

Abstract
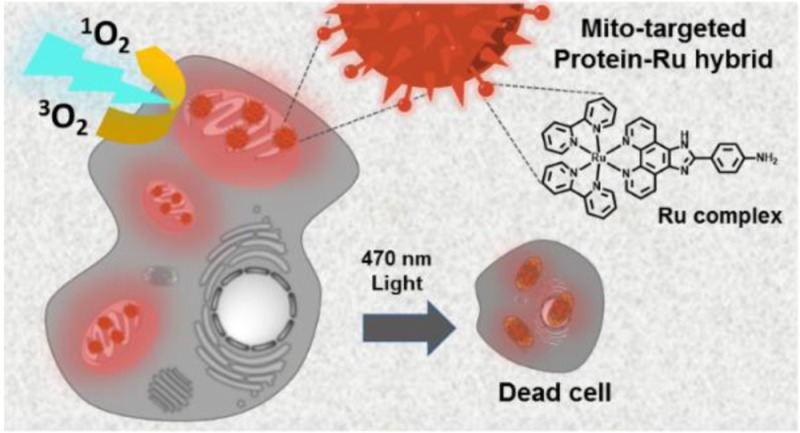
Organelle-targeted photosensitization represents a promising approach in photodynamic therapy where the design of the active photosensitizer (PS) is very crucial. In this work, we developed a macromolecular PS with multiple copies of mitochondria-targeting groups and ruthenium complexes that displays highest phototoxicity toward several cancerous cell lines. In particular, enhanced anticancer activity was demonstrated in acute myeloid leukemia cell lines, where significant impairment of proliferation and clonogenicity occurs. Finally, attractive two-photon absorbing properties further underlined the great significance of this PS for mitochondria targeted PDT applications in deep tissue cancer therapy.
Introduction
Singlet oxygen (1O2), the lowest-lying electronic excited states of molecular oxygen, has been envisioned as promising and highly effective cytotoxic agent in photochemical and photobiological research.1,2 Photodynamic therapy (PDT) emerges as a promising tool in organelle-directed, photoactivated and less-invasive medical technique3 for bacterial inactivation and regenerative medicine,4 where the production of reactive 1O2 induces cytotoxicity in the targeted region, leaving the surrounding biological environment undamaged. 1O2 is produced when energy transfer occurs between the triplet excited state of the photosensitizers (PS) and the ground state of molecular oxygen.5 However, the application of most synthesized PS molecules in biological media is limited by their low water-solubility resulting in extensive aggregate formation and consequently reduced quantum yields.6 In addition, poor selectivity in terms of target tissue and low extinction coefficients have reduced the efficiency of PDT in clinical trials.7 Thus, the preparation of efficient and water-soluble PS molecules that damage biological functions solely under irradiation but remain biocompatible in the dark state would be highly desirable.
Recently, ruthenium (Ru) complexes have attracted considerable recognition as PDT agents due to their unique photophysical and photochemical features as well as their DNA intercalation capacity and protein binding motifs.3,8,9 In particular, organo-ruthenium complexes coordinated by polypyridyl ligands exhibited promising anticancer activity when irradiated with light.10 Their high population of the triplet metal-to-ligand charge-transfer state (3MLCT), due to the heavy atom effect, produces large 1O2 yields, while the solubility of these complexes can be modified by adjusting the counterions. For example, the [Ru(bpy)3]2+ derivative TLD1433 recently entered phase I clinical trials as the first Ru-based PS, due to its potential in effectively producing 1O2.11 In combination with a targeting peptide providing high binding affinity for membrane proteins, a Ru-PS has been achieved with high selectivity for certain cancer cells.12 However, in order to further advance PDT for therapy, several limitations of the PS still need to be solved such as their low cellular uptake efficiency, low extinction coefficients, and only moderate cellular toxicity. Herein, we present a macromolecular approach to improve phototoxicity and efficacy of the PS by synergistic combination of Ru-complexes on a protein carrier scaffold decorated with subcellular mitochondria targeting groups.
Mitochondria, as indispensable organelles responsible for cell respiration, emerge as promising pharmacological target in clinical applications for the detection, inhibition and treatment of various diseases such as cancer or neurodegenerative diseases, due to their crucial role in mediating cell apoptosis.13,14 Until now, only little is known of the balance of reactive oxygen species in cancer cells and their survival mechanisms that effect mitochondria function. There have been many attempts to target cancer cells via signaling pathways.15 However, drug strategies targeting the mitochondrial metabolism are scarce and though present, treatment approaches were not achieved at low drug concentrations. The conjugation of PS with mitochondria targeting groups is considered an emerging strategy to enhance cellular toxicity by localizing the PS at the relevant site.16
Herein, we investigate efficient growth inhibition in an acute myeloid leukemia (AML) cell line by a macromolecular PS targeting mitochondria, which are known as “power house of the cell” and that are central organelles for tumor growth.17 AML is an aggressive disease which still leads to death in up to 8 of 10 patients outside of clinical trials. It is characterized by aberrant high proliferation and increase in immature blasts and progenitors due to blockade in cell differentiation. Leukemic cells initially respond to chemotherapy. However, relapse is common and in most cases fatal. Thus, there is an urgent need to develop innovative therapeutic concepts, which target leukemic cells, but spare normal hematopoietic stem and progenitor cells.
We propose a bioinspired strategy that converts the blood plasma protein serum albumin (HSA) into an efficient nanotransporter for phototoxic drug molecules,18−21 providing synergistic features due to the molecular design. The resultant nanotransporter denoted cHSA-PEO-TPP-Ru exhibited significantly improved photophysical properties and enhanced 1O2 quantum yields as compared to the bare Ru complex as well as excellent mitochondria-specific colocalization. Efficient phototoxicity of cHSA-PEO-TPP-Ru already at nanomolar concentrations were achieved, which was attributed to synergistic effects from the high number of Ru-complexes as well as organelle-targeting features of the biopolymer. To the best of our knowledge, cHSA-PEO-TPP-Ru reported herein displays the lowest IC50 value for cancerous cell lines and therefore highest cytotoxicity of a Ru-containing molecule reported to date in cellular studies. Efficient inhibition of growth in an AML cell line was observed, with preferential killing of leukemic cells compared to normal bone marrow cells, suggesting a therapeutic window for this compound in AML. Furthermore, Two-photon absorption features of cHSA-PEO-TPP-Ru underline its great potential as two-photon activated photosensitizer for in vivo PDT.
