

Abstract
Changes in synaptic strength and connectivity are thought to be a major mechanism through which many gene variants cause neurological disease. Hyperactivation of the PI3K-mTOR signaling network, via loss of function of repressors such as PTEN, causes epilepsy in humans and animal models, and altered mTOR signaling may contribute to a broad range of neurological diseases. Changes in synaptic transmission have been reported in animal models of PTEN loss; however, the full extent of these changes, and their effect on network function, is still unknown. To better understand the scope of these changes, we recorded from pairs of mouse hippocampal neurons cultured in a two-neuron microcircuit configuration that allowed us to characterize all four major connection types within the hippocampus. Loss of PTEN caused changes in excitatory and inhibitory connectivity, and these changes were postsynaptic, presynaptic, and transynaptic, suggesting that disruption of PTEN has the potential to affect most connection types in the hippocampal circuit. Given the complexity of the changes at the synaptic level, we measured changes in network behavior after deleting Pten from neurons in an organotypic hippocampal slice network. Slices containing Pten-deleted neurons showed increased recruitment of neurons into network bursts. Importantly, these changes were not confined to Pten-deleted neurons, but involved the entire network, suggesting that the extensive changes in synaptic connectivity rewire the entire network in such a way that promotes a widespread increase in functional connectivity.
SIGNIFICANCE STATEMENT Homozygous deletion of the Pten gene in neuronal subpopulations in the mouse serves as a valuable model of epilepsy caused by mTOR hyperactivation. To better understand how gene deletions lead to altered neuronal activity, we investigated the synaptic and network effects that occur 1 week after Pten deletion. PTEN loss increased the connectivity of all four types of hippocampal synaptic connections, including two forms of increased inhibition of inhibition, and increased network functional connectivity. These data suggest that single gene mutations that cause neurological diseases such as epilepsy may affect a surprising range of connection types. Moreover, given the robustness of homeostatic plasticity, these diverse effects on connection types may be necessary to cause network phenotypes such as increased synchrony.
Keywords: epilepsy, mTOR, neural synchrony, synaptic transmission
Introduction
Neurons that release glutamate or GABA send and receive the majority of synapses in the mammalian brain. The fast excitation and inhibition provided by these synapses are the building blocks of circuits throughout the brain, and the relative strength and timing of neurotransmission at these synapses regulate information flow through the circuits. Pathological changes in synaptic transmission are thought to underlie a range of neurological diseases, from epilepsy (Casillas-Espinosa et al., 2012; Euro EPINOMICS-RESC Consortium, 2014) and autism (Zoghbi and Bear, 2012) to schizophrenia (Sigurdsson, 2016); however, it is currently unknown how many disease phenotypes can be assigned to dysfunction at specific synapses, or whether distributed dysfunction over many synaptic motifs combine to cause an emergent network pathology (Nelson and Valakh, 2015).
Although glutamatergic and GABAergic neurons can be further subdivided into numerous specialized types, at a more basic level, there are four types of connections: (1) excitatory synapses onto other excitatory neurons, (2) excitatory synapses onto inhibitory neurons, (3) inhibitory synapses onto excitatory neurons, and (4) inhibitory synapses onto other inhibitory neurons. These synaptic motifs form the basis for microcircuit elements, such as feedforward, feedback, and lateral inhibition, which in turn, underlie network operations, such as rate and population coding. Gene variants that cause neurological disease potentially affect all of these connection motifs, and their ultimate consequences are likely a summation of the additive effects of these changes (Nelson and Valakh, 2015). However, studies of synaptic transmission in neurological disease models usually focus on one or two specific connections, due to both the difficulty of recording from multiple connection types or pairs in ex vivo preparations, as well as the quest to establish sufficiency for causing disease phenotypes.
Variants in the PTEN gene are associated with autism, megalencephaly, hemimegaencephaly, and epilepsy in humans (Butler et al., 2005; Crino, 2011; Mirzaa and Poduri, 2014), and numerous animal models with Cre-mediated Pten deletion in various neuronal subpopulations have been shown to recapitulate the phenotypes of these diseases (Lasarge and Danzer, 2014; Switon et al., 2017). In animal models, the effects of homozygous Pten deletion have been most intensely studied at excitatory-excitatory connections, where postsynaptic PTEN loss has been shown to increase synaptic strength (Weston et al., 2012; Xiong et al., 2012; Williams et al., 2015). However, the effects of homozygous Pten deletion on synaptic connections involving GABAergic neurons, and the effects of presynaptic PTEN loss, are not well understood (Weston et al., 2012; Vogt et al., 2015). Furthermore, most animal models of epilepsy due to mTOR hyperactivation use selective Pten deletion, and many human epilepsy patients have somatic PTEN mutations (Lee et al., 2012); therefore, these epileptic brains are comprised of a mixture of genetically normal and mutant neurons, and many synapses will be genetically abnormal only on either the presynaptic or postsynaptic side. Thus, how mosaic PTEN loss affects the diversity of fast connections found in the brain, how it affects presynaptic and postsynaptic transmission, and how these changes sum to affect the activity of the neurons and the function of the circuits in which they operate are key questions that do not have sufficient answers.
To better understand how genetic mutations that cause neurological disease affect synaptic and network function, and to dissect the presynaptic and postsynaptic mechanisms that dictate the altered neuronal behaviors downstream of these mutations, we adapted a two-neuron culture preparation that allowed us to test excitatory-excitatory, excitatory-inhibitory, inhibitory-excitatory, and inhibitory-inhibitory synaptic motif functions. Using Pten-deleted neurons as a model, we showed that the postsynaptic effects of Pten deletion were independent of the connection type, whereas other effects were connection-specific. Furthermore, using multicellular calcium imaging of hippocampal slice cultures in which Pten was deleted in a mosaic manner, we explored the effects on the spontaneous activity of neurons and their interactions, and found that PTEN loss led to an increase in functional connectivity that involved both genetically normal and mutant neurons.
Materials and Methods
Mice and cell culture.
Animal housing and use were in compliance with the National Institutes of Health Guide for the care and use of laboratory animals and were approved by the institutional animal care committees at Baylor College of Medicine and the University of Vermont. Experiments used Ptentm1Hwu/J mice that possess loxP sites on either side of exon 5 of the Pten gene (The Jackson Laboratory). The mice were crossed to C57BL/6 wild-type mice to generate Ptenwt/loxp mice, which were then bred to generate the Ptenwt/wt and Ptenloxp/loxp genotypes used for the study.
