

Abstract
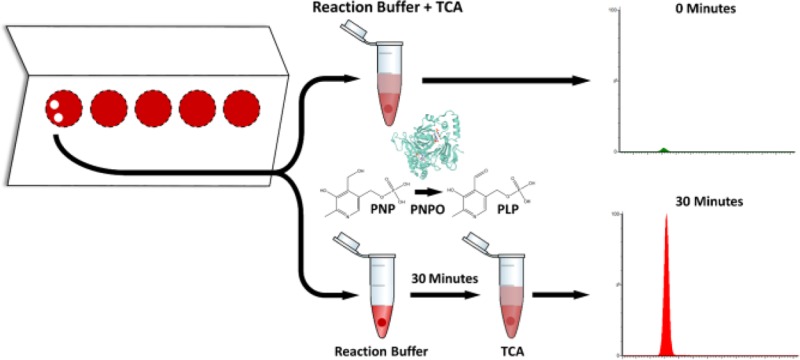
We report the development of a rapid, simple, and robust LC–MS/MS-based enzyme assay using dried blood spots (DBS) for the diagnosis of pyridox(am)ine 5′-phosphate oxidase (PNPO) deficiency (OMIM 610090). PNPO deficiency leads to potentially fatal early infantile epileptic encephalopathy, severe developmental delay, and other features of neurological dysfunction. However, upon prompt treatment with high doses of vitamin B6, affected patients can have a normal developmental outcome. Prognosis of these patients is therefore reliant upon a rapid diagnosis. PNPO activity was quantified by measuring pyridoxal 5′-phosphate (PLP) concentrations in a DBS before and after a 30 min incubation with pyridoxine 5′-phosphate (PNP). Samples from 18 PNPO deficient patients (1 day–25 years), 13 children with other seizure disorders receiving B6 supplementation (1 month–16 years), and 37 child hospital controls (5 days–15 years) were analyzed. DBS from the PNPO-deficient samples showed enzyme activity levels lower than all samples from these two other groups as well as seven adult controls; no false positives or negatives were identified. The method was fully validated and is suitable for translation into the clinical diagnostic arena.
Pyridox(am)ine 5′-phosphate oxidase (PNPO) deficiency (OMIM 610090) is an autosomal recessive seizure disorder caused by impaired activity of the PNPO enzyme, which catalyzes the oxidation of pyridoxamine 5′-phosphate (PMP) and pyridoxine 5′-phosphate (PNP) to pyridoxal 5′-phosphate (PLP), the active form of vitamin B6.
Dietary vitamin B6 is consumed as pyridoxine, pyridoxamine, and pyridoxal (PN, PM, and PL), their phosphorylated analogues, and the glucoside of PN.1−4 The hydrolyzed vitamers are absorbed, and PN and PM undergo phosphorylation prior to their conversion to PLP by PNPO (Figure 1). The Enzyme Commission (http://www.chem.qmul.ac.uk/iubmb/enzyme/) catalogues over 140 PLP-dependent enzymatic activities. In humans, PLP-dependent enzymes include several involved in the synthesis or degradation of amino acids or amines that serve as neurotransmitters or neuromodulators in the brain.
Figure 1.

Vitamin B6 metabolism. (a) Pyridoxine, (b) pyridoxal, (c) pyridoxamine, (d) pyridoxine 5′-phosphate, (e) pyridoxal 5′-phosphate, and (f) pyridoxamine 5′-phosphate. PK = pyridoxal kinase; PNPO = pyridox(am)ine 5′-phosphate oxidase.
Classical PNPO deficiency has been described as epileptic encephalopathy refractive to conventional anticonvulsants and pyridoxine but responsive to high doses (30–40 mg kg–1 day–1) of PLP.5 In recent years, however, considerable heterogeneity has been identified in the phenotype of affected patients with approximately 40%6 of PNPO-deficient patients showing some response to pyridoxine. Some of these individuals have not tolerated a switch in treatment to PLP with their seizure control worsening. In addition, patients have presented with seizures after the neonatal period.2,3 Patients that have severe neonatal epileptic encephalopathy but survive into infancy have a high disease burden with severe developmental delay, progressive seizures, and microcephaly.2,3,7 However, when diagnosed and treated early, patients can have a normal neurodevelopmental outcome.8
Diagnosing patients with PNPO deficiency can be challenging. There are many practical difficulties in relying upon a clinical response to PLP or PN as often neonates and infants can have multisystem pathology and the response to treatment may not be immediate and total. Additionally, there are other seizure disorders that respond to treatment with PLP and/or PN, which cannot be differentiated from PNPO deficiency using this approach.9 Seizures caused by pyridoxine-dependent epilepsy (PDE) can be indistinguishable from those caused by a deficiency of PNPO. Hyperprolinaemia type 2 (HPII) or hypophosphatasia can also cause perinatal B6-responsive seizures.1 Other neurological disorders known to benefit from PLP/pyridoxine supplementation include KCNQ2 deficiency and AADC deficiency.
Although biochemical findings that were initially thought to be characteristic of PNPO deficiency can be indicative,5 for some individuals these may be present only transiently or absent altogether. A more recent study has suggested that a high plasma PM concentration can be indicative of patients with PNPO deficiency irrespective of vitamin B6 supplementation and that this in conjunction with an elevated PM/pyridoxic acid (PA) ratio is a biomarker for the selective screening of PNPO deficiency.10 However, this study used a small sample size (n = 6), and it is currently still necessary to confirm these findings genetically. Current practice relies on the detection of mutations in the PNPO gene and subsequent expression studies to confirm pathogenicity of any variants detected.11,12 This is often carried out after the exclusion of other B6-responsive disorders such as pyridoxine-dependent epilepsy due to mutations in ALDH7A1 (PDE) and can cause a considerable delay in diagnosis.
Published assays for the determination of PNPO activity have shown that human erythrocytes convert pyridoxamine 5′-phosphate (PMP), a substrate of PNPO, to PLP. However, these assays are laborious, not suitable for routine clinical diagnosis, require large sample volumes, or use radiolabeled substrates.13−18 The use of dried blood spots (DBS) from a heel or finger-prick is an established method for sample collection, which was developed initially for the measurement of phenylalanine levels in the diagnosis of phenylketonuria in the 1960s.19 Subsequently, the use of DBS to assay enzymes present in the circulating blood has been reported for various metabolic disorders.20 The noninvasive collection and simple transport/storage are a major advantage of DBS.