Results and Discussion
Synthesis and Characterization of cHSA-PEO-TPP-Ru
The synthesis of the cHSA-PEO-TPP-Ru is depicted in Scheme 1. HSA serves as biocompatible and biodegradable backbone providing many reactive carboxylic acid, amino and hydroxyl groups originating from the respective amino acid side chains of HSA that can be further chemically functionalized. First, all accessible carboxylic acid groups were transformed into primary amino groups by applying ethylenediamine and 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide (EDC) to increase the total number of reactive amino groups for further conjugations and enhance interactions of the biopolymer with negatively charged cellular membranes as published previously.22 After purification through dialysis, globular, polycationic cHSA was obtained, which facilitates cellular uptake by Clathrin-mediated endocytosis.23 To impart water solubility and reduce nonspecific interactions, poly(ethylene oxide) (PEO-2000) side chains were introduced by reacting α-methoxy-ω-carboxylic acid succinimidyl ester polyethylene(oxide) (NHS-PEO) with cHSA and subsequent washing five times by ultrafiltration with vivaspin 20 (MWCO 30K) centrifuge tubes. According to Maldi-ToF, about 20 PEO chains were attached to cHSA.24 To achieve mitochondria targeting, multiple units of TPP were reacted to the free amino groups of cHSA-PEO. Briefly, we mixed EDC-NHS activated (3-carboxypropyl) triphenylphosphonium bromide (TPP) ester with cHSA-PEO to yield cHSA-PEO-TPP hybrid with approximately 34 TPP units attached. Finally, the resulting product was washed five times through vivaspin 20 (MWCO 30K) centrifugal concentrator for further usage.
Scheme 1.

(a) Synthetic scheme of the cHSA-PEO-TPP-Ru transporter based on HSA. Subsequent functionalities were conjugated at different reactive sites of the HSA backbone. (b) Schematic illustration of a part of the HSA polypeptide sequence exemplary with the PEO, TPP groups attached to, e.g., lysine and Ru conjugated to tyrosine residues.
Mannich reactions have been reported to modify phenol groups of tyrosine side chains of proteins with high selectivity.25 Therefore, the aniline-modified Ru-complex denoted “Ru1” was selected as it allows bioconjugation of these sterically demanding Ru-complexes following a Mannich-type reaction in aqueous solution. Compared to many known Ru-complexes, Ru1 reveals exceptional water solubility of 153 mg/mL making it well-suitable for protein modifications under mild conditions. HSA provides 18 tyrosine groups and half are located exposed to the surface according to computer simulations. A three-component Mannich-type coupling reaction was carried out applying a mixture of cHSA-PEO-TPP, formaldehyde (HCHO) and the Ru-complex (Ru1, SI) yielding the desired cHSA-PEO-TPP-Ru as yellowish-brown solution in 70% isolated yield. Excessive amounts of the reactants (e.g., unreacted Ru complex and HCHO) were removed by ultrafiltration with vivaspin 20 (MWCO 30K) centrifugation tubes until no free Ru-complex was detected in the elution media anymore. A control experiment was performed without formaldehyde and no Ru attachment was observed (Figure SI-1), indicating low tendency of Ru-complexes for unspecific adsorption into the hydrophobic pockets of HSA.
MALDI-ToF characterization of the chemically modified globular proteins was accomplished for each reaction step. The respective MALDI-ToF mass spectra are depicted in Figure SI-2, indicating that about 10 Ru-complexes were loaded to cHSA-PEO-TPP. The resulting cHSA-PEO-TPP-Ru shows solubility of >65 mg/mL as well as high stability at 4 °C for more than eight months (long-term storage studies are still ongoing). In order to evaluate the hydrodynamic radius of cHSA-PEO-TPP-Ru in solution and cell culture medium, DLS studies were accomplished. An average hydrodynamic radius of about 40 nm was obtained (Figure SI-3). Polymer sizes in this range are considered favorable for accumulation in tumor tissues via the EPR effect,26 although this effect is currently under debate.27 Zeta-potential of cHSA-PEO-TPP-Ru (Figure 1a) reveals a positive surface charge facilitating interactions with negatively charged cellular membranes to induce endocytosis.
Figure 1.

(a) Zeta potential of different bioconjugates. (b) Typical absorbance and emission spectra of cHSA-PEO-TPP-Ru, where characteristic peaks of the Ru complexes are preserved in cHSA-PEO-TPP-Ru. (c) Comparison of the photostability of the Ru1 and cHSA-PEO-TPP-Ru based on the absorbance decay under continuous irradiation over extended time periods. (d) 1O2 production yield of cHSA-PEO-TPP-Ru and bare Ru complex (Ru1), as obtained from the photobleaching of the characteristic absorption peak @380 nm of ABDA (100 μM) during irradiation with 470 nm LED light (∼20 mW/cm2, 5 min) in PBS (1×, pH 7.4) based on the same optical density in their first absorption peak. (e) Steady state emission spectra (λex = 460 nm) of Ru1 complex in water (red), Ru1 complex in simulated body fluid (orange), cHSA-PEO-TPP-Ru in water (blue), cHSA-PEO-TPP-Ru in simulated body fluid (green), and Ru(bpy)3 (black) as reference at same optical density. (f) Emission lifetime experiments upon excitation at 460 nm. For the compounds, the same color code is used and an artifact region between 1.2 and 1.35 μs has been removed.
Photostability and 1O2 Yield of cHSA-PEO-TPP-Ru
cHSA-PEO-TPP-Ru exhibited characteristic absorption and emission maxima at around 460 and 617 nm, respectively (Figure 1b), similar to the starting complex Ru1, cHSA-PEO-TPP-Ru revealed no alteration of the metal-to-ligand charge transfer band (MLCT) during the reaction. Furthermore, the emission spectrum of cHSA-PEO-TPP-Ru was insensitive to the composition of the solvent, i.e., the emissive excited state did not respond to environmental changes such as variations of the solvent composition. To evaluate the photostability of cHSA-PEO-TPP-Ru, photobleaching experiments were conducted in water.