Microisland astrocyte cultures were prepared according to published procedures (Burgalossi et al., 2012; Weston et al., 2012). Microislands were made by stamping collagen (0.7 mg/ml) and poly-d-lysine (0.1 mg/ml) on agarose-coated glass coverslips with a custom-built stamp to achieve uniform size (200 μm diameter). Astrocytes derived from wild-type neonatal cortices were grown on microislands for 1 week before plating the neurons. For the primary culture, the entire hippocampi from postnatal day 0–1 (P0–P1) mice of both sexes from a litter from Ptenwt/loxp × Ptenwt/loxp parents were removed and maintained in Neurobasal-A media (NBA, Thermo Fisher Scientific) while DNA was isolated from their cortices and used for genotyping to identify Ptenwt/wt and Ptenloxp/loxp pups. Once identified, the hippocampi from these genotypes were digested with papain (Worthington) for 45 min and then dissociated into cell suspensions containing 500–1000 neurons/μl. For mass cultures used to generate Figure 1F, neurons were plated directly into 6-well culture plates on a confluent layer of astrocytes at a density of 1 × 105 neurons per well. For two-neuron cultures, the cell suspensions were split into two tubes of 500 μl each. To one of these tubes, 2 μl of AAV8-hsyn-mCherry-Cre (titer: 2 × 1013 GC/ml, UNC Vector Core) was added, and the virus was left on for 3 h while the cell suspensions were gently shaken (500 rpm) at 37°C on a Thermomixer (Eppendorf). After 3 h, the tubes were centrifuged three times at 1500 rpm for 5 min on a benchtop centrifuge and resuspended in fresh NBA each time. After the third resuspension, the cells were counted. Neurons (2000–4000) were then added to the wells of a six-well plate to generate the Control pairs, which included the Ptenwt/wt and Ptenwt/wt plus virus groups, and the Mutant pairs, which included the Ptenwt/wt and Ptenloxp/loxp plus virus groups. The neurons were cultured in NBA medium supplemented with Glutamax and B-27 (Invitrogen).
Figure 1.
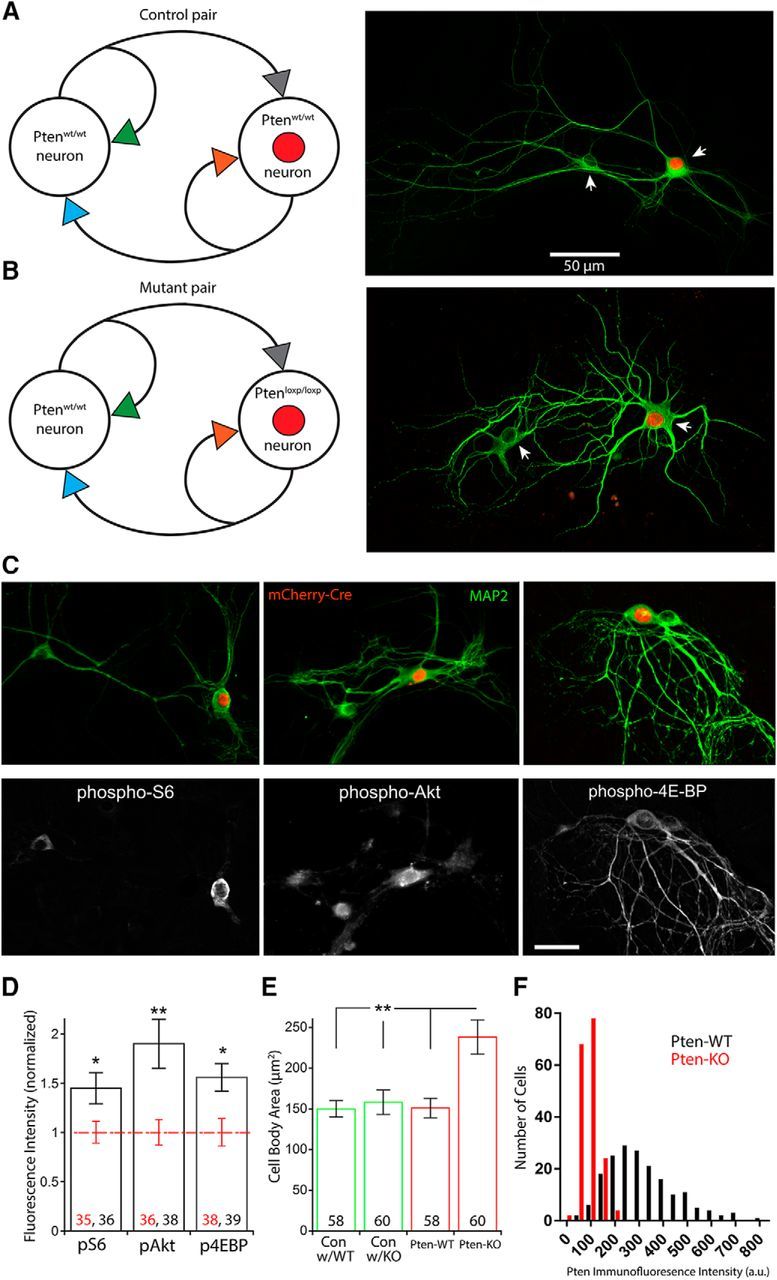
Loss of PTEN in the two-neuron culture system leads to selective hyperactivation of PI3K-mTOR signaling. A, Schematic of a “Control” pair (left), which consisted of a Ptenwt/wt neuron (Con) and a Ptenwt/wt neuron that expressed the mCherry-Cre protein in the nucleus (red circle) (Pten-WT). A representative image of a Control pair (right) shows the structure of the two neurons revealed by MAP2 immunofluorescence (green) and selective expression of the mCherry fluorescence (red). Arrows point to the cell bodies. B, Schematic of a “Mutant” pair (left), which consisted of a Ptenwt/wt neuron (Con) and a Ptenloxp/loxp neuron that expressed the mCherry-Cre protein in the nucleus (red circle) (Pten-KO). A representative image of a Mutant pair (right) shows the structure of the two neurons revealed by MAP2 immunofluorescence (green) and selective expression of the mCherry fluorescence (red). Arrows point to the cell bodies. C, Representative images of Mutant pairs (top row) show fluorescence from the mCherry-Cre protein (red) and MAP2 immunofluorescence (green), with corresponding images of pS6 (left), pAKT473 (center), and p4E-BP (right) immunofluorescence (bottom row). Scale bar, 25 μm. D, Bar graph represents the normalized increases in fluorescence intensity for pS6 (p = 0.024), pAKT (p = 0.002), and p4E-BP (p = 0.011) in Pten-KO neurons versus Pten-WT neurons (mean ± SEM). Black and red numbers indicate the number of Pten-KO and Pten-WT neurons analyzed, respectively. The values from these datasets were derived from three independent cultures and were normally distributed. *p < 0.05 (two-tailed Student's t test). **p < 0.01 (two-tailed Student's t test). E, Bar graph represents the increase in neuronal cell body area in Pten-KO neurons versus Pten-WT neurons and Con neurons (mean ± SEM). p < 0.01. Numbers on bars indicate the number of neurons analyzed. Kruskal–Wallis test followed by Dunn's multiple comparisons test. F, Histogram showing the Pten immunofluorescence intensity in Pten-WT (n = 180 neurons, black bars) and Pten-KO (n = 176 neurons, red bars) neurons.
Electrophysiology.
