


Abstract
Plant orthologs of the yeast sucrose non-fermenting (Snf1) kinase and mammalian AMP-activated protein kinase (AMPK) represent an emerging class of important regulators of metabolic and stress signalling. The catalytic α-subunits of plant Snf1-related kinases (SnRKs) interact in the yeast two-hybrid system with different proteins that share conserved domains with the β- and γ-subunits of Snf1 and AMPKs. However, due to the lack of a robust technique allowing the detection of protein interactions in plant cells, it is unknown whether these proteins indeed occur in SnRK complexes in vivo. Here we describe a double-labelling technique, using intron-tagged hemagglutinin (HA) and c-Myc epitope sequences, which provides a simple tool for co-immunopurification of interacting proteins expressed in Agrobacterium-transformed Arabidopsis cells. This generally applicable plant protein interaction assay was used to demonstrate that AKINβ2, a plant ortholog of conserved Snf1/AMPK β-subunits, forms different complexes with the catalytic α-subunits of Arabidopsis SnRK protein kinases AKIN10 and AKIN11 in vivo.
INTRODUCTION
The genome sequence of the model plant Arabidopsis thaliana has recently been completed and annotated providing a wealth of information for emerging proteomics and functional genomics studies (1,2). Construction of new platforms for transcript profiling aim at an integrated analysis of gene expression (3), whereas systematic protein interaction screens in the yeast two-hybrid system offer a means for characterisation of the proteome (4,5). As detection of protein interactions in the yeast two-hybrid system does not necessarily imply that the same proteins also interact in plants, there is a need for development of new techniques to facilitate the detection of in vivo protein interactions in plant cells. Current problems in the analysis of subunit composition of plant protein complexes are well illustrated by the example of Snf1-related plant protein kinases (SnRKs).
SnRKs belong to the conserved family of yeast sucrose non-fermenting (Snf1) kinase and animal AMP-activated kinase (AMPK) (6). These conserved protein kinases are heterotrimeric enzymes consisting of α-, β- and γ-subunits. The catalytic α-subunit carries an N-terminal serine/threonine protein kinase domain followed by C-terminal regulatory sequences which function as a kinase autoinhibitory domain (7). In the yeast Saccharomyces cerevisiae the α-subunit is encoded by the SNF1 gene, which is required for proper regulation of glycogen storage, sporulation and transcriptional derepression of glucose-repressed genes (8). In comparison, two different isoforms of α-subunit are known in mammals (9). The γ-subunit encoded by the SNF4 gene in yeast is involved in maintaining the active conformation of the α-subunit by binding to its autoinhibitory domain (7). The β-subunit mediates the formation of a heterotrimeric complex because it can independently interact with both the α- and γ-subunits (10). Co-transfection experiments indicate that the β-subunit is also required for reconstitution of AMPK kinase activity in animal cells (11). Remarkably, three genes (SIP1, SIP2 and GAL83) code for the β-subunits in yeast, whereas only two β-isoforms have been found so far in mammals (12). Recent studies in yeast demonstrate that, in addition to the assembly of active kinase complex, the β-subunits are required for specific recognition of diverse kinase substrates (13).
Orthologs of the Snf1/AMPK α-, β- and γ-subunits have been identified in several plant species (14–20). Phylogenetic analysis indicates a remarkable homology between the catalytic α-subunits of plant SnRKs and other members of the Snf1/AMPK family (21). According to this classification, plant protein kinases showing the highest similarity to Snf1 and AMPKs belong to the SnRK1 subfamily. In Arabidopsis two SnRK1 α-subunits are known which are capable of functionally complementing the yeast snf1 mutation (16). In addition, two different Arabidopsis proteins sharing homologous CβS (cystathione β synthase) domains with the Snf1/AMPK γ-subunits have been identified. However, only one of these putative Arabidopsis SnRK γ-subunits (AtSNF4) was observed to suppress the snf4 deficiency in yeast (17,19). Based on sequence homology, two Arabidopsis genes encoding potential orthologs of Snf1/AMPK β-subunits were identified and characterised by different patterns of transcriptional regulation (17). The interaction properties of these putative SnRK subunits have been studied in the yeast two-hybrid system and protein interaction assays in vitro (17,19). Nonetheless, it is still an open question whether the highly variable putative β- and γ-subunits indeed occur in vivo in common complexes with SnRK α-subunits, which are remarkably conserved in plant cells.
To characterise the interactions between SnRK subunits in vivo, we exploited an epitope tagging technique which was recently developed for facile detection and cellular localisation of proteins in Agrobacterium-transformed Arabidopsis cells (22). By generating fusions between intron-tagged epitope coding domains and plant cDNAs, this technique eliminates artificial expression of proteins in Agrobacterium. Thus, the expression of proteins in plant cells can be specifically and sensitively detected as early as 5 days after transformation. To verify the results of yeast two-hybrid tests, a co-immunopurification assay was developed based on co-expression of interacting proteins carrying hemagglutinin (HA) and c-Myc epitopes in plant cells. Using this generally applicable plant protein interaction assay, we demonstrate here that the catalytic α-subunits of Arabidopsis protein kinases AKIN10 and AKIN11 form different SnRK complexes with a regulatory AKINβ2 subunit in vivo.