Previous publications have reported that the photoinstability of Ru(II) polypyridyl complexes including [Ru(bpy)3]2+ limits their PDT applications28−30 and functionalized nanoparticles revealed higher photostability compared to the amine containing Ru(II) polypyridyl complexes.31Figure 1c shows the greatly improved photostability of cHSA-PEO-TPP-Ru compared to the Ru1, which was recorded under the same conditions. Even over greatly extended irradiation times (65 h), cHSA-PEO-TPP-Ru remained remarkably stable (with only 36% decrease) in comparison to Ru1. For the latter, significantly decreased absorbance of about 76% was detected after 18 h already (Figure SI-4). The luminescence of both compounds was observed over time and loss of emission intensity of about 68% and 89% occurred after 300 min for cHSA-PEO-TPP-Ru and Ru1, respectively. Obviously, photochemical stability of the Ru-complex in the biopolymer has significantly increased compared to Ru1.
PDT relies on efficient production of singlet oxygen in cellular environments. In order to monitor the generation of 1O2 in a quantitative fashion, we performed 1O2 production efficiency tests at four different LED sources, e.g., 770 nm, 625 nm, 525 nm and 470 nm as reported by us previously.12 The singlet oxygen sensor 9,10-anthracenediyl-bi(methylene)dimalonic acid (ABDA) was applied, which forms an endoperoxide of ABDA in the presence of 1O2, thus decreasing ABDA absorption and providing a valuable means of direct monitoring 1O2 production (Figure SI-5). According to these measurements, 470 nm proved to be most efficient excitation source (Figure SI-6). cHSA-PEO-TPP-Ru and cHSA-PEO-TPP as control were mixed separately with 100 μM of ABDA in PBS buffer and then irradiated with a 470 nm LED array (P = 20 ± 2 mW/cm2) for 5 min. As described in Figure SI-9a, cHSA-PEO-TPP-Ru produced 1O2 very efficiently, whereas cHSA- PEO-TPP remained inactive.
Equimolar concentrations of Ru in cHSA-PEO-TPP-Ru and Ru1 were used for all further experiments to compare the photophysical and biological features of Ru in the cHSA-PEO-TPP-Ru bioconjugate versus Ru1. As depicted in Figure 1d, the 1O2 production for Ru1 and cHSA-PEO-TPP-Ru was measured at the same optical density at 460 nm, e.g., at similar Ru concentrations. About ∼8-fold improved reduction of the ABDA absorption peak @380 nm has been achieved for a single Ru molecule attached to the biopolymer compared to a single Ru1 complex (details included in the Supporting Information). Again, molar concentrations have been used for comparison. We believe that the synergistic interaction between closely spaced multiple Ru1 in the lipophilic protein backbone might be the reason for this finding.
Photophysical Properties of cHSA-PEO-TPP-Ru
The photophysical properties of the model Ru1-complex and cHSA-PEO-TPP-Ru were investigated by steady state emission spectroscopy following a literature reported procedure.32,33 The photochemistry of Ru-complexes is highly sensitive to their respective environments. Emission spectra of both compounds were identical in water and simulated body fluid solutions (Figure 1e). A slightly higher emission quantum yield of cHSA-PEO-TPP-Ru (using Ru(bpy)3 as standard) was observed upon increasing the ionic strength of the solution, i.e., by comparing the simulated body fluid (ΦcHSA-PEO-TPP-Ru, body fluid = 6.7%) to water (ΦcHSA-PEO-TPP-Ru, water = 6.1%). On the contrary, this trend was not observed for the Ru1 (ΦRu, water = 4.8/ΦRu, body fluid = 4.5%)). The generally higher fluoresecence quantum yields of cHSA-PEO-TPP-Ru and Ru1 indicate that nonradiative deactivation paths were suppressed in the cHSA-PEO-TPP-Ru biopolymer. This might be attributed to a sterically hindered rotation of the imidazole-phenyl bond due to conjugation to the bulky HSA protein. In line with this assignment of blocking nonradiative decay paths, we observed the prolongation of the triplet excited state lifetime in cHSA-PEO-TPP-Ru compared to Ru1 (Figure 1f). Assuming a monoexponential decay to analyze the respective luminescence kinetics, the lifetime was increased from 300 ns for both solutions of Ru1 to 625/735 ns for cHSA-PEO-TPP-Ru in water/simulated body fluid.
Intracellular Optical Microscopy Imaging
We examined the intracellular localization of cHSA-PEO-TPP-Ru into a human cervical cancer cell line, HeLa cells, as model system, by laser scanning confocal microscopy. Typically, these cells were incubated with low concentrations of cHSA-PEO-TPP-Ru (500 nM) for about 4 h before images were recorded. To observe MLCT absorbance from the metal complex inside living cells, laser excitation at 458 nm was applied. The emission window was adjusted in the range of 530–710 nm. After staining with various subcellular organelle dyes (Figure 2) with selectivity for membranes (Cell Mask Deep Red Plasma Membrane Stain), the nucleus (Hoechst 33342 Solution), mitochondria (Mito Lite Blue FX490, Figure SI-7c), and lysosomes (Lyso Tracker Green DND-26), we found a clear colocalization of cHSA-PEO-TPP-Ru in mitochondria (Pearson’s coefficient 0.88), where they were mostly situated in the cytosol outside the nuclear region (Pearson’s coefficient 0.07). cHSA-PEO-TPP-Ru was transported rapidly across the membrane and accumulated in the cytosol and no localization in membranes (Pearson’s coefficient 0.2) or in lysosomes (Pearson’s coefficient 0.3) was observed.
Figure 2.