We have recently established a mass spectrometry-based method for quantification of the individual forms of vitamin B621 and have further developed this to enable us to assay PNPO activity in DBS. This highly sensitive method is more specific than published methods and allows quantification of PNPO activity without using large sample volumes or radiolabeled substrates. Here, we report the development and validation of this assay as well as its utility for the analysis of control and PNPO-deficient patient samples.
Experimental Section
Sample Collection and Storage of DBS
This study was approved by the National Research Ethics Service (NRES) Committee (London, Bloomsbury [REC ref no. 3/LO/0168]). The majority of control DBS were obtained from patients attending Great Ormond Street Hospital for Children, London, UK. Written informed consent was obtained for all subjects, and inclusion/exclusion criteria are discussed below. DBS samples were also received from patients with undiagnosed B6-responsive seizure disorders, after informed consent, from centers across Europe. DBS for validation of the measurement of PNPO activity were collected from healthy adults.
Whatman 903 filter paper was used for sample collection. Blood, either venous or capillary, was spotted in the middle of the preprinted circle of a Protein Saver card (GE Healthcare, Little Chalfont, Bucks, UK). The spotted blood was allowed to dry for 16–24 h at room temperature (22 °C) prior to the card being placed in a sealed foil zip-lock bag with desiccant (to protect from light and humidity) at room temperature for a maximum of 7 days. DBS samples were then stored at −20 or −80 °C until analysis.
Cards were visually inspected upon analysis and rejected if poor sampling protocol was suspected. Care was taken to avoid sample collection from recipients of blood transfusions within the preceding 100 days.
Materials
Trizma base, PLP, LC–MS/MS grade acetic acid, heptafluorobutyric acid (HFBA), PMP, PL, PM, d3-PM, PA, d2-PA, and PN were purchased from Sigma-Aldrich (Gillingham, Dorset, UK). Orthophosphoric acid was purchased from BDH Chemicals (Poole, Dorset, UK). PNP was purchased from Toronto Research Chemicals (Toronto, Canada). d2-PN and d3-PLP were purchased from CDN Isotopes (Thaxted, Essex, UK) and Buchem BV (Appeldoorn, The Netherlands), respectively. 6.1 N (=6.1 M) trichloroacetic acid (TCA) was purchased from MP Biomedicals (Solon, U.S.). Whatman desiccant was purchased from Scientific Laboratory Supplies (Nottingham, UK). Flavin mononucleotide (FMN) was purchased from Applichem (Darmstadt, Germany). All water used was purified by a Millipore Milli-Q Direct 8 system with a 0.22 μm filter.
Measurement of PNPO Activity
For each patient sample, two 3 mm punches (T0 and T30) were taken from the spotted blood card. These punches were rehydrated in a 1.5 mL polypropylene tube containing 60 μL of a 40 mmol/L Tris-phosphate buffer, prepared by adjusting a 40 mmol/L Tris solution to pH 7.6 using 8.5% orthophosphoric acid. The tubes were then sonicated in a Grant XUBA3 ultrasonic bath for 2 min. Subsequently, 60 μL of a reaction mix containing 800 nmol/L PNP substrate and 3 μmol/L FMN was added to each tube to a final concentration of 400 nmol/L and 1.5 μmol/L, respectively. 120 μL of a reaction stop mix (0.3 M TCA containing 50 nmol/L of the internal standard d3-PLP to a final concentration of 0.15 M and 25 nmol/L, respectively) was added immediately to tube T0 prior to incubation on ice for 45 min. Tube T30 was incubated for 30 min at 37 °C with shaking at 300 rpm in an Eppendorf Thermomixer C prior to the addition of 120 μL of reaction stop mix and incubation on ice for 45 min. After incubation with TCA, all samples were sonicated for 5 min prior to centrifugation at 16 000g for 10 min at 4 °C. The resulting supernatant, containing the B6 vitamers, was removed and stored at −20 °C until time of analysis. All samples were recentrifuged at 16 000g for 10 min at 4 °C after thawing, and the supernatants were transferred to 300 μL amber insert vials (Fisher Scientific). Because of the known photolability of PLP,22,23 care was taken at all times to avoid light exposure, and all standards were stored on ice during experimentation. For long-term storage, B6 vitamer stock solutions were kept in aqueous form at −80 °C. TCA addition and subsequent incubation was necessary for the precipitation of protein found in the DBS, release of protein-bound PLP into solution, and cessation of enzymatic activity. Acidic conditions are also known to stabilize the B6 vitamers in aqueous solution.24
Liquid Chromatography–Mass Spectrometry
Quantification of the B6 vitamers and pyridoxic acid was performed essentially as described in Footitt et al.21 with minor modifications. UPLC–MS/MS was carried out using a Waters Acquity H-Class UPLC system connected to a Waters Xevo TQ-S triple quadrupole mass spectrometer using electrospray ionization and multiple reaction monitoring (MRM). All compounds were detected in positive ion mode. Eight microliters of sample was flow-injected by the autosampler onto a Waters Acquity UPLC HSS T3 column (1.8 μm, 2.1 × 50 mm) protected by a 1.8 μm Acquity UPLC HSS T3 guard column. The mobile phase components consisted of (A) 3.7% acetic acid in water with 0.01% heptafluorobutyric acid (HFBA) and (B) 100% methanol. The gradient profile used is as detailed in Table S-1, and the flow rate was 0.4 mL/min. A representative chromatogram can be found in Figure S-1. The precursor and product ions used for the detection of PLP, PNP, PMP, PL, PN, PM, PA, d3-PLP, d2-PN, d3-PM, and d2-PA can be found in Table S-2. The retention times observed as well as the cone voltages and collision energies used for the analysis of these compounds are also detailed in Table S-2. The mass spectrometry settings were as follows: capillary voltage, 2.50 kV; source temperature, 150 °C; desolvation temperature, 600 °C; cone gas flow rate, 150 L/h; desolvation gas flow rate, 1200 L/h; dwell time, 17 ms. Data collection and analysis was performed using Waters MassLynx software.