MATERIALS AND METHODS
Plasmid constructs
Vectors carrying plant gene expression cassettes with an intron-tagged HA epitope coding domain were described previously (22). Similar vectors with an intron-tagged c-Myc epitope were constructed using a series of PCR reactions. A HindIII fragment of p35S GUS INT (23), containing intron IV2 of the potato gene ST-LS1 (24), was used as a template in combination with the primers MYCPIV1 (5′-GGAGATCTGAGCAAAAGTTGATTGTAAGTTTCTGCTTC TACCT-3′) and MYCPIV2 (5′-GGGTCGACAAGATCCTCCTCAGACTGCACATCAACAAATTTTG-3′), both of which carried five codons from the c-Myc epitope coding sequence (in bold). The PCR product was cloned as a BglII–SalI fragment in pPE1000 (25) and sequenced to yield the vector pMESHI. The MYCPIV2comp primer (5′-GGGTCGACTCAAGATCCTCCTCAGACTGCACATCAACAAATTTTG-3′) in combination with MYCPIV1 (see above) was used to similarly amplify and clone a PCR fragment in pPE1000 to obtain pLOLA. The vector pGIGI was obtained by SalI digestion and religation of plasmid pMESHI.
The intron-tagged c-Myc epitope coding domain was fused in multiple steps to the 3′-end AKIN11 coding region to create a vector for expression of AKIN11 tagged with a C-terminal c-Myc epitope. First, AKIN11 sequences in pGEM (19) were used as template for PCR amplification of a 3′ segment from the coding region by removing the stop codon using the primer pair 5′-CCCGGGAATTCCGTGTTCCAAGTGGCTATCT-3′ and 5′-GGGGTCGACGATATCCACACGAAGCTCGCGAAG-3′ (AKIN11 sequences in bold; restriction sites in italic). The obtained XbaI–SalI fragment, which encoded 175 C-terminal residues of AKIN11 without a stop codon, was used to substitute the corresponding AKIN11 fragment in pGEM. The modified AKIN11 coding region was cloned as a NcoI–EcoRV fragment in the NcoI and SmaI sites of pMESHI to obtain pAKIN11-myc. After filling in the ends with T4 DNA polymerase, a NotI fragment carrying the plant expression cassette from pAKIN11-myc was inserted into a filled-in HindIII site of binary vector pPCV002 (26), yielding pAKIN11-myc-pPCV002.
To generate a translational fusion between the c-Myc epitope and the N-terminus of AKIN10, first a blunt-ended NotI fragment, carrying a plant expression cassette from pLOLA, was inserted into the filled-in EcoRI and BamHI sites of pPCV002 to yield pPCV002-LOLA. The stop codon was removed from the AKIN10 cDNA as described above (T.Kleinow, unpublished results). The modified AKIN10 coding region was isolated from pGEM and, following treatment with T4 DNA polymerase, inserted as an EcoRI–SalI fragment into a filled-in BamHI site of pPCV002-LOLA to construct pMyc-AKIN10-pPCV002. To obtain a control pMyc-GUS construct, a SalI–EcoRI fragment, carrying a uidA/GUS gene from pHiA-GUS (22), was cloned in pPCV002-LOLA.
To label AKINβ2 with an N-terminal HA epitope, a full-length AKINβ2 cDNA was synthesised by PCR amplification using a cDNA clone isolated from an Arabidopsis cDNA library (see below) and primer pair 5′-GGGGTCGACGGCCATGGGCTCTGCTGCTTCTGATGG T-3′ and 5′-GGGGAATTCTCATCTAGACCTCTGCAGGGATTTGTA-3′ (coding sequences in bold; restriction sites in italic). The PCR product was digested with SalI and EcoRI and inserted in pPILY (22) to obtain pHA-AKINβ2. A blunt-ended NotI fragment from pHA-AKINβ2 was cloned into the T4 DNA polymerase-treated SmaI and SacI sites of pPCV812 (27) to construct pHA-AKINβ2-pPCV812.
To determine the frequency of co-transformation of Arabidopsis cells with two different Agrobacterium T-DNAs, the coding sequence of red fluorescent protein DsRed from Discosoma sp. was inserted downstream of a modified cauliflower mosaic virus (CaMV 2×35S) promoter and translational enhancer sequences (G.Jach, personal communication) in a plant gene expression cassette, which was cloned as a HindIII fragment in the Agrobacterium binary vector pR97 (28). Binary vector pBI121, carrying a plant expression cassette with the coding domain of green fluorescent protein mGFP4, has been described by Haseloff et al. (29). The pPCV binary vector constructs were introduced into Agrobacterium GV3101 pPMP90RK, whereas the pR97 and pBI121 vectors were transformed into GV3101 pMP90 by electroporation as described (30).
Cloning of AKINβ2 cDNA and its use in two-hybrid interaction tests with AKIN10 and AKIN11 baits
The last exon of the AKINβ2 sequence was PCR amplified using oligonucleotide primers PSA1 (5′-GACTATGTTCCTGAAGACATTCAAAGCATAT-3′) and PSA2 (5′-TCACCTCTGCAGGGATTTGTAGAGCACC-3′) and genomic DNA from A.thaliana (Col-0) as template. The purified PCR product was used as probe to screen 5 × 105 bacterial colonies from a cDNA library constructed in pACT2 (16). Among 96 clones characterised by sequencing, a cDNA was identified which carried a deletion of the first nine codons of the AKINβ2 coding domain in fusion with the GAL4 activation domain in pACT2. This construct was transformed into yeast strain Y187 carrying either pAS2-AKIN10, pAS2-AKIN11, pAS2-SNF1 or pAS2-NPK5 as bait (16,31) to perform two-hybrid interaction assays as described (7).