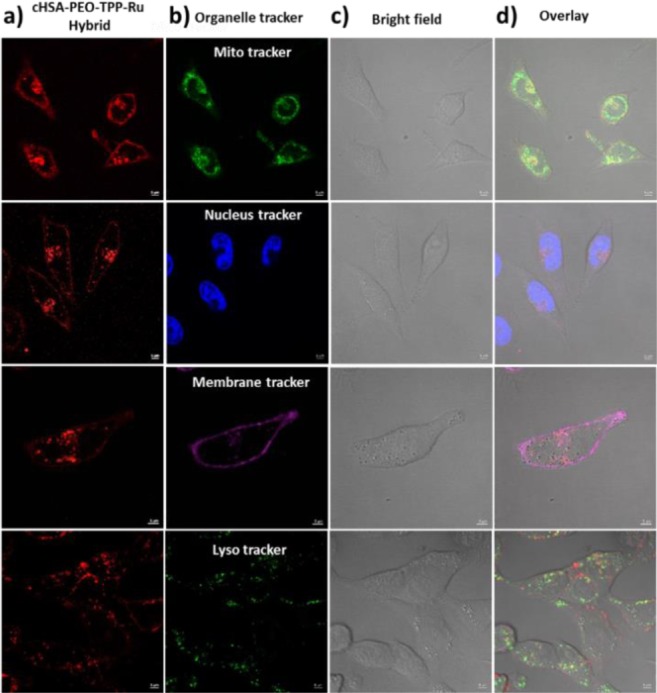
Confocal microscopy images of HeLa cells incubated with cHSA-PEO-TPP-Ru and treated with commercial organelle trackers. Overlay images and colocalization analysis of cells stained with mitochondria (0.88), nucleus (0.07), membrane (0.2), and lysosome (0.3) markers indicated that cHSA-PEO-TPP-Ru localized in mitochondria. (a) cHSA-PEO-TPP-Ru emission, (b) emission from the organelle trackers, (c) corresponding bright field images, and (d) overlay of all three images.
Light induced cellular toxicity evaluation
To evaluate the cellular uptake efficiency of cHSA-PEO-TPP-Ru in vitro, we incubated HeLa cells with the biopolymer over different time intervals ranging from 1–240 min. Flow cytometry measurements revealed that the fluorescence intensity of treated HeLa cells reached a saturation level after 200 min of biopolymer incubation (Figure SI-7). Thus, 240 min was selected as the appropriate incubation time to ascertain maximum cellular uptake. To identify the optimum concentration of cHSA-PEO-TPP-Ru, HeLa cells were incubated with 0–2 μM cHSA-PEO-TPP-Ru for 240 min after 5 min irradiation with 470 nm LED light (∼20 mW/cm2). Here, the applied light dose was comparable to the reported dosage of established photosensitizing drugs reported.34 Also, we used commercially available TOX-8 dye (Sigma-Aldrich), to obtain the number of viable cells quantitatively by means of spectrophotometric measurement in all cases of cell viability experiments, according to the manufacturer’s instructions.35 A concentration dependent cytotoxicity was observed in the photo irradiated sample, whereas cells incubated with of cHSA-PEO-TPP-Ru in the dark revealed almost no cellular toxicity (Figure 3a,b) over the entire concentration range. A very low IC50 value of 34.9 ± 2 nM was obtained for cHSA-PEO-TPP-Ru under light irradiation. In comparison, Ru1 had an IC50 value of only 7.7 ± 1.3 μM (Figure SI-8) indicating greatly enhanced cellular toxicity of cHSA-PEO-TPP-Ru by about 220-fold with minimal dark toxicity. Thus, attaching multiple Ru1 to one protein nanocarrier yielded a nanocarrier with surprisingly high cytotoxicity with an IC50 well below the value one would expect considering an just additive effect. Considering that 10 Ru chromophores were attached to cHSA-PEO-TPP-Ru, a calculated IC50 value of 0.349 μM could be estimated for each Ru-chromophore, which is considerably lower compared to Ru1 (IC50 value 7.7 μM) alone. Next, we studied the impact of mitochondria targeting TPP groups on carrier toxicity. Analogous phototlooxicity experiments were accomplished with cHSA-PEO-Ru without TPP groups and about 8-fold lower drug toxicity was obtained (Figure 3c).
Figure 3.
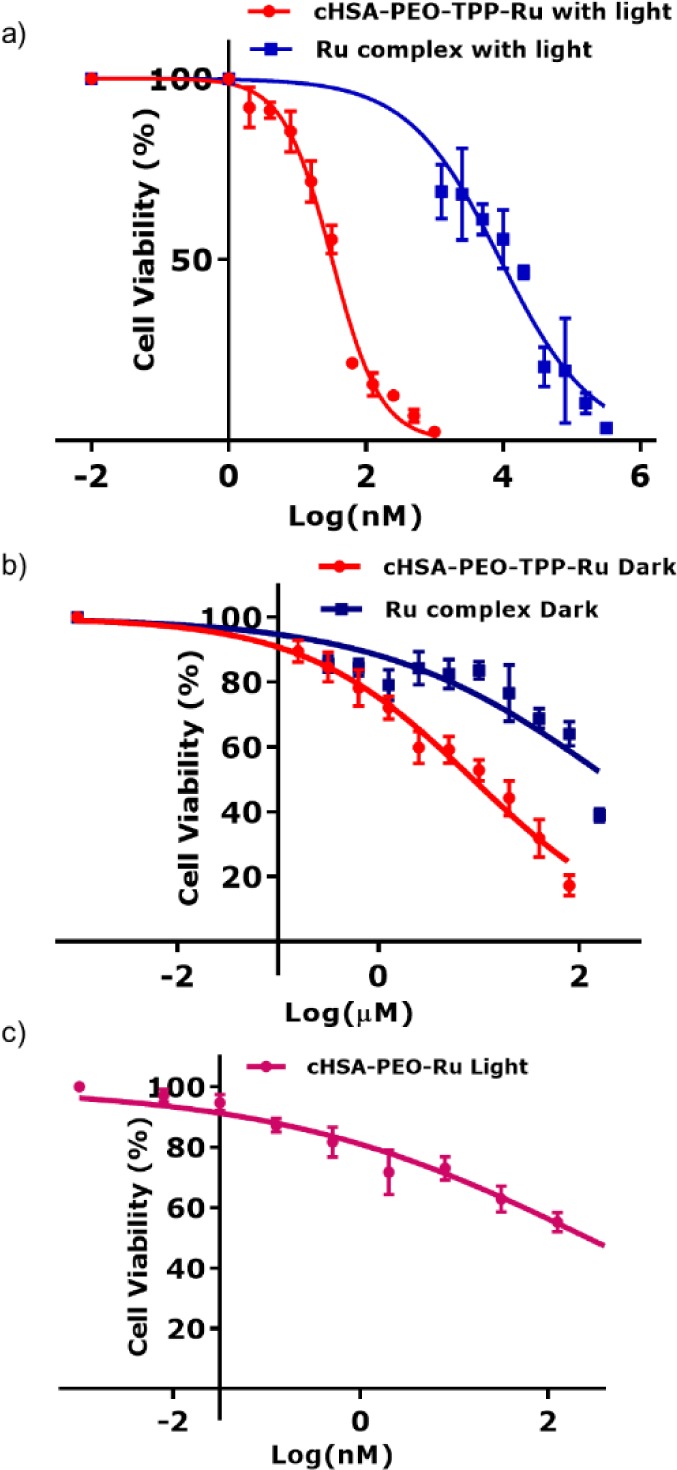
(a,b) Logarithmic fitting curve for cell viability of cHSA-PEO-TPP-Ru and bare Ru complex, over a broad concentration range with and without light. (c) Logarithmic fitting curve for cell viability of cHSA-PEO-Ru complex with light, where mitochondria targeting TPP group were absent. For all of the above experiments, HeLa cells were exposed to a 470 nm LED lamp (∼20 mW/cm2) for 5 min for light irradiation. cHSA-PEO-TPP-Ru reveals low dark toxicity (IC50 = 9 ± 2 μM) but very high phototoxicity (IC50 = 34.9 ± 2 nM) compared to Ru1 (dark IC50 = 203 ± 3 μM; photoirradiated IC50 = 7.7 ± 1.3 μM). In the absence of a TPP group, the phototoxic effect of the drug was reduced by ∼8 times (IC50 = 265 ± 1.2 nM).