PNPO activity was calculated by subtracting the endogenous PLP measured at 0 min in the T0 DBS punch from the amount of PLP present after 30 min of incubation of the DBS with PNP substrate in the T30 DBS punch. The enzyme activity was calculated and reported as pmol PLP (3 mm DBS)−1 h–1. To make comparisons with published data, we made the assumption that a 3 mm DBS punch contains 3.2 μL of blood,23 and a conversion factor of 0.3125 was used to convert this activity to pmol PLP μL blood–1 h–1. Although not used to quantify PNPO activity, concentrations of PNP were routinely quantified. During assay optimization, PMP, PL, PN, PM, and PA were also quantified. Despite having similar retention times, all analytes were well differentiated, and there was no cross talk between ion pairs except at high PNP concentrations as discussed below. Calibration curves from 1.25 to 200 nmol/L were constructed using the analyte:internal standard peak area ratios of PLP, PNP, and PMP:d3-PLP and were linear over this range (r2 > 0.99). A calibration curve, over the same range, was constructed for PM:d3-PM, PA:d2-PA, and PN:d2-PN; these were also found to be linear (r2 > 0.99). Pyridoxal was not quantified using a calibration curve or internal standard as measurement was only performed in early stages of method development. The area under the curve (AUC) upon LC–MS/MS elution was thus used to quantify pyridoxal levels.
Statistical Analysis
All analyses were carried out using GraphPad Prism version 6.05 for Windows (GraphPad Software, La Jolla, CA). Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test; ** = P < 0.01; **** = P < 0.0001.
Results and Discussion
PNPO Assay Optimization
The DBS assay was evaluated and optimized with regards to pH, buffer, incubation time, and cofactor/substrate concentrations.
PNPO enzyme activity in erythrocytes has been reported to be optimal at pH’s between pH 7.1 and 8.0.13,15 The pH dependence of the enzyme in the DBS was verified over this range, and it was found that there was little difference in PNPO activity when a 3 mm DBS from an adult control was incubated at pH’s at or between pH 7–8 (Table S-3). Analysis of the chromatogram of samples incubated at pHs > 7.8, however, revealed peak-splitting of PLP. Therefore, all subsequent incubation conditions were optimized while keeping the pH constant at 7.6.
The effects of different biological buffers on PNPO activity that were capable of buffering at pH 7–8 were investigated. A Tris-HCl buffer system, similar to that which we have used previously for analysis of PNPO activity in Chinese Hamster ovary cells overexpressing human PNPO,2 was considered initially. PNPO is regulated by feedback inhibition of its product, PLP, and because Tris complexes with PLP it would therefore be expected to minimize any such inhibition.25 However, it was evident that under the conditions used, approximately 12% of the PLP formed was subsequently hydrolyzed to PL (data not shown) presumably by the activity of multiple endogenous phosphatases that are present in blood, which hydrolyze the 5′ ester bond of PLP. Inorganic phosphate is known to be a product inhibitor of phosphatase activity;15,26 therefore, a custom 20 mmol/L Tris-phosphate buffer (pH 7.6) was adopted, and at the concentration of phosphate present negligible PLP hydrolysis was observed (Figure S-2). There was additional concern, particularly for the analysis of DBS from individuals on high-dose supplementation where the levels of B6 vitamers would be elevated, that any endogenous pyridoxal kinase activity present in the DBS would act on endogenous PL levels to form PLP and that PLP measured in the T30 DBS would not be solely representative of PNPO activity but of combined PNPO and pyridoxal kinase activity. Previous studies have shown, however, that pyridoxal kinase requires the presence of ATP and a divalent cation (e.g., Mg2+) for activity,27,28 cofactors that are not added to the PNPO reaction buffer. We have confirmed these findings in our DBS system. In the absence of exogenous ATP and Mg2+, PN phosphorylation to PNP was undetectable (Figure S-3).
PNP was chosen as a substrate for the assay rather than PMP because oxidation of PNP to PLP, under the conditions employed, was much more rapid than that observed when PMP was used as a substrate. The Vmax of PNP conversion to PLP upon the incubation of a 3 mm adult control DBS was found to be approximately twice that of PMP to PLP (39.5 vs 19.8 pmol DBS–1 h–1). Equally, the Km for PNP was 0.32 μmol/L as compared to 0.53 μmol/L for PMP (Figure S-4). The conversion of PMP (400 nmol/L) to PLP, when incubated for 30 min with an adult control DBS, was only 14.1% of that observed when the same concentration of PNP was used as a substrate (Figure S-5). In addition, the PMP standard utilized was found to contain a not insignificant (1–2%) proportion of PLP. This background of PLP could interfere with measurement of endogenous PLP in the DBS and lead to feedback inhibition.
A concentration of 400 nmol/L PNP was used for the assay for several reasons: (i) This concentration of PNP permitted analysis of the resulting supernatant by UPLC–MS/MS without further dilution steps. (ii) Upon the investigation of higher PNP concentrations for the determination of Michaelis–Menten kinetics, it was noted that there was a small amount of cross talk between the MRM channels for PNP and d3-PLP (<1%). Although at a concentration of 200 nmol/L upon analysis (as in the T0 repeats of the assay in its current form) the effect was negligible, at higher PNP concentrations (>1 μmol/L) interference was unacceptable and could affect results through aberrant PLP quantification. (iii) Saturation of the enzyme with substrate will not facilitate detection of PNPO deficiency in patients with mutations that cause an alteration in the Km of PNPO where the enzyme has residual activity and the Vmax and Kcat are relatively unaffected. By mildly limiting the substrate concentration but still achieving easily measurable conversion in controls, it is likely that the diagnosis of PNPO deficiency in patients with “milder” mutations is enabled.
There were concerns that the concentration of FMN, the cofactor for PNPO, in DBS samples may be dependent upon the vitamin B2 status of each subject. Indeed, activity in an adult control measured in the presence of endogenous FMN was only 61% of that seen when 1.5 μmol/L FMN was added to the reaction buffer, a concentration that we have used previously when studying PNPO activity in an overexpression system.2 To ensure that the concentration of FMN used in the assay was not limiting the activity of PNPO, punches from DBS collected from the same control subject were incubated with varying concentrations of FMN. No significant difference in PNPO activity was observed when 0.75–3 μmol/L FMN was included in the reaction buffer (Figure S-5). A cofactor concentration of 1.5 μmol/L was used for all subsequent experimentation.