Co-transformation of Arabidopsis cell suspensions with two Agrobacterium strains
Culture media and conditions for Agrobacterium-mediated transformation of Arabidopsis cell suspensions were described previously (22). For co-transformation with two different gene constructs, both Agrobacterium strains were grown in selective YEB medium (27) to an OD660 of 0.8 (1 × 109 cells/ml). The cells were collected by centrifugation and resuspended in plant cell culture medium (22) to obtain a suspension containing 1 × 1010 cells/ml. After mixing equal aliquots from both cultures, a maximum of 1 × 1010 Agrobacterium cells were used for infection of 50 ml of freshly subcultured Arabidopsis (Col-0) cell suspension as described (22). Arabidopsis cell line T87 (32) cultured in modified MS medium [4.7 g/l Murashige and Skoog medium (Sigma), 0.5 g/l MES, pH 5.7, 30% sucrose and 2.5 µM 1-naphtalene acetic acid] was similarly infected with Agrobacterium. After 3 days co-cultivation, either claforan (500 µg/ml; Hoechst) or a mixture of claforan (300 µg/ml) and tricarcillin/clavulanic acid (500 µg/ml; Duchefa) was added to arrest bacterial growth. Two days later the Arabidopsis cells were collected either to prepare protein extracts for immunoassays or to record the green and red fluorescence of mGFP4 and DsRed proteins, respectively, using epifluorescence and confocal laser scanning microscopy.
Preparation of protein extracts and co-immunopurification assays
To prepare crude protein extracts, plant cells were collected by filtration using a Millipore holder system, frozen in liquid nitrogen and stored at –70°C. The cells were homogenised to a powder in liquid N2 and resuspended in extraction buffer (50 mM Tris–HCl, pH 7.6, 10% glycerol, 1 mM EDTA, 1 mM DTT, 1% Igepal CA-630 and 20 µl/ml Sigma plant protease inhibitor mix). A cleared cell lysate was obtained by centrifugation (twice at 3000 g for 10 min at 4°C). Following the determination of protein concentration, the cleared lysate was subjected to immunoaffinity purification. The protein extract (1–2 mg) was supplemented with 150 mM NaCl and pre-cleared with 50 µl of protein G–Sepharose (Sigma) (equilibrated in IPW buffer, 20 mM Tris–HCl, pH 7.5, 150 mM NaCl and 0.1% Igepal CA-630). The pre-cleared lysate was incubated for at least 3 h at 4°C with 50 µl of affinity matrix carrying either mouse monoclonal anti-HA.11 IgG (clone 16B12; BAbCO) or anti-c-Myc IgG (clone 9E10; BAbCO). The matrix was placed on a 1 ml Mobicol (MobiTec) column and washed with a 100 bead volume (5 ml) of IPW buffer. The bound proteins were eluted by treating the beads twice with 1 bead volume (∼50 µl) of either HA or c-Myc peptide solution (Genosys) (1 mg/ml in 20 mM Tris–HCl, pH 7.5, 150 mM NaCl) for 15 min at 30°C. The eluted proteins were separated by SDS–PAGE and analysed by western blotting using Immobilon membrane (PVDF 0.45 µm; Millipore) as described (22). For immunodecoration, either a high affinity rat monoclonal anti-HA IgG (clone 3F10; Boehringer-Roche) or a mouse monoclonal anti-c-Myc IgG (clone 9E10; BAbCO) was used as primary antibody, followed by treatment with either peroxidase-conjugated anti-rat or anti-mouse IgG Fab fragment (Boehringer-Roche) and subsequent enhanced chemiluminescence detection (ECL; Pharmacia-Amersham).
Epifluorescence and confocal laser scanning microscopy
Fluorescence images were examined with a Zeiss Axiophot epifluorescence microscope equipped with fluorescence filter sets from AHF Analysentechnik (Tübingen, Germany). For DsRed a special filter was used (excitation HQ 545/30, dichroic mirror 565 LP and emission 600/40). GFP fluorescence was detected with an RS-GFP filter (excitation HQ 480/20, dichroic mirror 495 LP and emission HQ 510/20). A Leica TCS SP confocal laser scanning microscope (Heidelberg, Germany) fitted to a spectrophotometer for emission band wavelength selection was used with two laser sources (i.e. an argon ion laser emitting at 488 nm and a helium–neon laser emitting at 543 nm) to excite GFP and DsRed, respectively. For visualisation of GFP, the emission window was set at 495–535 nm, whereas DsRed was detected at an emission window of 570–660 nm. The double immunolabelled sections were studied by sequential excitation with each laser separately to avoid chlorophyll fluorescence in the red channel. The confocal image stacks were combined as x–y projection images.
RESULTS
Construction of vectors with intron-tagged HA and c-Myc epitope coding domains
To develop a co-immunopurification assay for detection of interacting proteins labelled with different epitopes in Agrobacterium-transformed plant cells, an intron from the potato gene ST-LS1 (23,24) was inserted in the coding domains of HA and c-Myc epitopes. This was necessary to eliminate artificial expression of epitope-tagged plant proteins in Agrobacterium, which could interfere with specific detection of epitope-labelled proteins in transformed plant cells. The construction of plant expression vectors pPILY and pMENCHU, carrying intron-tagged HA epitope coding domains, has been previously described (22). Using a similar approach, a set of vectors carrying intron-tagged c-Myc epitope coding domains were constructed to facilitate the labelling of plant proteins with either N- or C-terminal c-Myc peptide tags (see Materials and Methods). In the c-Myc epitope fusion vectors (Fig. 1) the polylinker sequences located 5′-upstream of the c-Myc coding domains are identical, but the cloning sites located 3′-downstream of the c-Myc epitope sequences are different. Plasmid pLOLA and pGIGI are compatible with pPILY and pMENCHU (22), respectively, allowing a fusion of protein coding sequences with HA and c-Myc epitopes using the same reading frames. Vector pMESHI provides a different reading frame to generate alternative translational fusions. Plasmid pGIGI is a derivative of pMESHI, which contains a BamHI site for generation of in-frame fusions with cDNAs derived from the yeast two-hybrid vectors pACT2 and pAS2 (Fig. 1). Plant gene expression cassettes in all HA and c-Myc epitope vectors carry a CaMV 35S promoter with a duplicated enhancer domain and 3′ transcription terminator sequences from the nopaline synthase gene. The plant gene expression cassettes are flanked by unique NotI sites to facilitate further cloning of epitope-tagged constructs in Agrobacterium binary vectors for plant transformation.