The phototoxic index (PI) of all compounds was calculated, which denotes the ratio of the dark and light-exposed IC50 values. The protein hybrid cHSA-PEO-TPP without Ru1 was light inactive (Figure SI-9a), whereas Ru1 only revealed a PI of 27. The PI of cHSA-PEO-Ru without TPP groups increased to 75 and cHSA-PEO-TPP-Ru had a significantly higher PI of 250. We believe this higher value was based on our molecular design, where the multiple molecular components contribute synergistically to the observed elevated cellular toxicity.
Furthermore, we examined the phototoxicity of cHSA-PEO-TPP-Ru toward various other cancerous cell lines such as CHO, MCF7 and A549. All tumor cells tested were proficiently damaged with low IC50 values in the nanomolar range, for instance, 135.2 ± 1 nM for CHO, 114.3 ± 1 nM for MCF7, and 119.1 ± 1 nM for the A549 cell line (Figures SI-10–12). As additional features, the HSA polypeptide backbone is fully biodegradable even after chemical modification, which should allow efficient elimination and reduced accumulation.
Colony Forming Cell (CFC) and Proliferation Assays
In order to test the efficacy of cHSA-PEO-TPP-Ru in a relevant primary cell assay, we treated an acute myeloid leukemia (AML) cell line, OCI-AML3, which reflects the biology of primary NPM1 mut - AML, comprising around 35% of all human AML cases36 and 60% of AML with normal karyotype.37 Irradiation of 2 and 5 min induced a significant (p < 0.005) reduction of 44% and 84.4% of colony growth, respectively, in comparison to the treated but nonirradiated control arm (dark) as assessed by colony number in the colony forming cell (CFC) assay (p < 0.005). In addition to the reduction in colony number, remaining colonies were smaller in size after exposure to cHSA-PEO-TPP-Ru (Figure 4b). Furthermore, we tested cHSA-PEO-TPP-Ru for its effect on proliferation with two different concentrations (75 and 37.5 nM). A significant decrease of the proliferative capacity was observed over a period of up to 72 h after exposure to light for 2 and 5 min, respectively, compared to the dark control (Figure 4c,d).
Figure 4.
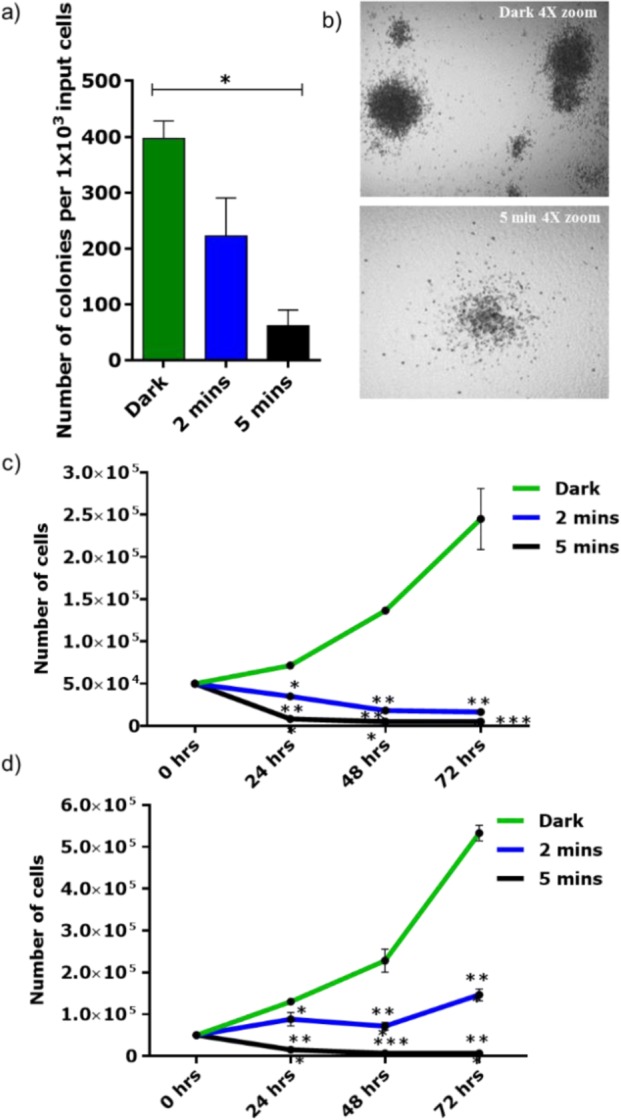
(a) Colony forming cell (CFC) assay of the OCI-AML3 (OA3) AML cell line (n = 2 in duplicates) showed reduction of colonies after 2 and 5 min exposure to light (470 nm) compared to the treated and nonexposed cells (dark). Bars indicate mean ± SEM. Significance calculated by Mann–Whitney test (*<0.05). (b) Morphology of the colonies in the control arm and 5 min exposure arm (4× magnification). (c,d) Proliferation assay of the OA3 cell line (n = 3) incubated with two different concentrations of cHSA-PEO-TPP-Ru (75 nM) and (37.5 nM) and exposed to light for 2 and 5 min or treated but not exposed to light. Significance calculated by two-way ANOVA multiple test comparisons test (*<0.05; ***<0.0001).