PNPO activity as a function of the amount of enzyme was established by incubating the substrate with an increasing weight of dried blood spot. Initial experiments showed that it was possible to accurately quantify PNPO activity using a 3 mm DBS punch, the weight of which is approximately 26 mg. PLP production was measured in the presence of varying amounts of DBS (0–80 mg) prepared using blood from a healthy adult volunteer. An approximately linear correlation was observed to 44 mg (Figure S-6). No PLP formation was observed when a 3 mm punch from a blank Whatman 903 card was incubated for 30 min under the final assay conditions; hence the enzyme activity in the DBS samples was not corrected.
The linearity of PLP production from PNP, under the conditions described above, was monitored in two healthy adult controls and one child hospital control (age 7 months) over 120 min. Product formation increased linearly for the first 60 min in all controls (Figure S-7). It was decided that an incubation time of 30 min would be used for all analyses as this provided adequate PLP formation for accurate quantification by LC-MS/MS while keeping the incubation time as short as possible to increase throughput in a clinical diagnostic arena. It also ensured linearity of the assay in subjects with particularly high PNPO activity.
Imprecision and Limits of Detection and Quantification
To validate assay precision, PNPO activity was measured in DBS from one healthy adult control. The intra-assay precision was 7.9% and was determined by measuring the PNPO activity in 10 different DBS punches from the same individual on 1 day. Interassay variability was determined by measuring enzyme activity in 10 different DBS punches from the same individual 5 times over a 4 week period. The observed interassay variability was 10.3%.
For quantification of PLP and PNP, the lower and upper limits of quantification (LLOQ and ULOQ) were set at the lower and upper bounds of the calibration curve created. This corresponds to concentrations of 1.25 and 200 nmol/L, respectively. At LLOQ, the signal/noise (S/N) ratio was greater than 10. The limit of detection (S/N greater than 3) was therefore lower than 1.25 nmol/L. It was ensured that, when back-calculated, the values at LLOQ and ULOQ never deviated from predetermined criteria specified by the European Medicines Agency analytical consideration guidelines (<20% imprecision for LLOQ and <15% for ULOQ). QC standards of PLP and PNP were analyzed alongside each LC–MS/MS run to determine precision of measurement. The %CVs of five repeated injections of QC standards of 5, 10, 100, and 175 nmol/L were 2.28%, 0.90%, 2.30%, and 3.45%, respectively.
While the concentration of endogenous PLP levels was below the limits of quantification (<1.25 nmol/L in the reaction/stop mixture, equivalent to 94 nmol/L whole blood) in controls not on B6 supplementation, in individuals on supplementation the concentration of endogenous PLP in the reaction/stop mixture ranged from 2.5–199 nmol/L. Accuracy of the extraction method of PLP from DBS was investigated to ensure that a high endogenous concentration of PLP in the T0 punch would not lead to discrepancies in measured PNPO activity. The intra and interassay %CVs for the extraction of endogenous PLP in a DBS card collected from a PNPO-deficient patient on supra-physiological doses (42 mg/kg/day) of B6 were 5.92 and 7.76, respectively.
Effect of the Presence of Supra-physiological Concentrations of B6 Vitamers on Pyridox(am)ine 5′-Phosphate Oxidase Activity
While DBS were incubated in a Tris-based buffer system to minimize any feedback inhibition that PLP may have on PNPO activity, there was concern that PLP present in DBS from patients on supra-physiological doses may inhibit PNPO activity. DBS from a healthy adult not on supplementation were incubated with increasing concentrations of PLP added to the reaction buffer (0–400 nmol/L) corresponding to concentrations of 0–200 nmol/L in the reaction/stop mixture after TCA precipitation (Table S-4). The PLP concentrations present in the reaction/stop mixture observed in controls and PNPO patients on B6 supplementation were 2.5–199 nmol/L. No product inhibition was seen in the presence of PLP concentrations ≤50 nmol/L. Product inhibition was observed at concentrations higher than this with a decrease in PNPO activity by 15–20% in the presence of 100–150 nmol/L PLP. The majority of patients analyzed receiving B6 supplements (13/18 PNPO; 14/16 others) had concentrations of PLP < 100 nmol/L in the T0 DBS reaction/stop mixture (data not shown).
PNPO-deficient patients have been reported to have high concentrations of PMP, and sometimes PNP, in plasma.21 Concentrations of endogenous PNP and PMP were therefore measured in the T0 DBS punches so that we could evaluate the effect of these vitamers on PNPO activity. In control samples from individuals not receiving/on high dose B6 supplementation (that were not PNPO deficient), the concentrations of PNP and PMP in the reaction/stop mixture were below the LLOQ (1.25 nmol/L). As compared to the concentration of PNP added to the assay (200 nmol/L), this is negligible. The concentration of PNP and PMP in the reaction/stop mixture of PNPO-deficient patients ranged from 0–137 and 0–121 nmol/L, respectively. The total concentration of substrate present in the reaction mixture (i.e., endogenous and added) of some PNPO-deficient patients will be higher therefore than that of control samples. However, as mentioned above, our data show that the rate of oxidation of PMP using the assay conditions employed was far lower than that of PNP. When a 3 mm DBS from an adult control was incubated with 400 nmol/L PMP in addition to 400 nmol/L PNP (equivalent to 200 nmol/L upon analysis of the reaction/stop mixture), a small decrease in PLP formation was identified; however, this was not statistically significant (Figure S-5). PMP is not thought to inhibit PNPO activity;25 therefore, high endogenous PMP levels should not significantly affect results.
Pyridox(am)ine 5′-Phosphate Oxidase Activity in Dried Blood Spots Following Storage
Stability studies were performed using DBS from a healthy adult control. Venous blood was collected and immediately spotted onto Whatman 903 Protein Saver cards. PNPO stability was evaluated after storage of the card at various temperatures. Previous studies have shown that ambient humidity can affect enzyme stability;29−31 hence, duplicate cards were stored, one without desiccant and the other in a sealed foil bag with desiccant after drying for 16–24 h. Enzyme activities observed after storage were compared to the PNPO activity present in a DBS after the 16–24 h drying period.