Figure 1.

Schematic map of intron-tagged c-Myc epitope vectors. The positions of restriction endonuclease cleavage sites are shown in parentheses. Functional elements of the plant gene expression cassette are highlighted by arrowheaded and striped boxes. The lower section shows polylinker sequences flanking the c-Myc epitope coding region which is interrupted by intron IV2 of the potato ST-LS1 gene. A BamHI site in pGIGI facilitates subcloning of cDNA sequences from the yeast two-hybrid vector pACT2 in-frame with the c-Myc epitope coding sequence. Polylinker sequences flanking the coding region of the c-Myc epitope in pLOLA and pGIGI are identical to those which flank the intron-tagged HA epitopes in the previously described vectors pPILY and pMENCHU (22), respectively. Amp, ampicillin resistance gene; f1 Ori and OriC, f1 phage and ColE1 plasmid replication origins, respectively; 35S, duplicated enhancer regions in the promoter of cauliflower mosaic virus 35S RNA gene (CaMV35S).
Optimisation of co-expression of HA and c-Myc epitope-tagged proteins in Arabidopsis cells by Agrobacterium-mediated transformation
To optimise the conditions for detection of c-Myc epitope-tagged proteins expressed in Agrobacterium-transformed plant cells, a β-glucuronidase (GUS) reporter protein was labelled with an N-terminal c-Myc epitope. A uidA/gusA coding sequence was inserted in pLOLA, then the plant gene expression cassette was moved into the Agrobacterium binary vector pPCV002 (26) and transformed into Arabidopsis cells as described (22). The amount of bacterial cells used for transformation was adjusted such that on average 80% of infected Arabidopsis cells showed staining with the histochemical GUS substrate X-Gluc 5 days after infection. From the transformed cells protein extracts were prepared, separated by SDS–PAGE and subjected to western blotting with different commercially available anti-c-Myc antibodies. In crude protein extracts prepared from control non-transformed Arabidopsis cells all anti-c-Myc IgGs tested in western blotting (including a BAbCO IgG from clone 9E10, which provided the highest sensitivity of detection) showed a non-specific cross-reaction with a protein of 100 kDa (Fig. 2, TOTAL). Nonetheless, the anti-c-Myc antibodies specifically detected a myc–GUS reporter protein with the expected molecular mass. Unlike western blotting, preclearing of protein extracts with protein G–Sepharose followed by immunoprecipitation with immobilised anti-c-Myc IgG resulted in specific detection of the myc–GUS protein, providing a reliable tool for development of a co-immunopurification assay (Fig. 2, IP).
Figure 2.
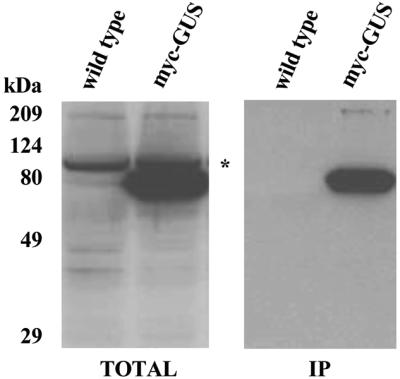
Western blotting and immunoprecipitation of myc–GUS reporter protein with monoclonal anti-c-Myc antibodies. Protein extracts (50 µg) prepared from wild-type and myc–GUS-expressing Arabidopsis cell lines were separated by SDS–PAGE and immunoblotted using a monoclonal anti-c-Myc antibody (left). An asterisk indicates non-specific cross-reaction of the antibody with a protein of 100 kDa present in both control wild-type and transformed cells. The protein extracts (1 mg) were immunaffinity purified on c-Myc IgG beads. The matrix-bound proteins were eluted with c-Myc peptide, separated by SDS–PAGE and subjected to western blotting with a monoclonal anti-c-Myc antibody (right).
To simultaneously express two different proteins in plant cells, the conditions for co-transformation were optimised using Agrobacterium strains which carried reporter gene constructs coding for the Discosoma red fluorescent protein DsRed in pR97 (28) and the green fluorescent protein mGFP4 in pBI121 (29). Titration of the inoculum size by infection of 50 ml aliquots of Arabidopsis cell cultures during the logarithmic growth phase indicated that the cultures could tolerate a maximum of 1 × 1010 Agrobacterium cells without losing their viability (data not shown). Using a total of 1 × 1010 bacterial cells for infection, the transformation frequencies ranged between 70 and 80% with a single Agrobacterium strain, whereas double infections with two strains yielded only 40–50% transformed Arabidopsis cells which displayed expression of either green or red fluorescent protein as visualised by epifluorescence microscopy (data not shown). To measure the co-transformation frequency, the proportion of cells showing both green (GFP) and red (DsRed) fluorescence was determined using confocal laser scanning microscopy. Sequential recording of green, red and merged yellow (GFP + DsRed) images provided a precise count for single and double transformed cells (Fig. 3). On average, 55% of cells growing in aggregates (i.e. microcalli) displayed both GFP and DsRed emission, indicating that it is feasible to use Agrobacterium-mediated co-transformation for expression of two different proteins in cultured Arabidopsis cells.
Figure 3.