Paired whole-cell voltage-clamp recordings were performed with a patch-clamp amplifier (MultiClamp 700B amplifier; Molecular Devices) under the control of Clampex 10.3 or 10.5 (Molecular Devices, pClamp, RRID:SCR_011323). Data were acquired at 10 kHz and low-pass filtered at 4 kHz. The series resistance was compensated at 70%, and only cells with series resistances <15 mΩ were analyzed. The pipette resistance was between 2 and 4 mΩ. Standard extracellular solution contained the following (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 MgCl2, and 2 CaCl2, pH 7.3 (305 mOsm). Internal solution contained the following: 136 mm KCl, 17.8 mm HEPES, 1 mm EGTA, 0.6 mm MgCl2, 4 mm ATP, 0.3 mm GTP, 12 mm creatine phosphate, and 50 U/ml phosphocreatine kinase. These concentrations set the chloride reversal potential high enough that GABA receptor-mediated synaptic responses resulted in an inward current. All experiments were performed at room temperature (22°C–23°C). Dual whole-cell recordings were performed on neuron pairs from control and mutant groups in parallel on the same day (day 7–10 in vitro). Only neuron pairs in which red fluorescence expression was visually identified in the nuclear region of one of the two neurons were targeted for patching. No effort was made to target specific subpopulations of glutamatergic or GABAergic neurons for recording; thus, our dataset likely includes glutamatergic pyramidal and granule cells, as well as the various classes of GABAergic interneurons that are found in the hippocampus.
For voltage-clamp experiments, neurons were held at −70 mV unless noted. Action potential-evoked EPSCs or IPSCs were triggered by a 2 ms somatic depolarization to 0 mV. The shape of the evoked response and the effect of receptor antagonists [3 mm kynurenic acid (KYN, Tocris Bioscience) or 20 μm bicuculline (BIC, Tocris Bioscience)] were applied to verify the glutamatergic or GABAergic identities of the neurons. Neurons were stimulated at 0.2 Hz (for EPSCs) or 0.1 Hz (for IPSCs) in standard external solution to measure basal-evoked synaptic responses. To determine the number of releasable synaptic vesicles onto each neuron in the glutamatergic-glutamatergic and GABAergic-GABAergic pairs, we measured the charge transfer of the transient synaptic current induced by a 4 s application of hypertonic sucrose solution directly onto the neuron pair and then divided the sucrose charge by the charge of the average miniature event onto the same neuron. To determine the number of releasable synaptic vesicles at each synaptic connection in the glutamatergic-GABAergic pairs, solutions of sucrose plus 3 mm KYN and sucrose plus 20 μm BIC were sequentially applied. The charge transfer recorded in the sucrose/KYN solution was then divided by the charge of the average miniature IPSC (mIPSC) onto that neuron to calculate the number of GABAergic synaptic vesicles for that connection, and the charge transfer recorded in the sucrose/BIC solution was divided by the charge of the average miniature EPSC (mEPSC) onto that neuron to calculate the number of GABAergic synaptic vesicles for the connection.
Miniature event detection.
Miniature synaptic potentials were recorded for 60–80 s in 3 mm KYN or 20 μm BIC to isolate GABAergic and glutamatergic events, respectively, with 500 nm TTX. For each cell, data were filtered at 1 kHz and analyzed using template-based miniature event detection algorithms implemented in the AxoGraph X 1.4 analysis software. The threshold for detection was set at three times the baseline SD from a template of 0.5 ms rise time and 3 ms decay for glutamatergic events and 0.5 and 20 ms for GABAergic events.
Immunocytochemistry.
Neurons were rinsed three times with PBS, fixed with 4% PFA for 30 min, and then washed with PBS three times again. Neurons were then placed in blocking solution (10% normal goat serum, 0.1% Triton X-100, and PBS) at room temperature for 1 h. The following primary antibodies in blocking solution were then applied to the neurons at 4°C overnight: MAP2 (mouse monoclonal, 1:1000 dilution, Synaptic Systems, catalog #188 011, RRID:AB_2147096), phospho-S6 Ribosomal Protein Ser240/244 (rabbit monoclonal, 1:1000 dilution, Cell Signaling Technology, catalog #5364, RRID:AB_10694233), phospho-AKT Ser473 (rabbit monoclonal, 1:1000 dilution, Cell Signaling Technology, catalog #4060, RRID:AB_2315049), phospho-4E-BP1 Thr37/46 (rabbit monoclonal, 1:1000 dilution, Cell Signaling Technology, catalog #2855, RRID:AB_560835), VGLUT1 (rabbit polyclonal, 1:5000 dilution, Synaptic Systems, catalog #135302, RRID:AB_887877), VGAT (guinea pig polyclonal, 1:1000 dilution, Synaptic Systems, catalog #131004, RRID:AB_887873), and PTEN (rabbit polyclonal 1:500 dilution, Cell Signaling Technology, catalog #9559, RRID: AB_390810.) Following primary antibody application, cells were washed again three times in PBS and then incubated in the following AlexaFluor secondary antibodies (Invitrogen) for 1 h at room temperature: goat anti-mouse 488 (1:1000, catalog #A-11017, RRID:AB_143160), goat anti-rabbit 647 (1:1000, catalog #A-21244, RRID:AB_141663), goat anti-mouse 555 (1:1000, catalog #A-21422, RRID:AB_141822), and goat anti-guinea pig 488 (1:1000 catalog #A-11073, RRID:AB_142018). Cells were then mounted to slides with Prolong Gold Antifade (Invitrogen) and were allowed to cure for 24 h.
Images (1024 × 1024 pixels) were obtained using a DeltaVision automated upright epifluorescent imaging system (Applied Precision/GE Life Sciences) attached to an Olympus IX70 microscope with a 20× oil objective, SoftWoRx software, and a CoolsnapHQ charge-coupled device digital camera (Photometrics) by an investigator blinded to the genotypes of the imaged groups. Images were acquired using equal exposure times between groups in stacks of 8–12 planes at 0.5 μm depth intervals and then deconvolved. Stacks were processed using Fiji software (Schindelin et al., 2012) to create maximum intensity projections. For neuron pair reconstructions, the MAP2 fluorescence in the cell bodies and dendritic trees was traced using the NeuronJ plugin for Fiji (Meijering et al., 2004). Dendritic branches that could not be definitively assigned to a Cre- (red fluorescence) or non–cre-expressing neuron were excluded from the tracing. Glutamatergic and GABAergic neurons in each pair were identified based on VGLUT1 and VGAT expression, respectively, at the cell soma (Hartman et al., 2006; Chang et al., 2014). The total number of glutamatergic and GABAergic synapses was quantified by manually counting the punctae from the fluorescence signals for VGLUT1 and VGAT, respectively. Only punctae with areas <2 μm2 and within 5 μm of an assigned dendrite were counted.
For analysis of the levels of mTOR effectors, image exposure times and settings were kept the same between groups in a culture and were optimized to ensure that there were no saturated pixels. Maximum intensity projections were created using Fiji, and the background was subtracted using the rolling ball method with a radius of 50 μm. Regions of interest (ROIs) were drawn around the cell body using the MAP2 channel, and then the mean fluorescence intensity and cell body area were measured for phospho-S6, phospho-AKT and phospho-4E-BP for each neuron imaged. Because the absolute values of the fluorescence intensity varied between cultures, the values were normalized to the mean value of the Pten-WT neurons for each culture.