To determine the differential effect of cHSA-PEO-TPP-Ru on normal versus leukemic primary bone marrow (BM) cells, we performed CFC assays on healthy murine BM cells as well as on a murine AML cell line derived from BM of a mouse transplanted with a truncated version of the leukemia-specific AML1-ETO fusion gene (AML1-ETO 9a (AE9a)).38 We could document a decrease in the colony forming capacity of the leukemic AE9a cell line by 37% and 88% reduction after 2 and 5 min exposure to light, respectively, compared to the nonirradiated control cells (Figure SI-13). In contrast, there was only a 10% and 28% reduction of colony growth, respectively, when normal BM cells were treated accordingly (Figure SI-14), indicating that cHSA-PEO-TPP-Ru significantly inhibits the proliferative and clonogenic potential of primary murine AML while sparing normal bone marrow stem and progenitor cells. Previously, Sieber and co-workers have successfully demonstrated photodynamic treatment of bone marrow/leukemic cell (L1210) mixtures with visible light of 410–500 nm.39 Even though significant reduction of the leukemic cell number was shown in vitro, subsequent in vivo studies in a mouse transplant were less successful. It was speculated that photoactive protoporphyrin IX (PpIX) had to be biosynthesized within the leukemic cells and leukemic cells within the resting phase did not perform this biosynthesis and therefore escaped this treatment. In our approach, cHSA-PEO-TPP-Ru conjugate exhibits very similar PDT effects, but due to the biopolymer design no biosynthesis of the photosensitizer is required for bioactivity.
Two-Photon Active Probe
For in vivo PDT, deep-tissue penetration would be relevant to reach also tumor cells located in deeper tissue. TP microscopy has evolved as an efficient tissue imaging and therapeutic platform due to its longer wavelength excitation laser light, which offers deep tissue penetration, reduced photodamage and 3D and high contrast imaging.10 We have measured the two-photon (TP) properties of cHSA-PEO-TPP-Ru to assess its suitability as TP probe. cHSA-PEO-TPP-Ru revealed almost five times higher TP action cross section compared to the Ru complex (Figure 5a). Improved TP features are of great relevance to trigger localized photochemical reactions beneath the skin with minimum off-target photodamage. The 5-times increased value clearly indicates that the design concept of combining multiple Ru complexes within one nanosized protein transporter allows achieving enhanced TP directed PDT applications. According to previous studies, the TPA cross section of 50 GM obtained for cHSA-PEO-TPP-Ru should be sufficient for its application as 2PA PDT drug candidate.40,41 Recent Ru-complexes providing high TP cross sections are based on sophisticated ligand designs or highly charged ligand substituents42,43 that influence cell uptake mechanisms and sub cellular distribution patterns as shown by Barton and Puckett.44 In case of cHSA-PEO-TPP-Ru, a clear three-dimensional (3D) distribution in the cytosol (red) was demonstrated in Figure 5b, where the location of the nucleus was stained with Hoechst dye (3D video, Video SI-V).
Figure 5.
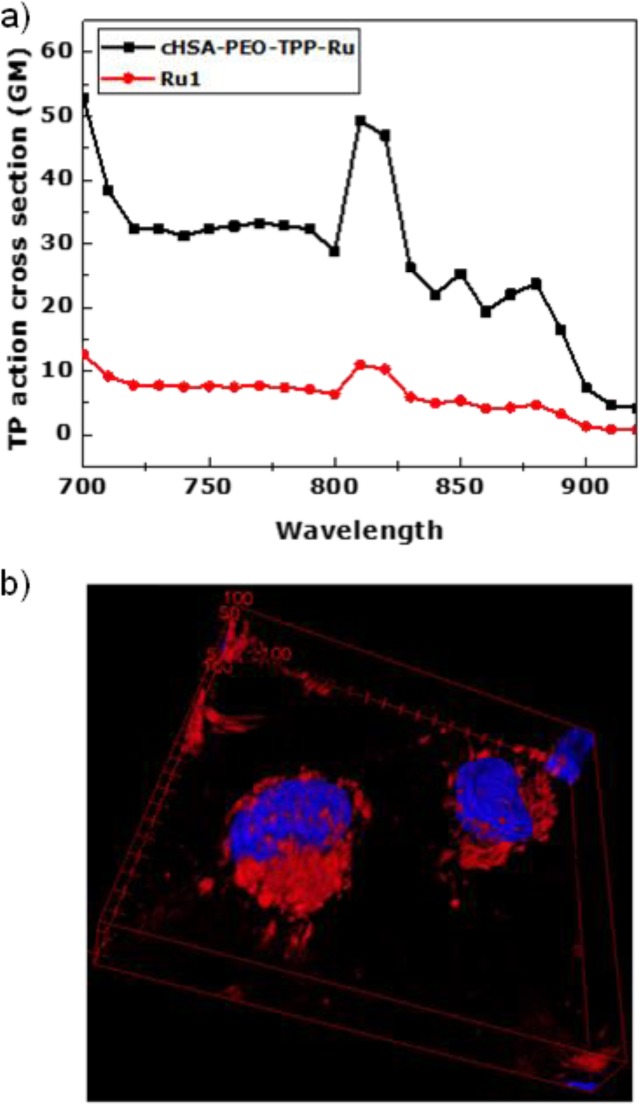
(a) Two-photon absorption cross section of cHSA-PEO-TPP-Ru and Ru1 measured in water at excitation wavelengths from 700 to 920 nm using Rhodamin B as reference. (b) Two-photon 3D surface projection z-stack confocal microscopy image of HeLa cells incubated with cHSA-PEO-TPP-Ru (red, excited at 810 nm) and nuclear staining dye Hoechst (blue, excited at 405 nm).
