) (Figure 1d). Similar results were obtained for LTR-III, c-myc, and un2 (Table 1). The hTel sequence showed a different behavior: in TEA/HFIP/IPA, around 62% of the peaks corresponded to 2K+-adducts and 27% to free DNA (Table 1). In contrast, in TMAA/IPA around 45% and 25% of peaks corresponded to 2K+-and 1K+-adducts, respectively, and no free DNA was detected, suggesting some conformational difference of hTel in the two buffers. In general the high charge peaks retained the G4-diagnostic K+, while aspecific salt-adducts were absent, expediting the assignments of the correct number of G-quartets. We ascribe the multicharge effect to the lower amount of R3NH+ ions in HFIP/TEA vs TMAA. In fact, TMAA at 120 mM pH 7.4 roughly consists of equimolar amounts of trimethylamine (pKb 4.2) and acetic acid (pKa 4.8); in contrast, the amount of triethylamine (pKb 3.3) necessary to neutralize a solution of 120 mM-HFIP (pKa 9.3) is considerably lower (<5 mM), yielding a relatively low concentration of triethylammonium salt. This low salt concentration on one hand reduces the neutralization of the phosphate groups and thus increases the DNA charge states detected by ESI-MS; on the other, it reduces signal suppression hence raising the ESI signal intensity.31 Thus, while in general terms multicharging of biomolecules depicts denaturation, in this case the salt effect increases G4 conformation and MS detection, as corroborated by CD analysis (Figure 1a) and MS detection of coordinated K+ ions (Figure 1b and Figure S2) and of G4-bound ligand (Figure 2a,b and Figure S3).
Figure 2.
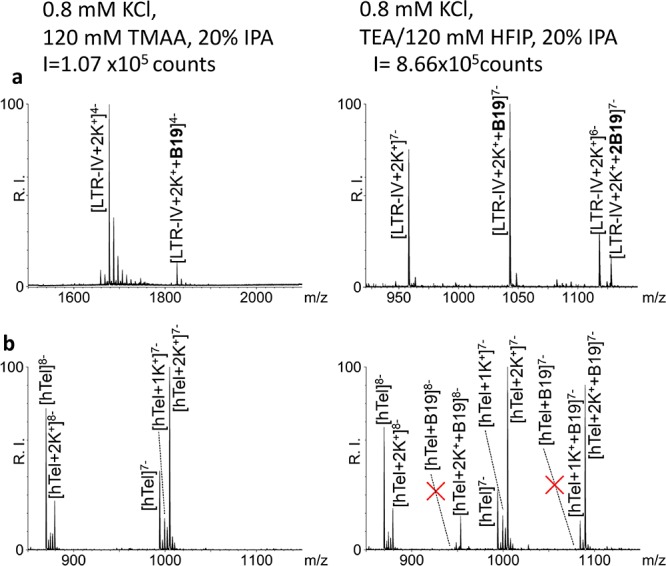
ESI-MS spectra of G4s in the presence of BRACO-19 in different buffers. (a) LTR-IV was incubated with 2 equivalents of B19 in 0.8 mM K+, 20% IPA, and either TMAA 120 mM pH 7.4 (left panel) or TEA/HFIP 120 mM pH 7.4 (right panel). (b) HTel 4 μM in the absence (left panel) or presence (right panel) of 1 equiv of B19 in TEA/HFIP 120 mM pH 7.4, 0.8 mM KCl, and 20% IPA. The red X symbols indicate the absence of adducts between the unstructured hTel and B19.