Hippocampal slice preparation.
Slice cultures of the hippocampus were made from P4-P5 Ptenwt/wt and Ptenloxp/loxp pups. The hippocampi were dissected free of adhering tissue and placed on a Teflon stage of a custom-built mechanical tissue chopper covered with filter paper and a small amount of L-15 media (Invitrogen). Transverse (375 μm) slices were cut and placed on prewarmed Millicell membrane inserts (Millipore) sitting in 35 mm Petri dishes filled with 1 ml of NBA medium supplemented with Glutamax and B-27 (Invitrogen). Two slices were put on each insert and arranged such that they did not contact one another. The culture media were changed at 2 day intervals. Approximately 4–5 h after slice preparation, AAV8-hsyn-mCherry-Cre and AAV9.Syn.GCaMP6m.WPRE.SV40M (titer: 4–5 × 1013 GC/ml, Penn Vector Core) were mixed in equal volumes and injected (three injection sites per slice, 25 nl per site) into slice cultures using a Nanoject II (Drummond Scientific). Expression of the fluorescent proteins was monitored daily and was usually apparent 2–3 d post-transduction.
Two-photon imaging.
After 7–10 d in vitro, the membrane inserts with the slices were transferred from the incubator to a custom-made chamber constantly perfused with oxygenated ACSF containing the following (in mm): 125 NaCl, 1.25 NaH2PO4, 2.5 KCl, 25 NaHCO3, 2 CaCl2, 1 MgCl2, and 15 glucose, and maintained at a temperature of 32°C–34°C with an automatic temperature controller (Warner Instruments). This chamber was mounted on the stage of a Zeiss 710 two-photon microscope (Carl Zeiss) equipped with a Chameleon Ti-Sapphire laser (Coherent). After 45 min recovery time, each slice was scanned using a 750 nm wavelength laser to identify regions with robust expression of both GCaMP and mCherry fluorescence. High-resolution z stacks of these regions were acquired to generate images as shown in Figure 6B. The laser wavelength was then adjusted to 920 nm, and time-lapse images of the CA1 or CA3 regions of the slices were acquired with a 20× (NA 1.0) water-immersion objective at 20 frames/s for 10 min using the Zen 2010 software. For loose-patch recordings, pipettes of 4–6 mΩ resistance were filled with ACSF and Alexa-594 (1:2000, Invitrogen). Pipettes were guided to GCaMP-expressing cells, and a low-resistance seal was established (≈100 mΩ) in voltage-clamp mode using a MultiClamp 700B amplifier. The holding potential was adjusted to achieve zero current at the resting potential.
Figure 6.
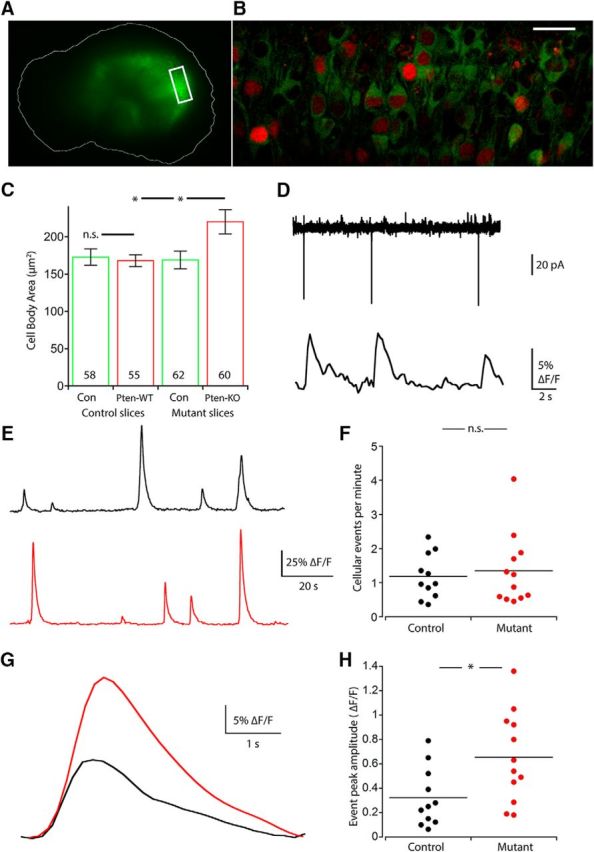
Multicellular calcium imaging shows that neurons in Mutant slices have greater event amplitude but not frequency than those of Controls. A, A stereoscope image shows GFP fluorescence from GCaMP6 expression in a hippocampal slice culture. An outline of the slice is drawn for reference. Box indicates the representative area selected for imaging. B, A two-photon image represents the fluorescence from GCaMP6 (green) and mCherry-Cre (red) coexpression in the principal cell layer of the hippocampal slice culture shown in A. Scale bar, 25 μm. C, Bar graph represents the increase in neuronal cell body area (mean ± SEM) in Pten-KO neurons versus Pten-WT and Con neurons. p = 0.015 versus Pten-WT (one-way ANOVA with Tukey's post test). p = 0.012 versus Con (one-way ANOVA with Tukey's post test). Numbers on bars indicate the number of neurons analyzed. D, Representative traces from a loose-patch recording (top) with the corresponding time-locked cellular GCaMP fluorescence from the patched neuron (bottom). E, Representative cellular fluorescence traces from a neuron from a Control (black) and Mutant (red) slice. F, Dot plot represents the lack of a change in event frequency (events per minute) in Mutant slices. Each dot represents the average event frequency of all individual neurons in one slice. G, Representative average event traces from Control (black) and Mutant (red) slices. H, A dot plot represents the increase in event amplitude (ΔF/F) in Mutant slices. Each dot represents the average event amplitude of all individual neurons in one slice. *p < 0.05 (Mann-Whitney test).
Multicellular calcium imaging data analysis.
Imaging files were opened with Fiji, and the ROIs were manually drawn around all identifiable cell bodies. In cases of mild drift in the x-y plane, the StackReg plugin (Thévenaz et al., 1998) was used to align ROIs, and a Kallman Stack Filter was applied. The fluorescence change versus time for each cell was measured by averaging all pixels within the ROI, and these values were exported to AxoGraph X 1.4 for further analysis. Calcium events were detected via a template-based event detection algorithm. The threshold for event detection was set at 4–5 times the baseline SD from a template of 0.2 s rise time and 1.5 s decay time. Large amplitude events that were sometimes missed because of slower decay or rise times were manually detected, and the cells with no detected events were considered not active. ΔF/F0 was calculated as (F − F0)/F0, where F0 is the baseline fluorescence signal averaged over a 1 s period immediately before the event onset. To detect network events, the total number of event onsets within a sliding 1 s window was calculated for each set of images. A network event was considered to occur when the number of event onsets within a given window was significantly greater (p < 0.001) than expected by chance based on the number of active neurons in the slice and their spontaneous event frequency. The number of neurons participating in the network event was the number of neurons that had an event onset within that window. To find significantly correlated cell pairs, time series were exported to MATLAB (The MathWorks) and analyzed using the Mic2net software package (Smedler et al., 2014).