DBS collected for short-term stability studies were stored in the dark at 22 °C for 3, 5, 7, 14, and 28 days with/without desiccant (Figure 2). All cards were then stored with desiccant at −80 °C before being assayed on day 28. When stored at room temperature without desiccant for 3 days, a residual activity of 83% was seen. This decreased to 52% over the 28-day period studied. As expected, the decrease in activity was mitigated when DBS were stored with desiccant in sealed foil bags after the initial 16–24 h drying period. Residual activity under these conditions was 92% and 74% after 3 and 28 days, respectively.
Figure 2.

Effect of humidity on the short-term stability of the PNPO enzyme in dried blood spots. Error bars indicate SEM. DBS were stored in sealed foil bags with desiccant (▲) or under ambient conditions (■) at room temperature (22 °C). Data points represent the mean percentage of activity as compared to that found after 1 day (n = 2).
Long-term stability of the enzyme in DBS was determined after storage for 1, 4, 8, and 12 weeks at 4, −20, and −80 °C, again after an initial 16–24 h drying period at 22 °C (Figure 3). After storage for 12 weeks, a reduction in activity of 27% was measured in those spots stored at 4 °C. In DBS stored at −20 and −80 °C, activity was 92.7% and 87.0%, respectively, as compared to baseline levels after 12 weeks. Under these conditions, minimal difference was seen in the activity of the enzyme after storage of the DBS without desiccant.
Figure 3.

Effect of temperature (4 °C [●], −20 °C [■], and −80 °C [▲]) on the long-term stability of the PNPO enzyme in dried blood spots (DBS). Data points represent the mean percentage of PNPO activity relative to measurement at time zero. Error bars indicate SEM. No significant difference was seen in activities of samples with/without desiccant; the data from DBS stored both with (n = 2) and without (n = 2) desiccant at each temperature have therefore been combined (n = 4).
Analysis of Clinical Samples
PNPO enzyme activity was measured in DBS from 37 child hospital controls (age range 5 days–15 years) and 7 healthy adult controls. These individuals did not have seizures. Sixteen samples were also collected from children with other seizure disorders receiving vitamin B6 supplementation in whom either PNPO deficiency had been excluded by Sanger sequencing or another conclusive genetic diagnosis had been made; this included 2 individuals with mutations in ALDH7A1, 1 subject with PROSC deficiency,32 7 subjects with other genetic seizure disorders, and 6 individuals in whom the etiological basis of their seizures is unknown (age range 1 month–16 years). Eighteen samples were collected from subjects (age range 1 day–25 years) with known variants in the PNPO gene (Table S-5). Three of these samples were from individuals homozygous for the PNPO variant p.R116Q. We were interested in looking at the effect of p.R116Q as it is predicted to affect FMN binding and binding of pyridox(am)ine 5′-phosphate oxidase monomers to form the dimeric form of the enzyme; however, previous in vitro studies have suggested that this variant has high residual activity and indeed it is present in the ExAC database with an allele frequency of 0.0558, suggesting it could be a polymorphism.33 We have concluded previously,2 because of its higher incidence in B6-responsive epilepsy patients relative to the general population, that it contributes to the pathogenesis of epilepsy and that it may be a common mutation, perhaps responsible for the susceptibility locus for genetic generalized epilepsy on 17q21.32 (close to rs72823592).34 Two of the individuals homozygous for p.R116Q (subjects 3 and 13) receive B6 for the treatment of their epilepsy; however, subject 4, the sibling of subject 3, has never had a seizure and is not on B6 supplementation. DBS from the parents of the two siblings homozygous for p.R116Q were also analyzed. All DBS samples were transferred into sealed foil bags after drying at room temperature for 16–24 h after collection. They were kept for a maximum period of 1 week at room temperature prior to storing at −20 °C or temperatures lower than this.
The mean activity of PNPO in DBS samples from hospital controls was 41.7 pmol DBS–1 h–1 (range: 10.0–95.0 pmol DBS–1 h–1). The activity of PNPO-deficient patients (range: 0.0–4.6 pmol DBS–1 h–1; mean 1.1 pmol DBS–1 h–1) was significantly lower than the controls (p < 0.0001). This PNPO-deficient cohort includes the 3 samples from the individuals homozygous for the p.R116Q variant in PNPO. The range for patients with other seizure disorders receiving vitamin B6 supplementation, similar to that of the hospital controls, was significantly higher than that of the PNPO-deficient patients (23.0–85.9 pmol DBS–1 h–1; mean = 56.0 pmol DBS–1 h–1). Seven healthy adult controls displayed activity from 13.8–44.0 pmol DBS–1 h–1 with a mean of 28.3 pmol DBS–1 h–1. The p.R116Q heterozygous parents of subjects 3 and 4 had intermediate PNPO activities of 7.1 and 10.8 pmol DBS–1 h–1 (Figure 4). Patients with mutations in PNPO could be clearly delineated from all of the control groups with the lowest control having a PNPO activity more than 2-fold greater than that of the PNPO patient with highest residual activity. Differences between the groups were shown to be statistically significant (P values shown in Figure 4). No false positives were identified. The large range of PNPO activity seen in controls could be representative of population differences reported previously.13,14 There was no direct correlation of PNPO activity with age in the samples from control individuals not receiving B6 supplementation (age range 5 days–15 years and adult samples). In the future, it will be important to expand this age range and measure PNPO activity in DBS of babies born prematurely as Kang et al.35 have reported that fetal expression of PNPO is relatively low as compared to that of adults. The mean PNPO activity in 5 control neonates (<1 month of age) not receiving B6 supplementation measured in this study was 52.7 pmol DBS–1 h–1 (range 41.3–66.3 pmol DBS–1 h–1). This is not significantly different from the PNPO activity seen in controls (>1 month of age) irrespective of whether they are receiving B6 supplementation or not. The youngest neonate (5 days of age) had PNPO activity of 64.3 pmol DBS–1 h–1.
Figure 4.

DBS PNPO enzyme activity in PNPO-deficient patients and control samples. Box plots indicate range, interquartile range, and median. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparisons test; ** = P < 0.01; **** = P < 0.0001.