We next compared detection of G4-ligand complexes in TEA/HFIP vs TMAA. BRACO-19 (B19) was used as a reference ligand, since it has been shown to interact with most G4s and, in particular, with all those used in this study.8,12,33,34 When B19 was added to LTR-IV at 1:2 G4/B19 molar ratio in 0.8 mM K+, 20% IPA, and 120 mM TEA/HFIP, molecular masses corresponding to LTR-IV with both one and two bound B19 molecules were detected. The base peak corresponding to LTR-IV/2K+/1B19 had a charge state 7– (Figure 2a and Figure S3b). In contrast, in 120 mM TMAA, only the complex with one B19 was observed (LTR-IV/2K+/1B19), the base peak charge state was 4– and the intensity ∼10-fold lower (Figure 2a and Figure S3a). We next checked the degree of specific binding obtainable in TEA/HFIP: we tested the binding of B19 to hTel, which in TEA/HFIP is present both as G4-folded and unfolded DNA. In the presence of B19, only K+-adducts (1K+ and 2K+) bound to B19 were found, while no B19 was detected bound to the free unstructured DNA, indicating that a stringent specificity of binding was maintained in TEA/HFIP (Figure 2b and Figure S3c). Because signal intensity in TEA/HFIP was very high, we tested the possibility to analyze diluted samples: LTR-IV + 2K+ either free or bound to one molecule of B19 was detected at G4 concentration as low as 40 nM (Figure S4c). At higher concentration (i.e., 400 nM, 4 μM) LTR-IV + 2K+ with two molecules of B19 was also detected (Figure S4a,b). The increased sensitivity in TEA/HFIP allows decreasing the concentration of both target molecule and ligand minimizing aspecific binding, if present.
Conclusions
The possibility to increase MS signal intensity without affecting physiological folding enables the analysis of G4s and G4-small molecule adducts in the submicromolar range. This feature can be exploited for high-throughput screening of complex mixtures of ligands on a folded nucleic acid scaffold and therefore for the development of drug molecules effective against significant human diseases.
Acknowledgments
This work was supported by the Bill and Melinda Gates Foundation (GCE Grant Numbers OPP1035881, OPP1097238) and the European Research Council (ERC Consolidator Grant 615879). Helpful discussions with Dr. M. Bellini and Dr. D. Dalzoppo are gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01282.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Williamson J. R.; Raghuraman M. K.; Cech T. R. Cell 1989, 59, 871–880. 10.1016/0092-8674(89)90610-7. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Patel D. J. Biochemistry 1992, 31, 8112–8119. 10.1021/bi00150a002. [DOI] [PubMed] [Google Scholar]
- Phan A. T.; Kuryavyi V.; Patel D. J. Curr. Opin. Struct. Biol. 2006, 16, 288–298. 10.1016/j.sbi.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert J. L.; Balasubramanian S. Nucleic Acids Res. 2007, 35, 406–413. 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaume N.; Pathak R.; Yadav V. K.; Kota S.; Misra H. S.; Gautam H. K.; Chowdhury S. Nucleic Acids Res. 2013, 41, 76–89. 10.1093/nar/gks1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metifiot M.; Amrane S.; Litvak S.; Andreola M.-L. Nucleic Acids Res. 2014, 42, 12352–12366. 10.1093/nar/gku999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone R.; Nadai M.; Poe J. A.; Frasson I.; Palumbo M.; Palù G.; Smithgall T. E.; Richter S. N. PLoS One 2013, 8, e73121. 10.1371/journal.pone.0073121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone R.; Nadai M.; Frasson I.; Poe J. A.; Butovskaya E.; Smithgall T. E.; Palumbo M.; Palù G.; Richter S. N. J. Med. Chem. 2013, 56, 6521–6530. 10.1021/jm400914r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. P. Nature 2014, 507, 175–177. 10.1038/nature13067. [DOI] [PubMed] [Google Scholar]