Experimental design and statistical analysis.
Data were analyzed offline with AxoGraph X 1.4 (AxoGraph Scientific, RRID:SCR_014284), KaleidaGraph 4.5 (Synergy Software), and Prism 7 (GraphPad Prism, RRID:SCR_002798). Values for electrophysiological analysis were always pooled from at least four independent cultures and ranged from 18 to 27 cells per group per connection type. Experiments were conducted and analyzed with the knowledge of the genotype of the animal except where noted elsewhere in the Methods. The n used to calculate means and errors for both electrophysiological analysis and immunostaining analysis was the number of neurons. The n used to calculate means and errors for the multicellular calcium imaging experiments are the number of slices. All numbers for each experiment are indicated in corresponding figures, figure legends, and in Table 1. Normality was tested by the D'Agostino and Pearson normality test; however, because values from the electrophysiological analysis were often not normally distributed, the nonparametric Mann–Whitney test was used to assess statistical significance. For other experiments, the statistical test used is stated in the figure legend that corresponds to each experiment.
Table 1.
Summary of the measurements of evoked synaptic strength in all pair typesa
| Eself1 | Epartner1 | Epartner2 | Eself2 | |
|---|---|---|---|---|
| E1-E2 (Fig. 2) | ||||
| Control, n = 25 | 3.84 ± 0.71 | 2.28 ± 0.52 | 2.47 ± 0.43 | 3.32 ± 0.68 |
| Mutant, n = 27 | 4.37 ± 0.73 | 2.21 ± 0.64 | 4.43 ± 0.67 | 7.49 ± 1.41 |
| p | 0.637 | 0.353 | 0.016 | 0.008 |
| I-E (Fig. 3) | Iself | Ipartner | Epartner | Eself |
| Control, n = 20 | 4.05 ± 0.69 | 1.72 ± 0.24 | 7.91 ± 1.48 | 3.37 ± 0.997 |
| Mutant, n = 23 | 8.84 ± 1.54 | 2.08 ± 0.33 | 16.48 ± 2.51 | 8.12 ± 1.71 |
| p | 0.029 | 0.436 | 0.005 | 0.044 |
| I1-I2 (Fig. 4) | Iself1 | Ipartner1 | Ipartner2 | Iself2 |
| Control, n = 20 | 5.21 ± 1.25 | 2.49 ± 0.65 | 2.58 ± 0.56 | 5.22 ± 1.49 |
| Mutant, n = 20 | 5.33 ± 0.85 | 2.97 ± 0.77 | 4.54 ± 0.80 | 13.92 ± 2.07 |
| p | 0.414 | 0.705 | 0.033 | 0.002 |
| E-I (Fig. 5) | Eself | Epartner | Ipartner | Iself |
| Control, n = 21 | 3.10 ± 0.97 | 8.71 ± 1.35 | 2.28 ± 0.32 | 4.42 ± 0.69 |
| Mutant, n = 18 | 2.35 ± 0.70 | 4.74 ± 0.81 | 4.81 ± 0.73 | 9.92 ± 2.01 |
| p | 0.278 | 0.024 | 0.007 | 0.014 |
aData are mean ± SEM of the peak synaptic current (nA) in response to a 2 ms depolarization to 0 mV delivered at 0.1–0.2 Hz. Measurements in the first column correspond to green traces, second column to blue traces, third column to gray traces, and fourth column to orange traces in Figures 2–5. p values were obtained from Mann–Whitney tests.
Results
Loss of PTEN in two-neuron cultures leads to selective hyperactivation of PI3K-mTOR signaling
To test for changes in synaptic connectivity and strength due to PTEN loss, we used a two-neuron microcircuit culture in which we systematically varied the neuron type (glutamatergic or GABAergic) and the genetic identity (Pten-KO or WT) of each neuron (Fig. 1A,B). This system allowed us to test the effects of PTEN loss in the presynaptic or postsynaptic compartment, and whether they were unique to specific synaptic motifs. Control neuron pairs consisted of one untransduced Ptenwt/wt neuron (Con) and one Ptenwt/wt neuron transduced with an adeno-associated virus (AAV) expressing the mCherry-Cre fusion protein (Pten-WT) (Fig. 1A). We compared these Control pairs to Mutant pairs, which consisted of one untransduced Ptenwt/wt neuron (Con) and one Ptenloxp/loxp neuron transduced with an AAV expressing the mCherry-Cre fusion protein (Pten-KO) (Fig. 1B).
To confirm that Pten-KO neurons show selective increases in PI3K-mTOR activity, we performed quantitative immunofluorescence analysis for three markers of the pathway's activity: phospho-S6 (S240/244), phospho-AKT (S473), and phospho-4E-BP (T37/46). The mean fluorescence intensities of all three markers in the cell bodies were increased in Pten-KO neurons relative to Pten-WT neurons (Fig. 1C,D). In addition to biochemical markers, increased cell size is a reliable readout of mTOR pathway hyperactivation. We therefore measured cell body area in the two-neuron cultures and found that the mean area of Pten-KO neurons was ∼1.6-fold larger than that of Pten-WT and both groups of Con neurons, a change qualitatively similar to previous findings in brain slices (1.5- to 2-fold increases) (Ljungberg et al., 2009; Vogt et al., 2015; Williams et al., 2015) (Fig. 1E). These experiments demonstrate that loss of PTEN in the two-neuron cultures causes an increase in PI3K-mTOR signaling that is specific to Ptenloxp/loxp neurons expressing the mCherry-Cre fusion protein. Finally, to assess whether expression of mCherry-Cre was sufficient to cause recombination in the majority of neurons in which it was visible, we performed immunostaining against the PTEN protein in dissociated cultures of Pten-WT and Pten-KO neurons. Plotting the values of PTEN fluorescence intensity for each neuron on a histogram (Fig. 1F) demonstrated a strong leftward shift in the distribution of PTEN fluorescence intensity in the Pten-KO group and a lack of a second peak to the right, suggesting that recombination of the Pten gene by the mCherry-Cre fusion protein is very efficient.
Loss of PTEN in E-E two-neuron microcircuits increases glutamatergic input onto the Pten-KO cell but does not increase its output
Synapses originating from excitatory neurons onto other excitatory neurons form the basis of the canonical hippocampal circuit. We therefore first tested the effects of PTEN loss on evoked synaptic strength between pairs of excitatory hippocampal neurons (E-E). In these E-E pairs, each neuron forms a synaptic connection with its partner, as well as with itself, meaning four distinct synaptic connections are present in each pair (Fig. 2A). We evoked action potentials in each of the two neurons, recorded the peak amplitude of the responses in voltage-clamp mode, and compared the amplitudes recorded from Control pairs with those from Mutant pairs (Fig. 2B; Table 1). The response amplitudes of the two synapses onto the Pten-KO neurons in Mutant pairs were increased compared with those onto the Pten-WT neurons in Control pairs (Fig. 2B,C, orange and gray symbols). However, the response amplitudes of the two synapses onto the Con neurons in Mutant pairs were not different from those onto Con neurons in Control pairs (Fig. 2B,C, green and blue symbols). These data suggest two things: (1) postsynaptic loss of PTEN in glutamatergic neurons is sufficient to increase excitatory synaptic input; and (2) presynaptic loss of PTEN does not increase glutamatergic output, at least onto other excitatory neurons that are Pten-WT.