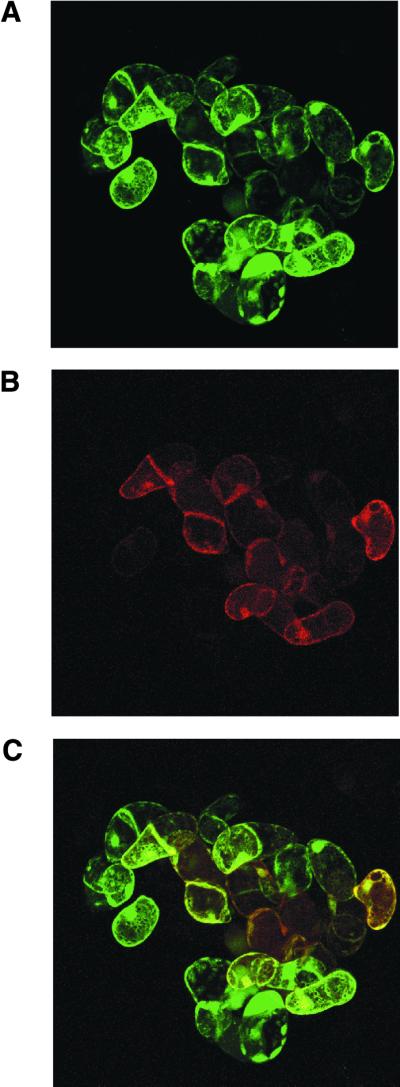
Optimisation of co-transformation using green and red fluorescent reporter proteins. Arabidopsis cell suspensions were co-transformed with Agrobacterium strains carrying mGFP4 and DsRed expression vectors. The number of cells showing green, red and both green and red fluorescence was determined 5 days after transformation by confocal laser scanning microscopy. Images from the same sections were sequentially examined for GFP and DsRed fluorescence. (A) Cells expressing green mGFP4; (B) red DsRed-expressing cells; (C) merged images.
AKINβ2, an ortholog of the Sip2 β-subunit of yeast Snf1 kinase, forms different complexes with Arabidopsis SnRK α-subunits AKIN10 and AKIN11 in vivo
To identify potential orthologs of Snf1/AMPK β-subunits, different domains of yeast Sip2 protein were used to search for homologous sequences in the Arabidopsis database using the TBLAST program. The best hit identified a gene (accession no. gb:ATFCA6, chromosome 4, ESSA I contig fragment no. 6) which coded for a protein showing partial homology to conserved kinase-interacting sequences (i.e. KIS domains) of β-subunits of yeast Snf1 and animal AMPKs. The same gene was recently identified as encoding a putative SnRK subunit designated AKINβ2 (17). A cDNA encoding AKINβ2 in fusion with the Gal4 activation domain was isolated from a cDNA library made in the yeast two-hybrid vector pACT2 (16) using a specific probe obtained by PCR amplification of the last exon. This cDNA clone was used as prey in the yeast strain Y187 to test for interaction of AKINβ2 with the Arabidopsis SnRK1 α-subunits AKIN10 and AKIN11 used as baits in two-hybrid assays. Quantitative measurement of β-galactosidase enzyme levels, reflecting activation of a LacZ reporter gene by the bait–prey interactions, suggested that AKINβ2 is a putative binding partner for both AKIN10 and AKIN11 (Fig. 4). The specificity of observed two-hybrid interactions was controlled using distantly related SnRK1 α-subunits, such as yeast Snf1 and tobacco NPK5, self-activation tests with yeast strains carrying the AKIN10 and AKIN11 baits alone and interaction assays with several unrelated preys, including pVA3 and pLAM (Clontech) (data not shown).
Figure 4.

Quantitative measurement of two-hybrid interactions of AKINβ2 with different SnRK1 α-subunits using a β-galactosidase assay. Yeast strain 187 was co-transformed with a pACT2-AKINβ2 prey and either pAS2-AKIN10, pAS2-AKIN11, pAS2-NPK5 or pAS2-SNF1 as bait. Yeast strains carrying the baits AKIN10 and AKIN11 alone were used as self-activation controls. Three colonies selected on minimal SD medium lacking tryptophan and leucine were grown independently in liquid SD medium to mid exponential phase, then the cells were collected for determination of β-galactosidase activity. Bars indicate standard deviations.
To verify the results of two-hybrid interactions, AKINβ2 was co-expressed with either AKIN10 or AKIN11 in Arabidopsis cells using the Agrobacterium-mediated co-transformation method described above. AKIN10 and AKIN11 were labelled with N- and C-terminal c-Myc epitopes, respectively, whereas the N-terminus of AKINβ2 was fused to a HA epitope as described (see Materials and Methods). First, the epitope-tagged constructs were independently transformed into Arabidopsis cells to evaluate the expression level and stability of modified AKIN10, AKIN11 and AKINβ2 proteins. Following 5 days of Agrobacterium infection, protein extracts were prepared from the transformed cells, resolved by SDS–PAGE and immunoblotted using monoclonal anti-HA and anti-c-Myc antibodies. Both c-Myc-tagged AKIN10 and AKIN11 proteins (both 61 kDa) were detected at apparently low levels (as compared to an HA epitope-tagged GUS reporter protein; data not shown). As noted earlier, western blotting revealed a non-specific cross-reaction of monoclonal anti-c-Myc antibody with a protein of 100 kDa in all samples prepared from transformed and non-transformed Arabidopsis cells. Intriguingly, AKIN11–myc was typically detected as a doublet, suggesting post-translational modification or specific degradation of this protein (Fig. 5A). In comparison to myc–AKIN10 and AKIN11–myc, HA–AKINβ2 was produced at an apparently higher level, but appeared to be less stable because, in addition to its intact form, some degradation products were also detected with the anti-HA antibody (Fig. 5B). These characteristics observed by immunoblotting of epitope-tagged proteins did not change when HA–AKINβ2 was co-expressed with either myc–AKIN10 or AKIN11–myc (Fig. 6A). The apparent reduction in the intensity of the bands with respect to the single transformations probably reflects a reduced number of transformed cells when performing co-transformation experiments.
Figure 5.