- Murat P.; Zhong J.; Lekieffre L.; Cowieson N. P.; Clancy J. L.; Preiss T.; Balasubramanian S.; Khanna R.; Tellam J. Nat. Chem. Biol. 2014, 10, 358–364. 10.1038/nchembio.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artusi S.; Perrone R.; Lago S.; Raffa P.; Di Iorio E.; Palù G.; Richter S. N. Nucleic Acids Res. 2016, 44, 10343–10353. 10.1093/nar/gkw968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artusi S.; Nadai M.; Perrone R.; Biasolo M. A.; Palù G.; Flamand L.; Calistri A.; Richter S. N. Antiviral Res. 2015, 118, 123–131. 10.1016/j.antiviral.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidzinska J.; Cimino-Reale G.; Zaffaroni N.; Folini M. Molecules 2013, 18, 12368–12395. 10.3390/molecules181012368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone R.; Butovskaya E.; Daelemans D.; Palu G.; Pannecouque C.; Richter S. N. J. Antimicrob. Chemother. 2014, 69, 3248–3258. 10.1093/jac/dku280. [DOI] [PubMed] [Google Scholar]
- Perrone R.; Doria F.; Butovskaya E.; Frasson I.; Botti S.; Scalabrin M.; Lago S.; Grande V.; Nadai M.; Freccero M.; Richter S. N. J. Med. Chem. 2015, 58, 9639–9652. 10.1021/acs.jmedchem.5b01283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosu F.; De Pauw E.; Gabelica V. Biochimie 2008, 90, 1074–1087. 10.1016/j.biochi.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Rosu F.; Gabelica V.; Houssier C.; Colson P.; Pauw E. D. Rapid Commun. Mass Spectrom. 2002, 16, 1729–1736. 10.1002/rcm.778. [DOI] [PubMed] [Google Scholar]
- Li H.; Liu Y.; Lin S.; Yuan G. Chem. - Eur. J. 2009, 15, 2445–2452. 10.1002/chem.200801922. [DOI] [PubMed] [Google Scholar]
- Smargiasso N.; Rosu F.; Hsia W.; Colson P.; Baker E. S.; Bowers M. T.; De Pauw E.; Gabelica V. J. Am. Chem. Soc. 2008, 130, 10208–10216. 10.1021/ja801535e. [DOI] [PubMed] [Google Scholar]
- Pierce S. E.; Wang J.; Jayawickramarajah J.; Hamilton A. D.; Brodbelt J. S. Chem. - Eur. J. 2009, 15, 11244–11255. 10.1002/chem.200901047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans S. E.; Mendez M. A.; Turner K. B.; Keating L. R.; Grimes R. T.; Melchoir S.; Szalai V. A. JBIC, J. Biol. Inorg. Chem. 2007, 12, 1235–1249. 10.1007/s00775-007-0292-0. [DOI] [PubMed] [Google Scholar]
- Marchand A.; Gabelica V. J. Am. Soc. Mass Spectrom. 2014, 25, 1146–1154. 10.1007/s13361-014-0890-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nicola B.; Lech C. J.; Heddi B.; Regmi S.; Frasson I.; Perrone R.; Richter S. N.; Phan A. T. Nucleic Acids Res. 2016, 44, 6442–6451. 10.1093/nar/gkw432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Patel D. J. Structure 1993, 1, 263–282. 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- Siddiqui-Jain A.; Grand C. L.; Bearss D. J.; Hurley L. H. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11593–11598. 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randazzo A.; Spada G. P.; da Silva M. W. In Quadruplex Nucleic Acids; Chaires J. B., Graves D., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, Germany, 2012; Vol. 330, pp 67–86. [Google Scholar]
- Vorlíčková M.; Bednářová K.; Kypr J. Biopolymers 2006, 82, 253–260. 10.1002/bip.20488. [DOI] [PubMed] [Google Scholar]
- Smirnov I. V.; Shafer R. H. Biopolymers 2007, 85, 91–101. 10.1002/bip.20609. [DOI] [PubMed] [Google Scholar]
- Zaccaria F.; Paragi G.; Fonseca Guerra C. Phys. Chem. Chem. Phys. 2016, 18, 20895–20904. 10.1039/C6CP01030J. [DOI] [PubMed] [Google Scholar]
- Lim K. W.; Amrane S.; Bouaziz S.; Xu W.; Mu Y.; Patel D. J.; Luu K. N.; Phan A. T. J. Am. Chem. Soc. 2009, 131, 4301–4309. 10.1021/ja807503g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apffel A.; Chakel J. A.; Fischer S.; Lichtenwalter K.; Hancock W. S. Anal. Chem. 1997, 69, 1320–1325. 10.1021/ac960916h. [DOI] [PubMed] [Google Scholar]
- Basiri B.; van Hattum H.; van Dongen W. D.; Murph M. M.; Bartlett M. G. J. Am. Soc. Mass Spectrom. 2017, 28, 190–199. 10.1007/s13361-016-1500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read M.; Harrison R. J.; Romagnoli B.; Tanious F. A.; Gowan S. H.; Reszka A. P.; Wilson W. D.; Kelland L. R.; Neidle S. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (9), 4844–4849. 10.1073/pnas.081560598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemarteleur T.; Gomez D.; Paterski R.; Mandine E.; Mailliet P.; Riou J.-F. Biochem. Biophys. Res. Commun. 2004, 323 (3), 802–808. 10.1016/j.bbrc.2004.08.150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.