Figure 2.
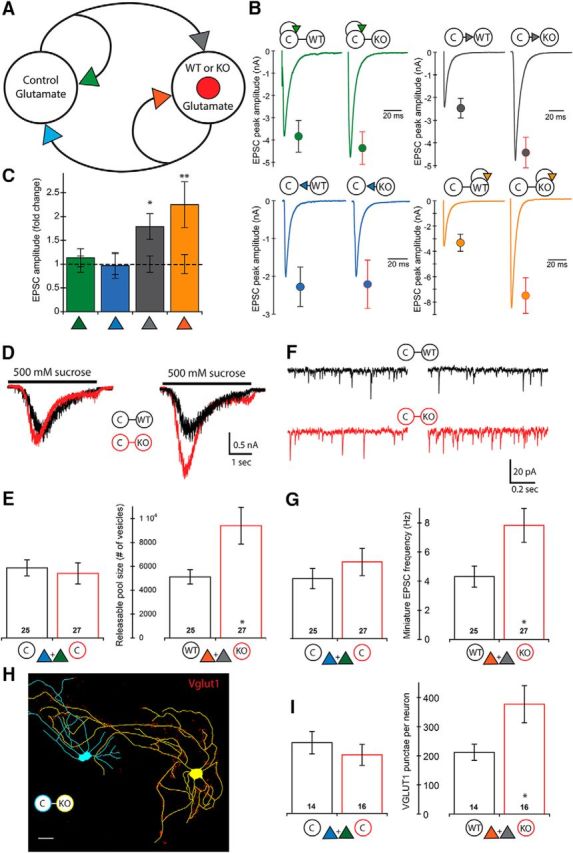
Loss of PTEN in E-E pairs leads to increased synaptic strength onto the postsynaptic neuron only. A, Schematic of an E-E pair shows the four synaptic connections tested (color-coded arrowheads). B, Representative traces and mean ± SEM values for Control (left traces and circles) and Mutant (right traces and circles) pairs are shown for each of the four connections. Color code matches the connections shown in A. A summary of all values, numbers of neurons tested, and statistical results are in Table 1. C, Bar graph represents the normalized mean ± SEM EPSC peak amplitude values for each of the four connections [Mutant pair relative to Control pair (dotted line)]. D, Representative traces indicate the current response to the 500 mm sucrose application used to assay the size of the readily releasable pool of synaptic vesicles onto each neuron in Control (black traces) and Mutant (red traces) pairs. E, Bar graphs represent the increase in the number of available synaptic vesicles onto the Pten-KO neurons (numbers on bars indicate the number of neurons analyzed). Bars represent color-matched traces directly above them. F, Representative traces indicate the mEPSC activity recorded in each neuron in Control (black traces) and Mutant (red traces) pairs. G, Bar graphs represent the increase in the mEPSC frequency recorded in the Pten-KO neurons (numbers on bars indicate the number of neurons analyzed). Bars represent color-matched traces directly above them. H, Representative tracing of a Con (blue) and Pten-KO (yellow) E-E Mutant pair showing VGLUT1 staining (red punctae). Scale bar, 25 μm. I, Bar graphs represent the increase in the number of VGLUT1 punctae on Pten-KO neurons in E-E pairs (numbers on bars indicate the number of neurons analyzed). *p < 0.05 (Mann-Whitney test). **p < 0.01 (Mann-Whitney test).
The increase in glutamatergic input in E-E pairs is due to increased synaptic connectivity
Previous studies of glutamatergic connections onto glutamatergic Pten-KO neurons, both in brain slice and culture preparations, have found increases in synaptic strength and indicate that the primary driver of these increases is an increase in the number of synapses formed. To test for an increase in synaptic connectivity, we used three measures, two physiological and one morphological. For the first physiological assay, we applied 500 mm sucrose to the pairs, which causes the neurons to release their full complement of fusion-competent synaptic vesicles (Fig. 2D). The number of vesicles released onto the Pten-KO neurons in Mutant pairs was increased by 1.84-fold over the number released onto Pten-WT neurons in Control pairs (Fig. 2E, right bars). The number of vesicles released onto Con neurons was not significantly different in Mutant versus Control pairs (Fig. 2E, left bars). For the second physiological assay, we recorded mEPSC frequencies (Fig. 2F). The mEPSC frequency recorded in the Pten-KO neurons in Mutant pairs was increased by 1.81-fold over the frequency recorded in Pten-WT neurons in Control pairs (Fig. 2G, right bars), whereas the frequency recorded in Con neurons was not significantly different in Mutant versus Control pairs (Fig. 2G, left bars).
The results from the two physiological assays indicate an increase in the number of synaptic vesicles available for release, which could be caused by two possibilities: (1) more vesicles per synapse or (2) more synapses. To distinguish between these possibilities, we analyzed two-neuron cultures immunostained with antibodies against VGLUT1 to mark glutamatergic synapses, and MAP2 to visualize dendrites. We then quantified the number of VGLUT1 punctae along dendrites that could be definitely assigned to either the Con or Pten-WT neuron in Control pairs, or the Con or Pten-KO neuron in Mutant pairs (Fig. 2H). The number of glutamatergic synapses onto Con neurons was not different in Control versus Mutant pairs (Fig. 2I, left bars), whereas the number of glutamatergic synapses onto Pten-KO neurons was increased 1.65-fold over the number onto Pten-WT neurons (Fig. 2I, right bars). This result indicates that the increased synaptic strength and vesicle number at synaptic connections onto Pten-KO neurons is largely due to an increase in the number of synapses, and that postsynaptic loss of PTEN is sufficient to cause this increase.
Loss of PTEN in the glutamatergic neuron in I-E pairs increases glutamatergic and GABAergic input onto the Pten-KO cell, as well as self-innervation by the GABAergic neuron
Because principal neurons in the hippocampus are strongly regulated by local GABAergic interneurons, we next tested how interactions between excitatory and inhibitory neurons are affected by PTEN loss. To do this, we assayed synaptic strength in pairs consisting of one GABAergic and one glutamatergic hippocampal neuron, in which Pten was deleted specifically in the glutamatergic neuron (I-E) (Fig. 3A). Similar to the E-E pairs, loss of PTEN in the postsynaptic glutamatergic neuron was sufficient to increase the strength of both synaptic connections onto that neuron. The peak amplitude of the glutamatergic synapse onto the Pten-KO neuron was increased 2.31-fold over the same connection onto the Pten-WT neuron (Fig. 3B,C, orange symbols), whereas the peak amplitude of the GABAergic synaptic connection was increased 2.13-fold (Fig. 3B,C, gray symbols), providing evidence that postsynaptic loss of PTEN can increase both glutamatergic and GABAergic synaptic strength.
Figure 3.