Expression of epitope-tagged SnRK subunits in transformed Arabidopsis cell lines. (A) Protein extracts (50 µg) were prepared from Arabidopsis cells expressing myc–AKIN10 and AKIN11–myc, separated by SDS–PAGE and immunoblotted with anti-c-Myc antibody. (B) Immunoblotting of protein extracts from a cell line expressing the HA–AKINβ2 protein using an anti-HA antibody. Control protein samples prepared from a non-transformed Arabidopsis cell line were used to monitor the specificity of antibodies. An asterisk in (A) indicates non-specific cross-reaction of the anti-c-Myc antibody.
Figure 6.

AKINβ2 is detected in different protein complexes with SnRK α-subunits AKIN10 and AKIN11 in vivo. Arabidopsis cell suspensions were transformed with Agrobacterium strains to express myc–AKIN10, AKIN11–myc and HA–AKINβ2 separately. In co-transformation experiments, HA–AKINβ2 was expressed together with either myc–AKIN10 or AKIN11–myc. (A) Immunoblotting of protein extracts (50 µg) from cell lines co-expressing HA–AKINβ2 with c-Myc-tagged SnRK α-subunits using anti-c-Myc and anti-HA antibodies. (B) From each single and double transformed cell line 1 mg protein extract was subjected to immunoaffinity purification on either an anti-HA IgG or anti-c-Myc IgG matrix. Proteins eluted from the anti-HA IgG and anti-c-Myc IgG matrices using HA and c-Myc peptides, respectively, were immunoblotted and detected with anti-HA and anti-c-Myc antibodies. The symbols + and – indicate the presence or absence of constructs in the transformation assays. IP and ID refer to the antibodies used for immunoprecipitation (IP) and immunodetection (ID), respectively.
To examine whether HA–AKINβ2 occurred in common complexes with the c-Myc epitope-tagged SnRK α-subunits, protein extracts were prepared from control non-transformed cells and cultures transformed separately with each construct, as well as from cells co-transformed with HA–AKINβ2 and either myc–AKIN10 or AKIN11–myc. The extracts were subjected to immunaffinity purification on immobilised anti-HA and anti-c-Myc IgG matrices. Proteins eluted from the anti-HA and anti-c-Myc IgG matrices by HA and c-Myc peptides, respectively, were immunoblotted with anti-HA and anti-c-Myc antibodies (Fig. 6B). HA–AKINβ2 was detected by the anti-HA antibody after immunaffinity binding of both myc–AKIN10 and AKIN11–myc. Interestingly, only full-length HA–AKINβ2 was recovered in complex with the immunopurified SnRK α-subunits, suggesting a lack of interaction of C-terminally truncated AKINβ2 derivatives with myc–AKIN10 and AKIN11–myc. Similarly, both c-Myc-tagged SnRK α-subunits were detected in association with HA–AKINβ2 after immunoaffinity purification on anti-HA IgG. The specificity of these protein interactions was corroborated by control co-transformation experiments which revealed no interaction of HA–AKINβ2 with an affinity purified myc–GUS protein (data not shown). Thus, these data demonstrated that AKINβ2 is a β-subunit of two different SnRK protein kinase complexes in Arabidopsis.
DISCUSSION
Recent annotation and comparative sequence analysis of the Arabidopsis genome reveals an unexpectedly high level of redundancy. A comparison of the chromosome 2 and 4 sequences indicates that ∼75% of all predicted gene products show a significant similarity to another protein encoded elsewhere in the genome (1). A typical example of this potential functional redundancy is provided by components of the SnRK family in Arabidopsis. Until completion of the genome sequence, two Arabidopsis genes coding for SnRK catalytic α-subunits had been identified and demonstrated to complement the corresponding yeast snf1 mutation (16). Similarly, two different genes were identified to code for putative homologs of Snf1/AMPK γ-subunits based on sequence homology searches and yeast two-hybrid interaction studies (17,19). In addition, two different proteins showing domain homology to β-subunits of yeast Snf1 (Sip/Gal83 family) and animal AMPKs were identified and tested for interaction with an Arabidopsis SnRK α-subunit in the two-hybrid system (17). However, our survey of the completed Arabidopsis genome sequence indicates the presence of additional genes coding for putative SnRK subunits. Using the coding sequence of AKINβ2 in a BLASTP search, in addition to AKINβ1 we have identified five sequences (accession nos At2g28060, AT3g01510, At1g27070, AT5g39790 and AT3g52180) which show an overall similarity and significant domain homology to members of the yeast Sip protein family (P values of 1.3e–20, 4.8e–08, 4.8e–06, 8.4e–04 and 2.3e–02, respectively). A similar BLASTP search with the coding region of AKIN10 reveals a new member of the highly conserved α-subunit gene family (16) (AT5g39440, P = 2.9e–196), as well as three additional genes for predicted proteins (AT4g24400, AT5g25110 and AT5g10930, P values of 3.7e–74, 2.0e–71 and 1.4e–70, respectively) showing significant homology to the regulatory domains of AKIN10 and AKIN11 catalytic α-subunits. As conserved sequences of regulatory domains of α-subunits are used as characteristic modules to identify members of the SnRK1 subfamily, it is likely that these proteins represent novel SnRK1 α-subunits. This surprising redundancy of genes coding for putative SnRK1 subunits suggests that many alternative kinase complexes could be formed by combinations of different subunits in plants. Determination of the subunit composition of SnRK complexes thus represents a demanding task for future studies.
Among the currently available techniques, the yeast two-hybrid system offers a simple and reliable means for sorting of protein interactions. Nonetheless, the results of two-hybrid interaction assays often require confirmation. Therefore, control in vitro protein binding studies are usually performed with recombinant proteins in combination with [35S]methionine labelling of their binding partners in various coupled transcription–translation systems. In these in vitro binding assays the interacting proteins are assembled in heterologous systems which lack potentially necessary endogenous factors (such as other subunits of a protein complex) and post-translational modifications. These evident problems call for development of new protein interaction assays which facilitate the characterisation of protein complexes in vivo.