Conclusion
We have converted the plasma protein HSA into a highly phototoxic, biodegradable macromolecular PS by controlling its solubility, subcellular targeting pathways, and toxicity. The mitochondria targeted macromolecular PS reported herein revealed significantly enhanced photophysical and chemical properties as well as greatly improved 1O2 quantum yields. To the best of our knowledge, the observed phototoxicity was the highest (indicated by the lowest IC50 value) ever reported for Ru-complexes. We believe that high Ru-loading capacity, enhanced cellular uptake efficiency, and localization in mitochondria combined with high photostability and 1O2 generation ability contributed to the greatly enhanced cytotoxicity of cHSA-PEO-TPP-Ru. Moreover, effectively blocked cell proliferation and clonogenic potential of the myeloid leukemic cell line OCI-AML3 further underlines the strong antileukemic activity. Intriguingly, preliminary experiments demonstrated less toxicity to normal BM cells, possibly indicating that this biopolymer preferentially targets leukemic cells. These results open the attractive opportunity of treating AML with cHSA-PEO-TPP-Ru-like conjugates using a photodynamic purging step in autologous treatment concepts such as autologous hematopoietic stem cell grafts.45,46 The observed TP features of the cHSA-PEO-TPP-Ru biopolymer further provide important prospects for PDT in vivo. The presented strategy to merge a multifunctional protein scaffold and diverse synthetic entities into a versatile nanotransporter platform with tailor-made and potentially synergistic molecular properties could be of great relevance for the preparation of more efficient diagnostic and therapeutic tools in biomedicine.
Acknowledgments
The authors are grateful to the financial support of the German Research Foundation (DFG) under P3246029, the ERC Synergy Grant 319130-BioQ, the Horizon2020 project “Hyperdiamond”, and LGFG scholarship from Ulm University.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b13399.
The authors declare no competing financial interest.
Supplementary Material
References
- Ogilby P. R. Chem. Soc. Rev. 2010, 39, 3181–3209. 10.1039/b926014p. [DOI] [PubMed] [Google Scholar]
- DeRosa M. C.; Crutchley R. J. Coord. Chem. Rev. 2002, 234, 351–371. 10.1016/S0010-8545(02)00034-6. [DOI] [Google Scholar]
- Mari C.; Pierroz V.; Ferrari S.; Gasser G. Chem. Sci. 2015, 6, 2660–2686. 10.1039/C4SC03759F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin M. R.; Jori G.. Photodynamic Inactivation of Microbial Pathogens; Hamblin M. R., Jori G., Eds.; Comprehensive Series in Photochemical & Photobiological Sciences; The Royal Society of Chemistry: London, 2011. [Google Scholar]
- Wilkinson F.; Helman W. P.; Ross A. B. J. Phys. Chem. Ref. Data 1993, 22, 113–262. 10.1063/1.555934. [DOI] [Google Scholar]
- Lovell J. F.; Liu T. W. B.; Chen J.; Zheng G. Chem. Rev. 2010, 110, 2839–2857. 10.1021/cr900236h. [DOI] [PubMed] [Google Scholar]
- Cheng L.; Wang C.; Feng L.; Yang K.; Liu Z. Chem. Rev. 2014, 114, 10869–10939. 10.1021/cr400532z. [DOI] [PubMed] [Google Scholar]
- Sun B.; Guan J. X.; Xu L.; Yu B.-L.; Jiang L.; Kou J. F.; Wang L.; Ding X. D.; Chao H.; Ji L. N. Inorg. Chem. 2009, 48, 4637–4639. 10.1021/ic900102r. [DOI] [PubMed] [Google Scholar]
- Marin V.; Holder E.; Hoogenboom R.; Schubert U. S. Chem. Soc. Rev. 2007, 36, 618–635. 10.1039/B610016C. [DOI] [PubMed] [Google Scholar]
- Liu J.; Chen Y.; Li G.; Zhang P.; Jin C.; Zeng L.; Ji L.; Chao H. Biomaterials 2015, 56, 140–153. 10.1016/j.biomaterials.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Arenas Y.; Monro S.; Shi G.; Mandel A.; McFarland S.; Lilge L. Photodiagn. Photodyn. Ther. 2013, 10, 615–625. 10.1016/j.pdpdt.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Wang T.; Zabarska N.; Wu Y.; Lamla M.; Fischer S.; Monczak K.; Ng D. Y. W.; Rau S.; Weil T. Chem. Commun. 2015, 51, 12552–12555. 10.1039/C5CC03473F. [DOI] [PubMed] [Google Scholar]
- Wongrakpanich A.; Geary S. M.; Joiner M. L.; Anderson M. E.; Salem A. K. Nanomedicine (London, U. K.) 2014, 9, 2531–2543. 10.2217/nnm.14.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaudin D.; Marzo I.; Brenner C.; Kroemer G. Int. J. Oncol. 1998, 12, 141–152. 10.3892/ijo.12.1.141. [DOI] [PubMed] [Google Scholar]
- Downward J. Nat. Rev. Cancer 2003, 3, 11–22. 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Wang B.; Galliford C. V.; Low P. S. Nanomedicine 2014, 9, 313–330. 10.2217/nnm.13.175. [DOI] [PubMed] [Google Scholar]