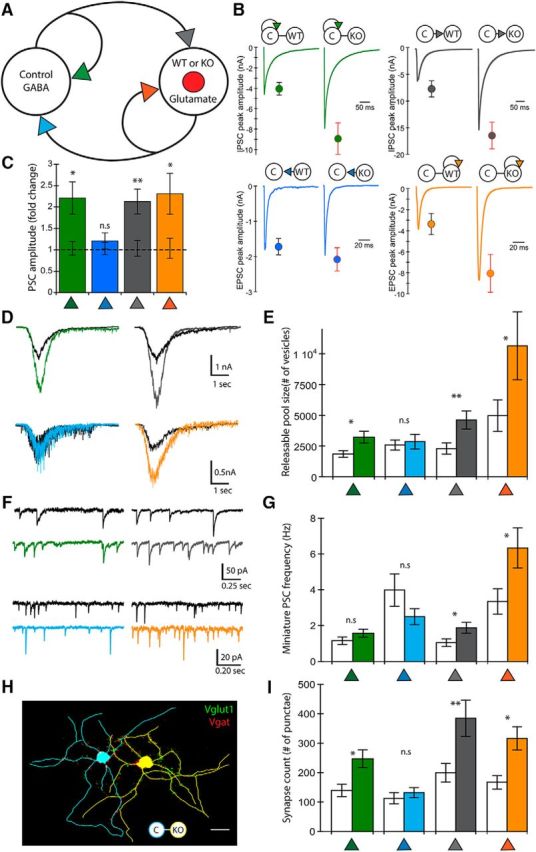
Loss of PTEN in I-E pairs leads to increased synaptic strength at three of the four connections. A, Schematic of an I-E pair shows the four synaptic connections tested (color-coded arrowheads). B, Representative traces and mean ± SEM values for Control (left traces and circles) and Mutant (right traces and circles) pairs are shown for each of the four connections. Color code matches the connections shown in A. A summary of all values, numbers of neurons tested, and statistical results are in Table 1. C, Bar graph represents the normalized mean ± SEM PSC peak amplitude values for each of the four connections [Mutant pair relative to Control pair (dotted line)]. D, Representative traces indicate the current response to the 500 mm sucrose application used to assay the size of the readily releasable pool of synaptic vesicles at each of the four connections. The traces from Control pairs are in black and those from Mutant pairs are colored to match the schematic in A. E, Bar graph represents the increase in the number of available synaptic vesicles in the three connections that showed increased synaptic strength. Control pair data (n = 20) are indicated by clear bars and Mutant pair data (n = 23) by colored bars to match the schematic in A. F, Representative traces of mEPSC and mIPSC activity. The traces from Control pairs are in black and those from Mutant pairs are colored to match the schematic in A. G, Bar graph represents the increase in the mEPSC frequency at two of the four connections. Control pair data (n = 20) are indicated by clear bars and Mutant pair data (n = 23) by colored bars to match the schematic in A. H, Representative tracing of a Con GABA (blue) and Pten-KO glutamate (yellow) I-E Mutant pair shows VGLUT1 (green punctae) and VGAT (red punctae) immunofluorescence. Scale bar, 25 μm. I, Bar graph represents the increase in the number of VGLUT1 and VGAT punctae on the Pten-KO neurons, and an increase in VGAT punctae on the Con neurons, in I-E pairs. Control pair data (n = 17) are indicated by clear bars and Mutant pair data (n = 19) by colored bars to match the schematic in A. *p < 0.05 (Mann-Whitney test). **p < 0.01 (Mann-Whitney test).
The strength of the glutamatergic synapses originating from the Pten-KO cell was again unchanged compared with that originating from the Pten-WT cell (Fig. 3B,C, blue symbols), suggesting that presynaptic loss of PTEN does not affect baseline synaptic output. In contrast to the results from E-E pairs, in which only the synaptic strength onto the Pten-KO neuron was increased, loss of PTEN in the glutamatergic neuron in the I-E pairs also increased the strength of the GABAergic synapses that the GABAergic neuron made with itself in Mutant pairs (Fig. 3B,C, green symbols). This result suggests that loss of PTEN in hippocampal glutamatergic neurons also increases inhibition of inhibition via a transsynaptic mechanism.
The increase in glutamatergic and GABAergic synaptic strength input in I-E pairs is due to increased synaptic connectivity
In contrast to the E-E pairs, in which only the sum of the two inputs onto a neuron can be assayed, in I-E pairs the synaptic connectivity of each of the four synapses can be tested separately using glutamate or GABA receptor antagonists. We first assayed the number of synaptic vesicles available for release at each connection by combining 500 mm sucrose with 3 mm KYN to block glutamate receptors, or 20 μm BIC to block GABA receptors (Fig. 3D). The number of available vesicles was increased in Mutant pairs at each of the three synapses that showed increased strength, but not at the one that did not (Fig. 3E). Next, we combined the synaptic blockers with TTX and measured the miniature current frequencies for each connection. The mIPSC (Fig. 3F,G, gray symbols) and mEPSC frequencies (Fig. 3F,G, orange symbols) onto the glutamatergic Pten-KO neuron were both increased compared with those onto the Pten-WT neuron. The mean mIPSC frequency of the GABAergic self-synapses (Fig. 3F,G, green symbols), though elevated, was not significantly greater in Mutant pairs than in Control pairs (Control: 1.15 ± 0.22 Hz vs Mutant: 1.57 ± 0.22 Hz, p = 0.20).
To test whether changes in synapse number cause the observed changes in synaptic strength, we simultaneously visualized glutamatergic and GABAergic synapses by immunostaining with antibodies against VGLUT1 and VGAT, respectively, and MAP2. We reconstructed the dendrites of each neuron in Control and Mutant pairs and quantified the number of VGLUT1 and VGAT punctae along dendrites that could be definitively assigned to either the Con or Pten-WT neuron in Control pairs, or the Con or Pten-KO neuron in Mutant pairs (Fig. 3H). There was an increase in the number of glutamatergic and GABAergic synapses onto Pten-KO neurons (Fig. 3I, gray and orange bars). Also, in agreement with the change in vesicle number, the number of GABAergic synapses onto Con neurons when they were paired with a Pten-KO neuron was significantly greater (p = 0.027) than when paired with a Pten-WT neuron (Fig. 3I, green bar). Thus, postsynaptic loss of PTEN in hippocampal excitatory neurons is sufficient to increase glutamatergic and GABAergic synaptic connectivity onto these neurons, and the increase in the number of GABAergic synapses is not limited to those made onto the Pten-KO neuron.