With the aim of detecting in vivo protein interactions between subunits of SnRK protein kinases in plant cells, we have developed a simple co-immunopurification assay based on a previously described intron-tagged epitope-labelling technique and efficient transformation of Arabidopsis cell suspensions by Agrobacterium. In our previous studies we have exploited an intron-tagged HA epitope coding domain to affinity purify proteins and determine their localisation in plant cells using either transient expression or stable transformation (22). In the present work we have optimised the application of another epitope in order to establish a reliable assay for monitoring the interaction of plant proteins labelled with HA and c-Myc epitopes in Arabidopsis cells. We used this technique to determine whether a recently described putative Arabidopsis SnRK β-subunit (AKINβ2; 17) occurs in vivo in complexes with two previously characterised different α-subunits, AKIN10 and AKIN11 (16). We have observed that both AKIN10 and AKIN11 interact with the putative AKINβ2 regulatory subunit in the two-hybrid system. To verify the results of two-hybrid assays, AKINβ2 was labelled with an HA epitope and co-expressed with c-Myc epitope-labelled AKIN10 and AKIN11 proteins in Arabidopsis cells. A considerable difference was noted between the expression levels of c-Myc-tagged α-subunits and HA-tagged AKINβ2 protein by immunoblotting. However, this observation must be treated with necessary caution due to likely differences in the sensitivity and specificity of anti-HA and anti-c-Myc antibodies. Whereas western blotting suggested apparently low level expression, both myc–AKIN10 and AKIN11–myc proteins were isolated in association with comparable amounts of HA–AKINβ2 protein using immunoaffinity binding to anti-HA and anti-c-Myc IgG beads.
Recent studies suggest that formation of different Snf1 kinase enzyme complexes with the Sip/Gal83 β-subunits may account for specific targeting of the Snf1 kinase to different substrates in yeast (13). Using the in planta protein interaction assay presented here, it is feasible now to label as pair-wise combinations all putative subunits of plant SnRK1 kinases with epitope tags. In addition to immunoaffinity purification of SnRK complexes, this approach could help to decipher the combinations of different subunits and determine their role in the recognition of different substrates and SnRK-binding signalling proteins (16). Although our present results are confined to suspension cultured cells, the described epitope-tagged constructs can also be used for generation of transgenic plants to facilitate the identification of multiprotein complexes that may show different subunit compositions in diverse cell types and phases of plant development.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge Elmon Schmelzer (Central Microscopy Service, MPIZ, Köln, Germany) for his help with fluorescence microscopy and Monica Pons (Servei de Microscòpia Confocal, IBMB-CSIC, Barcelona, Spain) for her excellent technical assistance in the confocal analysis. Special thanks are due to Pablo Leivar Rico and Albert Boronat (Facultad Quimicas, Universidad Barcelona, Barcelona, Spain) for providing support and facilities to perform the co-transformation experiments with Arabidopsis cell line T87. This work was supported by grants from the Deutsche Forschungsgemeinschaft (KO1483/3-2) and HSFP (RG0162/2000-M). A.F.T. acknowledges support from CICYT-BIO-99-453.
References
- 1.The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature, 408, 796–815. [DOI] [PubMed] [Google Scholar]
- 2.Pandey A. and Mann,M. (2000) Proteomics to study genes and genomes. Nature, 405, 837–846. [DOI] [PubMed] [Google Scholar]
- 3.Wisman E. and Ohlrogge,J. (2000) Arabidopsis microarray service facilities. Plant Physiol., 124, 1468–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fields S. and Song,O.K. (1989) A novel genetic system to detect protein-protein interactions. Nature, 340, 245–246. [DOI] [PubMed] [Google Scholar]
- 5.Fields S. and Sternglanz,R. (1994) The two-hybrid system: an assay for protein-protein interactions. Trends Genet., 10, 286–292. [DOI] [PubMed] [Google Scholar]
- 6.Hardie D.G., Carling,D. and Carlson,M. (1998) The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem., 67, 821–855. [DOI] [PubMed] [Google Scholar]
- 7.Jiang R. and Carlson,M. (1996) Glucose regulates protein interactions within the yeast Snf1 protein kinase complex. Genes Dev., 10, 3105–3115. [DOI] [PubMed] [Google Scholar]
- 8.Carlson M. (1999) Glucose repression in yeast. Curr. Opin. Microbiol., 2, 202–207. [DOI] [PubMed] [Google Scholar]
- 9.Kemp B.E., Mitchelhill,K.I., Stapleton,D., Michell,B.J., Chen,Z.-P. and Witters,L.A. (1999) Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem. Sci., 24, 22–25. [DOI] [PubMed] [Google Scholar]
- 10.Jiang R. and Carlson,M. (1997) The Snf1 protein kinase and its activating subunit, Snf4, interact with distinct domains of the Sip1/Sip2/Gal83 component in the kinase complex. Mol. Cell. Biol., 17, 2099–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyck J.R.B., Gao,G., Widmer,J., Stapleton,D., Fernandez,C.S., Kemp,B.E. and Witters,L.A. (1996) Regulation of 5′-AMP-activated protein kinase activity by the noncatalytic β and γ subunits. J. Biol. Chem., 271, 17798–17803. [DOI] [PubMed] [Google Scholar]