The results obtained using the DBS assay were compared to those from previous assays of erythrocyte PNPO activity described in the literature. Anderson et al.14 reported mean PNPO activity levels of approximately 10 nmol PLP gHb–1 h–1, and similar activity levels were reported more recently by Mushtaq et al.36 To compare our results directly, two assumptions had to be made: (1) a 3 mm punch from a DBS contains 3.2 μL of whole blood, and (2) the mean hemoglobin level of our sample cohort was 14 gHb/dL (hemoglobin levels were not available for our subjects). If these assumptions are accepted, a conversion factor of 2.2 can be used to convert pmol PLP DBS–1 h–1 to nmol PLP gHb–1 h–1. This gave mean PNPO activity levels of 62.2 and 91.8 nmol gHb–1 h–1 in our adult and child control cohorts, respectively (those not receiving B6 supplementation). Although these values are significantly higher than those reported previously, there are major differences in methodology that can account for this difference. First, we have used PNP rather than PMP as a substrate. As mentioned previously, the oxidation of PMP to PLP under the conditions used is only 14.1% of that observed when PNP is used as a substrate, that is, 7-fold less. Equally, we have shown that upon use of PNP as substrate, a lower Km and higher Vmax are identified, when compared to PMP. Second, methods previously published have not added FMN to their assay and rely on endogenous FMN present in the samples, and both our results and previous work13 have shown that addition of FMN increases PNPO activity in vitro.
Recently, Mathis et al.10 have shown that a raised plasma PM/PA ratio can be used as a diagnostic marker of PNPO deficiency. PM/PA ratios in dried blood spots of 18 PNPO deficient subjects and 13 individuals from the cohort of controls receiving B6 supplementation were measured, although it was not possible to ascertain recovery of these vitamers due to interconversion in spiked whole blood by endogenous enzymes. Ratios were found to range from 0.00–0.95 (mean 0.28) and from 0.00–0.74 (mean 0.09) in subjects with mutations in PNPO and controls receiving B6 supplementation, respectively (Figure S-8). Although the PM/PA ratios in patients with mutations in PNPO were significantly higher (p = 0.0059), it was not possible to use the PM/PA ratio in dried blood spots to differentiate PNPO-deficient individuals from controls receiving B6 for seizure control with certainty.
There are several factors that may explain the different findings between the two studies. Our study (n = 18 PNPO patients) has investigated the ratio in DBS, while that of Mathis et al.10 (n = 6) studied plasma. Plasma samples were not available in our study. Comparison of whole blood and plasma B6 vitamer concentrations will be important in the future. The PNPO genotype may also be influential, as PNPO-deficient patients investigated previously were homozygous for either the c.674G > A (n = 4), c.263 + 2T > C (n = 1), and c.416A > C (n = 1). In this study, we have studied the ratios of subjects with 14 different genotypes (Table S-5), only one of which was studied previously (c.263 + 2T > C; subject 18). The individual with this genotype was found to have a raised PM/PA ratio in both studies. Equally, in those patients for whom supplementation type was known, a larger proportion of our subjects were receiving PLP monotherapy for the treatment of their seizures as opposed to PN: 19/23 overall, 14/17 PNPO deficient versus 2/37 overall, and 2/6 PNPO deficient in Mathis et al.10 Type of B6 supplementation and dosage could be important if using PM/PA ratios for diagnosis of PNPO deficiency.
The three subjects (3, 4, and 13) homozygous for the p.R116Q variant had extremely low PNPO activities (<0.2 pmol DBS–1 h–1), while the two parents who were heterozygous for the p.R116Q PNPO variant had intermediate PNPO activities of 7.1 and 10.8 pmol DBS–1 h–1. This contrasts to our previous findings where variants had been investigated in a human in vitro HeLa cell lysate expression system, in which p.R116Q was shown to result in 83% activity as compared to wild-type PNPO activity.2 We have previously suggested that p.R116Q contributes to the pathogenesis of epilepsy. The results of the DBS assay confirm this. The difference observed between the two systems with regard to this variant may be due to how PNPO utilizes PNP as a substrate, as we have used in this DBS assay, rather than PMP, as used in the in vitro overexpressed system. Indeed, a patient homozygous for p.R116Q has previously been reported to have a normal plasma PM/PA ratio.10 In this study, 2/3 subjects homozygous for p.R116Q also had low DBS PM/PA ratios. Why subject 4, the sibling homozygous for this variant, exhibits negligible activity and has not presented with seizures has yet to be explained. It may be that there are other environmental or genetic factors that are implicated. The mother of this subject received multivitamin tablets containing PN during pregnancy, and it is possible that this increased the PLP and PL in her plasma/breast milk to concentrations high enough to provide adequate intracranial PLP concentrations in the subject as a fetus/neonate. Accordingly, it is also possible that a “second hit” is required at another locus for an epileptic phenotype to become apparent. Further work will be needed to clarify this and to investigate the effects of p.R116Q on PNPO activity.
There have been reports of liver dysfunction in individuals receiving high doses (>30 mg kg–1 d–1) of PLP for the treatment of seizures. In particular, this has been noted in PNPO-deficient patients.37,38 Transiently raised liver function tests have also been reported in 14/28 patients with infantile spasms when receiving 30–50 mg kg–1 d–1 PLP.39 A clear mechanism behind this pathology has yet to be elucidated, but it would be prudent to minimize patient exposure to high PLP doses where possible.40 This DBS assay can identify individuals receiving PLP supplementation in whom PNPO activity is normal and could therefore help to assess which patients would be more likely to tolerate a switch to PN supplementation while maintaining seizure control. This would help to eliminate potential liver damage in these individuals. An alternative treatment approach could be pyridoxal supplementation; however, this has not been tested clinically.
This DBS assay will also be useful for the diagnosis of patients in whom only one heterozygous pathogenic variant has been identified in the PNPO gene upon molecular genetic investigation. Subject 6 is heterozygous for the PNPO variant c.[641dupA]; however, no other pathogenic variant has been identified. They had undetectable PNPO activity using our DBS assay, confirming subject 6 is PNPO deficient.
Currently, in patients with neonatal/infantile seizures, if CSF analysis shows alterations in neurotransmitter amine metabolites (e.g., raised 3-O-methyldopa; low homovanillic acid, 5-hydroxyindoleacetic acid) or amino acids (e.g., raised glycine, threonine) indicative of PLP deficiency or low CSF PLP, urinary α-aminoadipic semialdehyde (α-AASA) is measured to exclude ALDH7A1 deficiency. This DBS assay could, in combination with the clinical presentation of these individuals, be used for the simultaneous assessment of PNPO activity alongside urinary α-AASA measurement. This will aid the development of protocols for the rapid diagnosis and treatment of neonatal seizures.