- Dayem A. A.; Choi H.-Y.; Kim J.-H.; Cho S.-G. Cancers 2010, 2, 859–884. 10.3390/cancers2020859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D. Y. W.; Wu Y.; Kuan S. L.; Weil T. Acc. Chem. Res. 2014, 47, 3471–3480. 10.1021/ar5002445. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Pramanik G.; Eisele K.; Weil T. Biomacromolecules 2012, 13, 1890–1898. 10.1021/bm300418r. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Chakrabortty S.; Gropeanu R. a; Wilhelmi J.; Xu Y.; Er K. S.; Kuan S. L.; Koynov K.; Chan Y.; Weil T. J. Am. Chem. Soc. 2010, 132, 5012–5014. 10.1021/ja909570v. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Eisele K.; Doroshenko M.; Algara-Siller G.; Kaiser U.; Koynov K.; Weil T. Small 2012, 8, 3465–3475. 10.1002/smll.201200409. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Ihme S.; Feuring-Buske M.; Kuan S. L.; Eisele K.; Lamla M.; Wang Y.; Buske C.; Weil T. Adv. Healthcare Mater. 2013, 2, 884–894. 10.1002/adhm.201200296. [DOI] [PubMed] [Google Scholar]
- Eisele K.; Gropeanu R. A.; Zehendner C. M.; Rouhanipour A.; Ramanathan A.; Mihov G.; Koynov K.; Kuhlmann C. R. W.; Vasudevan S. G.; Luhmann H. J. Biomaterials 2010, 31, 8789–8801. 10.1016/j.biomaterials.2010.07.088. [DOI] [PubMed] [Google Scholar]
- Palesch D.; Boldt F.; Müller J. A.; Eisele K.; Stürzel C. M.; Wu Y.; Münch J.; Weil T. ChemBioChem 2016, 17, 1504–1508. 10.1002/cbic.201600193. [DOI] [PubMed] [Google Scholar]
- Joshi N. S.; Whitaker L. R.; Francis M. B. J. Am. Chem. Soc. 2004, 126, 15942–15943. 10.1021/ja0439017. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Wu J.; Sawa T.; Matsumura Y.; Hori K. J. Controlled Release 2000, 65, 271–284. 10.1016/S0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- Jabir N. R.; Tabrez S.; Ashraf G. M.; Shakil S.; Damanhouri G. A.; Kamal M. A. Int. J. Nanomed. 2012, 7, 4391–4408. 10.2147/IJN.S33838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann L.; Staehle R.; Pfeifer M.; Reichardt C.; Sorsche D.; Wächtler M.; Popp J.; Dietzek B.; Rau S. Chem. - Eur. J. 2016, 22, 8240–8253. 10.1002/chem.201505113. [DOI] [PubMed] [Google Scholar]
- Fong J.; Kasimova K.; Arenas Y.; Kaspler P.; Lazic S.; Mandel A.; Lilge L. Photochem. Photobiol. Sci. 2015, 14, 2014–2023. 10.1039/C4PP00438H. [DOI] [PubMed] [Google Scholar]
- Neugebauer U.; Pellegrin Y.; Devocelle M.; Forster R. J.; Signac W.; Moran N.; Keyes T. E. Chem. Commun. 2008, 2, 5307–5309. 10.1039/b810403d. [DOI] [PubMed] [Google Scholar]
- Lu D.; Gai F.; Qiao Z.-A.; Wang X.; Wang T.; Liu Y.; Huo Q. RSC Adv. 2016, 6, 51591–51597. 10.1039/C6RA02917E. [DOI] [Google Scholar]
- Stephenson M.; Reichardt C.; Pinto M.; Wächtler M.; Sainuddin T.; Shi G.; Yin H.; Monro S.; Sampson E.; Dietzek B.; Mcfarland S. A. J. Phys. Chem. A 2014, 118, 10507–10521. 10.1021/jp504330s. [DOI] [PubMed] [Google Scholar]
- Reichardt C.; Pinto M.; Wächtler M.; Stephenson M.; Kupfer S.; Sainuddin T.; Guthmuller J.; McFarland S. A.; Dietzek B. J. Phys. Chem. A 2015, 119, 3986–3994. 10.1021/acs.jpca.5b01737. [DOI] [PubMed] [Google Scholar]
- Breskey J. D.; Lacey S. E.; Vesper B. J.; Paradise W. A.; Radosevich J. A.; Colvard M. D. Photomed. Laser Surg. 2013, 31, 398–407. 10.1089/pho.2013.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. Bull. Environ. Contam. Toxicol. 1981, 26, 145–149. 10.1007/BF01622068. [DOI] [PubMed] [Google Scholar]
- Fey M. F.; Buske C. Ann. Oncol. 2013, 24, 1–6. [DOI] [PubMed] [Google Scholar]
- Quentmeier H.; Martelli M. P.; Dirks W. G.; Bolli N.; Liso a; Macleod R. a F.; Nicoletti I.; Mannucci R.; Pucciarini a; Bigerna B.; Martelli M. F.; Mecucci C.; Drexler H. G.; Falini B. Leukemia 2005, 19, 1760–1767. 10.1038/sj.leu.2403899. [DOI] [PubMed] [Google Scholar]
- Vegi N. M.; Klappacher J.; Oswald F.; Mulaw M. A.; Mandoli A.; Thiel V. N.; Bamezai S.; Feder K.; Martens J. H. A.; Rawat V. P. S.; Mandal T.; Quintanilla-Martinez L.; Spiekermann K.; Hiddemann W.; Döhner K.; Döhner H.; Stunnenberg H. G.; Feuring-Buske M.; Buske C. Cell Rep. 2016, 16, 498–507. 10.1016/j.celrep.2016.05.094. [DOI] [PubMed] [Google Scholar]
- Cunderlíková B. C.; Vasovič V.; Sieber F.; Furre T.; Nesland J.; Peng Q. Bone Marrow Transplant. 2010, 45, 1553–1561. 10.1038/bmt.2009.382. [DOI] [PubMed] [Google Scholar]
- Boca S. C.; Four M.; Bonne A.; van der Sanden B.; Astilean S.; Baldeck P. L.; Lemercier G. Chem. Commun. 2009, 4590–4592. 10.1039/b907143a. [DOI] [PubMed] [Google Scholar]
- Boreham E. M.; Jones L.; Swinburne A. N.; Blanchard-Desce M.; Hugues V.; Terryn C.; Miomandre F.; Lemercier G.; Natrajan L. S. Dalton Trans. 2015, 44, 16127–16135. 10.1039/C5DT01855B. [DOI] [PubMed] [Google Scholar]
- Huang H.; Yu B.; Zhang P.; Huang J.; Chen Y.; Gasser G.; Ji L.; Chao H. Angew. Chem., Int. Ed. 2015, 54, 14049–14052. 10.1002/anie.201507800. [DOI] [PubMed] [Google Scholar]
- Mongin O.; Four M.; Chevreux S.; Blanchard-Desce M.; Lemercier G. Chimia 2015, 69, 666–669. 10.2533/chimia.2015.666. [DOI] [PubMed] [Google Scholar]
- Puckett C. A.; Barton J. K. J. Am. Chem. Soc. 2007, 129, 46–47. 10.1021/ja0677564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traul D. L.; Sieber F. J. Photochem. Photobiol., B 2015, 153, 153–163. 10.1016/j.jphotobiol.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum L. G.; Yamagami M.; Giddings B. R.; Joshi I.; Schober S. L.; Sensenbrenner L. L.; Sieber F. Blood 1991, 77, 2701–2706. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.