- 12.Thornton C., Snowden,M.A. and Carling,D. (1998) Identification of a novel AMP-activated protein kinase β subunit isoform that is highly expressed in skeletal muscle. J. Biol. Chem., 273, 12443–12450. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt M.C. and McCartney,R.R. (2000) β-Subunits of Snf1 kinase are required for kinase function and substrate definition. EMBO J., 19, 4936–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alderson A., Sabelli,P.A., Dickinson,J.R., Cole,D., Richardson,M., Kreis,M., Shewry,P.R. and Halford,N.G. (1991) Complementation of snf1, a mutation affecting global regulation of carbon metabolism in yeast, by a plant protein kinase cDNA. Proc. Natl Acad. Sci. USA, 88, 8602–8605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muranaka T., Banno,H. and Machida,Y. (1994) Characterization of tobacco protein kinase NPK5, a homolog of Saccharomyces cerevisiae Snf1 that constitutively activates expression of the glucose-repressible SUC2 gene for a secreted invertase of S. cerevisiae. Mol. Cell. Biol., 14, 2958–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhalerao R.P., Salchert,K., Bakó,L., Ökrész,L., Szabados,L., Murakana,T., Machida,Y., Schell,J. and Koncz,C. (1999) Regulatory interaction of PRL1 WD protein with Arabidopsis SNF-like protein kinases. Proc. Natl Acad. Sci. USA, 96, 5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouly J.-P., Gissot,L., Lessard,P., Kreis,M. and Thomas,M. (1999) Arabidopsis thaliana proteins related to the yeast SIP and SNF4 interact with AKINα1, an SNF1-like protein kinase. Plant J., 18, 541–550. [DOI] [PubMed] [Google Scholar]
- 18.Lakatos L., Klein,M., Höfgen,R. and Bánfalvi,Z. (1999) Potato StubSNF1 interacts with StubGAL83: a plant protein kinase complex with yeast and mammalian counterparts. Plant J., 17, 569–574. [DOI] [PubMed] [Google Scholar]
- 19.Kleinow T., Bhalerao,R., Breuer,F., Umeda,M., Salchert,K. and Koncz,C. (2000) Functional identification of an Arabidopsis Snf4 ortholog by screening for heterologous multicopy suppressors of snf4 deficiency in yeast. Plant J., 23, 115–122. [DOI] [PubMed] [Google Scholar]
- 20.Lumbreras V., Albá,M., Kleinow,T., Koncz,C. and Pagés,M. (2000) Domain fusion between SNF1-related kinase subunits during plant evolution. EMBO Rep., 2, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halford N.G. and Hardie,D.G. (1998) SNF1-related protein kinases: global regulators of carbon metabolism in plants? Plant Mol. Biol., 37, 735–748. [DOI] [PubMed] [Google Scholar]
- 22.Ferrando A., Farràs,R., Jasik,J., Schell,J. and Koncz,C.(2000) Intron-tagged epitope: a tool for facile detection and purification of proteins expressed in Agrobacterium-transformed plant cells. Plant J., 22, 1–8. [DOI] [PubMed] [Google Scholar]
- 23.Vancanneyt G., Schmidt,R., O’Connor-Sanchez,A., Willmitzer,L. and Rocha-Sosa,M. (1990) Construction of an intron-containing marker gene: splicing of the intron in transgenic plants and its use in monitoring early events in Agrobacterium-mediated plant transformation. Mol. Gen. Genet., 220, 245–250. [DOI] [PubMed] [Google Scholar]
- 24.Eckes P., Rosahl,S., Schell,J. and Willmitzer,L. (1986) Isolation and characterization of a light-inducible, organ-specific gene from potato and analysis of its expression after tagging and transfer into tobacco and potato shoots. Mol. Gen. Genet., 205, 14–22. [Google Scholar]
- 25.Hancock K.R., Phillips,L.D., White,D.W.R. and Ealing,P.M. (1997) pPE1000: a versatile vector for the expression of epitope tagged foreign proteins in transgenic plants. Biotechniques, 22, 861–865. [DOI] [PubMed] [Google Scholar]
- 26.Koncz C. and Schell,J. (1986) The promoter of TL-DNA gene 5 controls the tissue specific expression of chimaeric genes carried by a novel type of Agrobacterium binary vector. Mol. Gen. Genet., 204, 383–396. [Google Scholar]
- 27.Koncz C., Martini,N., Szabados,L., Hrouda,M., Bachmair,A. and Schell,J. (1994) Specialized vectors for gene tagging and expression studies. In Gelvin,S.B., Schilperoort,R.A. and Verma,D.P.S. (eds), Plant Molecular Biology Manual. Kluwer Academic, Dordrecht, The Netherlands, Vol. B2, pp. 1–22.
- 28.Szabados L., Charrier,B., Kondorosi,A., de Bruijn,F.J. and Ratet,P. (1995) New plant promoter and enhancer testing vectors. Mol. Breeding, 1, 419–423. [Google Scholar]
- 29.Haseloff J., Siemering,K.R., Prasher,D.C. and Hodge,S. (1997) Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc. Natl Acad. Sci. USA, 94, 2122–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maing G.D., Reynolds,S. and Gartland,J.S. (1995) Electroporation protocols for Agrobacterium. In Gartland,K.M.A and Davey,M.R. (eds), Methods in Molecular Biology: Agrobacterium Protocols. Humana Press, Totowa, NJ, Vol. 44, pp. 405–412. [DOI] [PubMed]
- 31.Németh K., Salchert,K., Putnoky,P., Bhalerao,R., Koncz-Kálmán,Z., Stankovic-Stangeland,B., Bakó,L., Mathur,J., Ökrész,L., Stabel,S., Geigenberger,P., Stitt,M., Rédei,G.P., Schell,J. and Koncz,C. (1998) Pleiotropic control of glucose and hormone responses by PRL1, a nuclear WD protein, in Arabidopsis. Genes Dev., 12, 3959–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Axelos M., Curie,C., Mazzolini,L., Bardet,C. and Lescure,B. (1992) A protocol for transient gene expression in Arabidopsis thaliana protoplasts isolated from cell suspension cultures. Plant Physiol. Biochem., 30, 123–128. [Google Scholar]