Conclusions
We have developed a novel LC–MS/MS-based assay for the determination of PNPO activity, which would be suitable for translation into the clinical diagnostic setting. The method is simple, robust, and clearly separates PNPO-deficient patients from individuals with normal PNPO activity. It does not require prior enzyme extraction from the DBS nor derivatization, requires minimal sample preparation, and can be performed in 1.5 h, followed by a 6.5 min LC–MS/MS analytical run. All previously published assays for measuring PNPO activity have not been suitable for the clinical setting and often require large sample volumes not possible to obtain from severely ill neonates. This new rapid DBS PNPO assay will now allow clinicians to distinguish PNPO deficiency from other disorders, potentially in combination with the plasma or DBS PM/PA ratio. This will be a powerful tool for clinicians to help diagnose these patients.
Acknowledgments
We would like to thank Dr. F. Feillet, Dr. O. Maier, Dr. N. Gayatri, Dr. M. Taylor, Dr. H. Mundy, Dr. E. Del Giudice, Dr. L. Delfiner, Dr. C. Freihuber, Dr. P. Bala, Dr. Livingston, and Dr. I. Kern for their involvement in the study and Dr. E. S. Reid for her technical help and support. M.P.W., P.T.C., and P.B.M. are supported by Great Ormond Street Children’s Charity (GOSHCC). This project was funded by GOSHCC. P.B.M., P.T.C., P.G., and S.J.R.H. are supported by the National Institute for Health Research Biomedical Research Centre at GOSH for Children NHS Foundation Trust and University College London. P.G. was supported by a Wellcome Trust Senior Clinical Fellowship (WT095662MA). We are indebted to the patients and their families for participating in this study.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01358.
Tables and figures showing experimental data collected during optimization of enzyme assay conditions as well as more detailed information on the PNPO-deficient subjects analyzed during method development (PDF)
Author Present Address
○ C.G.: Dana Farber Cancer Institute, 440 Brookline Avenue, Boston, Massachusetts 02215, United States.
Author Contributions
△ P.T.C. and P.B.M. contributed equally. M.P.W. undertook all laboratory analyses supervised principally by P.B.M. P.T.C., S.J.R.H., K.A.M., and P.G. contributed valuable intellectual support and discussion and were involved in experimental design. E.J.F., A.P., M.-L.U., R.P., D.C.S., C.G., T.S., K.R., M.S., M.B., and D.B. obtained patient samples and supplied clinical details. The manuscript was written by M.P.W., P.T.C., and P.B.M. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Clayton P. T. J. Inherited Metab. Dis. 2006, 29, 317–326. 10.1007/s10545-005-0243-2. [DOI] [PubMed] [Google Scholar]
- Mills P. B.; Camuzeaux S. S.; Footitt E. J.; Mills K. A.; Gissen P.; Fisher L.; Das K. B.; Varadkar S. M.; Zuberi S.; McWilliam R.; Stodberg T.; Plecko B.; Baumgartner M. R.; Maier O.; Calvert S.; Riney K.; Wolf N. I.; Livingston J. H.; Bala P.; Morel C. F.; Feillet F.; Raimondi F.; Del Giudice E.; Chong W. K.; Pitt M.; Clayton P. T. Brain 2014, 137, 1350–1360. 10.1093/brain/awu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plecko B.; Paul K.; Mills P.; Clayton P.; Paschke E.; Maier O.; Hasselmann O.; Schmiedel G.; Kanz S.; Connolly M.; Wolf N.; Struys E.; Stockler S.; Abela L.; Hofer D. Neurology 2014, 82, 1425–1433. 10.1212/WNL.0000000000000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey A. D.; McMahon R. J.; Townsend J. H.; Gregory J. F. III. J. Nutr. 2004, 134, 842–846. [DOI] [PubMed] [Google Scholar]
- Mills P. B.; Surtees R. A.; Champion M. P.; Beesley C. E.; Dalton N.; Scambler P. J.; Heales S. J.; Briddon A.; Scheimberg I.; Hoffmann G. F.; Zschocke J.; Clayton P. T. Hum. Mol. Genet. 2005, 14, 1077–1086. 10.1093/hmg/ddi120. [DOI] [PubMed] [Google Scholar]
- Guerin A.; Aziz A. S.; Mutch C.; Lewis J.; Go C. Y.; Mercimek-Mahmutoglu S. J. Child Neurol 2015, 30, 1218–1225. 10.1177/0883073814550829. [DOI] [PubMed] [Google Scholar]
- van Karnebeek C. D.; Tiebout S. A.; Niermeijer J.; Poll-The B. T.; Ghani A.; Coughlin C. R. 2nd; Van Hove J. L.; Richter J. W.; Christen H. J.; Gallagher R.; Hartmann H.; Stockler-Ipsiroglu S. Pediatric Neurology 2016, 59, 6–12. 10.1016/j.pediatrneurol.2015.12.013. [DOI] [PubMed] [Google Scholar]
- Hatch J.; Coman D.; Clayton P.; Mills P.; Calvert S.; Webster R. I.; Riney K. JIMD Rep 2015, 26, 91–97. 10.1007/8904_2015_482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl P. L.; Gospe S. M. Jr. Neurology 2014, 82, 1392–1394. 10.1212/WNL.0000000000000351. [DOI] [PubMed] [Google Scholar]
- Mathis D.; Abela L.; Albersen M.; Burer C.; Crowther L.; Beese K.; Hartmann H.; Bok L. A.; Struys E.; Papuc S. M.; Rauch A.; Hersberger M.; Verhoeven-Duif N. M.; Plecko B. J. Inherited Metab. Dis. 2016, 39 (5), 733–741. 10.1007/s10545-016-9955-8. [DOI] [PubMed] [Google Scholar]
- Rodan L. H.; Gibson K. M.; Pearl P. L. Pediatric Neurology 2015, 53, 277–286. 10.1016/j.pediatrneurol.2015.04.016. [DOI] [PubMed] [Google Scholar]
- Ng J.; Papandreou A.; Heales S. J.; Kurian M. A. Nat. Rev. Neurol. 2015, 11, 567–584. 10.1038/nrneurol.2015.172. [DOI] [PubMed] [Google Scholar]
- Clements J. E.; Anderson B. B. Biochim. Biophys. Acta 1980, 613, 401–409. 10.1016/0005-2744(80)90094-7. [DOI] [PubMed] [Google Scholar]
- Anderson B. B.; Giuberti M.; Perry G. M.; Salsini G.; Casadio I.; Vullo C. Am. J. Clin. Nutr. 1993, 57, 666–672. [DOI] [PubMed] [Google Scholar]
- Ubbink J. B.; Schnell A. M. J. Chromatogr., Biomed. Appl. 1988, 431, 406–412. 10.1016/S0378-4347(00)83110-2. [DOI] [PubMed] [Google Scholar]
- Bates C. J.; Powers H. J. Hum. Nutr. Clin. Nutr. 1985, 39, 107–115. [PubMed] [Google Scholar]
- DePecol M. E.; McCormick D. B. Anal. Biochem. 1980, 101, 435–441. 10.1016/0003-2697(80)90210-9. [DOI] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Wang E. Methods Enzymol. 1986, 122, 110–116. 10.1016/0076-6879(86)22156-4. [DOI] [PubMed] [Google Scholar]
- Guthrie R.; Susi A. Pediatrics 1963, 32, 338–343. [PubMed] [Google Scholar]
- Ombrone D.; Giocaliere E.; Forni G.; Malvagia S.; la Marca G. Mass Spectrom. Rev. 2016, 35, 71–84. 10.1002/mas.21463. [DOI] [PubMed] [Google Scholar]
- Footitt E. J.; Clayton P. T.; Mills K.; Heales S. J.; Neergheen V.; Oppenheim M.; Mills P. B. J. Inherited Metab. Dis. 2013, 36, 139–145. 10.1007/s10545-012-9493-y. [DOI] [PubMed] [Google Scholar]
- Reiber H. Biochim. Biophys. Acta, Gen. Subj. 1972, 279, 310–315. 10.1016/0304-4165(72)90148-1. [DOI] [PubMed] [Google Scholar]
- Saidi B.; Warthesen J. J. J. Agric. Food Chem. 1983, 31, 876–880. 10.1021/jf00118a051. [DOI] [Google Scholar]
- Shephard G. S.; Labadarios D. Clin. Chim. Acta 1986, 160, 307–311. 10.1016/0009-8981(86)90198-1. [DOI] [PubMed] [Google Scholar]
- Choi J. D.; Bowers-Komro M.; Davis M. D.; Edmondson D. E.; McCormick D. B. J. Biol. Chem. 1983, 258, 840–845. [PubMed] [Google Scholar]
- Jang Y. M.; Kim D. W.; Kang T. C.; Won M. H.; Baek N. I.; Moon B. J.; Choi S. Y.; Kwon O. S. J. Biol. Chem. 2003, 278, 50040–50046. 10.1074/jbc.M309619200. [DOI] [PubMed] [Google Scholar]
- Laine-Cessac P.; Allain P. Enzyme Protein 1996, 49, 291–304. [DOI] [PubMed] [Google Scholar]
- Chern C. J.; Beutler E. Am. J. Hum. Genet. 1976, 28, 9–17. [PMC free article] [PubMed] [Google Scholar]
- Adam B. W.; Hall E. M.; Sternberg M.; Lim T. H.; Flores S. R.; O’Brien S.; Simms D.; Li L. X.; De Jesus V. R.; Hannon W. H. Clin. Biochem. 2011, 44, 1445–1450. 10.1016/j.clinbiochem.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe B. J.; Blanchard S.; Sadilek M.; Scott C. R.; Turecek F.; Gelb M. H. Anal. Chem. 2011, 83, 1152–1156. 10.1021/ac102777s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcenas M.; Xue C.; Marushchak-Vlaskin T.; Scott C. R.; Gelb M. H.; Turecek F. Anal. Chem. 2014, 86, 7962–7968. 10.1021/ac501994b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darin N.; Reid E.; Prunetti L.; Samuelsson L.; Husain R. A.; Wilson M.; El Yacoubi B.; Footitt E.; Chong W. K.; Wilson L. C.; Prunty H.; Pope S.; Heales S.; Lascelles K.; Champion M.; Wassmer E.; Veggiotti P.; de Crecy-Lagard V.; Mills P. B.; Clayton P. T. Am. J. Hum. Genet. 2016, 99, 1325–1337. 10.1016/j.ajhg.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M.; Karczewski K. J.; Minikel E. V.; Samocha K. E.; Banks E.; Fennell T.; O’Donnell-Luria A. H.; Ware J. S.; Hill A. J.; Cummings B. B.; et al. Nature 2016, 536, 285–291. 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens M.; Leu C.; Ruppert A. K.; Zara F.; Striano P.; Robbiano A.; Capovilla G.; Tinuper P.; Gambardella A.; Bianchi A.; et al. Hum. Mol. Genet. 2012, 21, 5359–5372. 10.1093/hmg/dds373. [DOI] [PubMed] [Google Scholar]
- Kang J. H.; Hong M. L.; Kim D. W.; Park J.; Kang T. C.; Won M. H.; Baek N. I.; Moon B. J.; Choi S. Y.; Kwon O. S. Eur. J. Biochem. 2004, 271, 2452–2461. 10.1111/j.1432-1033.2004.04175.x. [DOI] [PubMed] [Google Scholar]
- Mushtaq S.; Su H.; Hill M. H.; Powers H. J. Am. J. Clin. Nutr. 2009, 90, 1151–1159.F. 10.3945/ajcn.2009.28338. [DOI] [PubMed] [Google Scholar]
- Sudarsanam A.; Singh H.; Wilcken B.; Stormon M.; Arbuckle S.; Schmitt B.; Clayton P.; Earl J.; Webster R. JIMD Rep 2014, 17, 67–70. 10.1007/8904_2014_338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coman D.; Lewindon P.; Clayton P.; Riney K. JIMD Rep 2015, 25, 71–75. 10.1007/8904_2015_456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma Y.; Seki T. J. Child Neurol 1996, 11, 35–40. 10.1177/088307389601100109. [DOI] [PubMed] [Google Scholar]
- Mohamed-Ahmed A. H.; Wilson M. P.; Albuera M.; Chen T.; Mills P. B.; Footitt E. J.; Clayton P. T.; Tuleu C. J. Pharm. Pharmacol. 2017, 69 (4), 480–488. 10.1111/jphp.12701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.