Loss of PTEN in I-I pairs increases GABAergic input onto the Pten-KO cell but does not increase its output
The increase in autaptic inhibition observed in the I-E pairs raises the possibility that PTEN loss may also increase the synaptic strength of connections between GABAergic neurons. Although regulation of principal cell activity is the more studied function of GABAergic neurons in the hippocampus, GABAergic neurons also form extensive contacts with each other to regulate the flow of information through hippocampal and cortical circuits (Chamberland and Topolnik, 2012; Jiang et al., 2015). We therefore tested the effects of PTEN loss on evoked synaptic strength between pairs of inhibitory hippocampal neurons (I-I) (Fig. 4A). We evoked action potentials in each of the two neurons and found, similar to the E-E pairs, that the response amplitudes of the two synapses onto the Pten-KO neurons in Mutant I-I pairs were increased compared with the response amplitudes onto the Pten-WT neurons in Control pairs (Fig. 4B,C, orange and gray symbols). Also, similar to E-E pairs, the response amplitudes of the two synapses onto the Con neurons were not different in Control versus Mutant pairs (Fig. 4B,C, green and blue symbols). These data indicate that, in the two-neuron culture system, postsynaptic loss of PTEN is sufficient to increase synaptic strength onto the Pten-KO neuron, but does not increase Pten-KO neuron output, and that these effects are not dependent on the neurotransmitter phenotype of the Pten-KO neurons. Moreover, these data suggest that the increased GABAergic self-inhibition seen in the I-E pairs (Fig. 3B,C, green symbols) requires that the Pten-KO neuron be a glutamatergic neuron.
Figure 4.
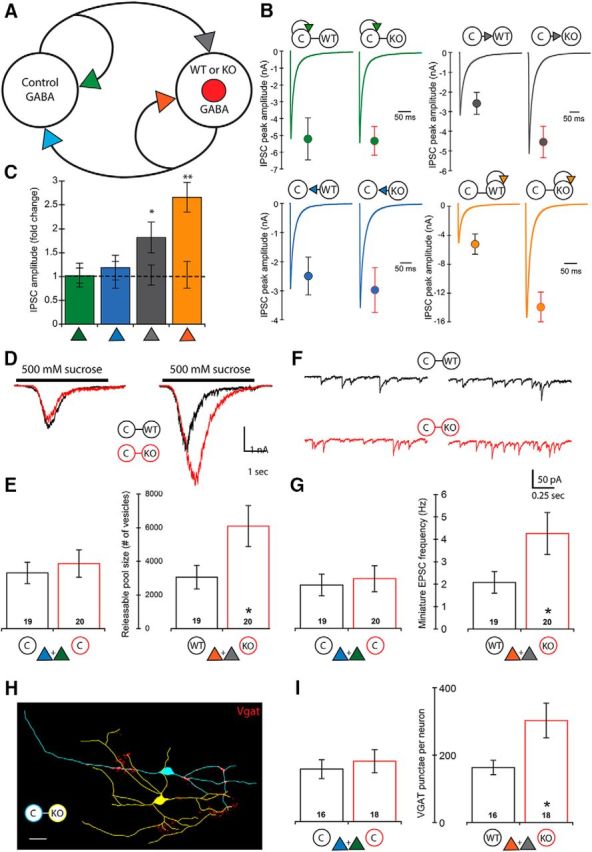
Loss of PTEN in I-I pairs leads to increased synaptic strength onto the postsynaptic neuron only. A, Schematic of an I-I pair shows the four synaptic connections tested (color-coded arrowheads). B, Representative traces and mean ± SEM values for Control (left traces and circles) and Mutant (right traces and circles) pairs are shown for each of the four connections. Color code matches the connections shown in A. A summary of all values, numbers of neurons tested, and statistical results are in Table 1. C, Bar graph represents the normalized mean ± SEM IPSC peak amplitude values for each of the four connections [Mutant pair relative to Control pair (dotted line)]. D, Representative traces indicate the current response to the 500 mm sucrose application used to assay the size of the readily releasable pool of synaptic vesicles onto each neuron in Control (black traces) and Mutant (red traces) pairs. E, Bar graphs represent the increase in the number of available synaptic vesicles onto the Pten-KO neurons (numbers on bars indicate the number of neurons analyzed). Bars represent color-matched traces directly above them. F, Representative traces indicate the mEPSC activity recorded in each neuron in Control (black traces) and Mutant (red traces) pairs. G, Bar graphs represent the increase in the mEPSC frequency recorded in the Pten-KO neurons (numbers on bars indicate the number of neurons analyzed). Bars represent color-matched traces directly above them. H, Representative tracing of a Con (blue) and Pten-KO (yellow) I-I Mutant pair showing VGAT staining (red punctae). Scale bar, 25 μm. I, Bar graphs represent the increase in the number of VGAT punctae on Pten-KO neurons in I-I pairs (numbers on bars indicate the number of neurons analyzed). *p < 0.05 (Mann-Whitney test). **p < 0.01 (Mann-Whitney test).
The increase in GABAergic input in I-I pairs is due to an increased number of synapses
To test whether an increase in synaptic connectivity also causes the increased GABAergic synaptic strength in I-I pairs, we measured synaptic vesicle numbers and mIPSC frequencies. The number of vesicles released by sucrose application onto the Pten-KO neurons in Mutant I-I pairs was increased by twofold over the number released onto Pten-WT neurons in Control pairs (Fig. 4D,E, right bars). The number of vesicles released onto Con neurons was not significantly different in Mutant versus Control pairs (Fig. 4D,E, left bars). The mIPSC frequency recorded in the Pten-KO neurons in Mutant pairs was increased by 2.05-fold over the frequency recorded in Pten-WT neurons in Control pairs (Fig. 4F,G, right bars), whereas the frequency recorded in Con neurons was not significantly different in Mutant versus Control pairs (Fig. 4F,G, left bars).
To visualize GABAergic synapses, we immunostained with antibodies against VGAT and MAP2. We then quantified the number of VGAT punctae along dendrites that could be definitively assigned to either the Con or Pten-WT neuron in Control pairs, or the Con or Pten-KO neuron in Mutant pairs (Fig. 4H). The number of GABAergic synapses onto Con neurons was not different in Control versus Mutant pairs (Fig. 4I, left bars), whereas the number of GABAergic synapses onto Pten-KO neurons was increased 1.79-fold over the number onto Pten-WT neurons (Fig. 4I, right bars). Thus, postsynaptic loss of PTEN in hippocampal inhibitory neurons is sufficient to increase GABAergic synaptic connectivity onto these neurons.
Loss of PTEN in the GABAergic neuron in E-I pairs increases glutamatergic and GABAergic input onto the Pten-KO cell but decreases its output onto the glutamatergic cell
Finally, we tested the effects of PTEN loss in GABAergic neurons on synaptic strength and connectivity between pairs of glutamatergic and GABAergic neurons, in which Pten was deleted selectively in the GABAergic neuron (E-I) (Fig. 5A). Similar to PTEN loss in glutamatergic neurons in the I-E pairs, postsynaptic loss of PTEN in the GABAergic neuron was sufficient to increase both excitation and inhibition onto that neuron (Fig. 5B,C, gray and orange symbols). The strength of the control neuron's self-input was unchanged (Fig. 5B,C, green symbols). We observed a unique effect on the output of the Pten-KO GABAergic neuron, however, as the strength of this connection was decreased by >50% (Fig. 5B,C, blue symbols), indicating that PTEN loss in GABAergic neurons can cause a decrease in inhibitory output onto glutamatergic neurons.
Figure 5.
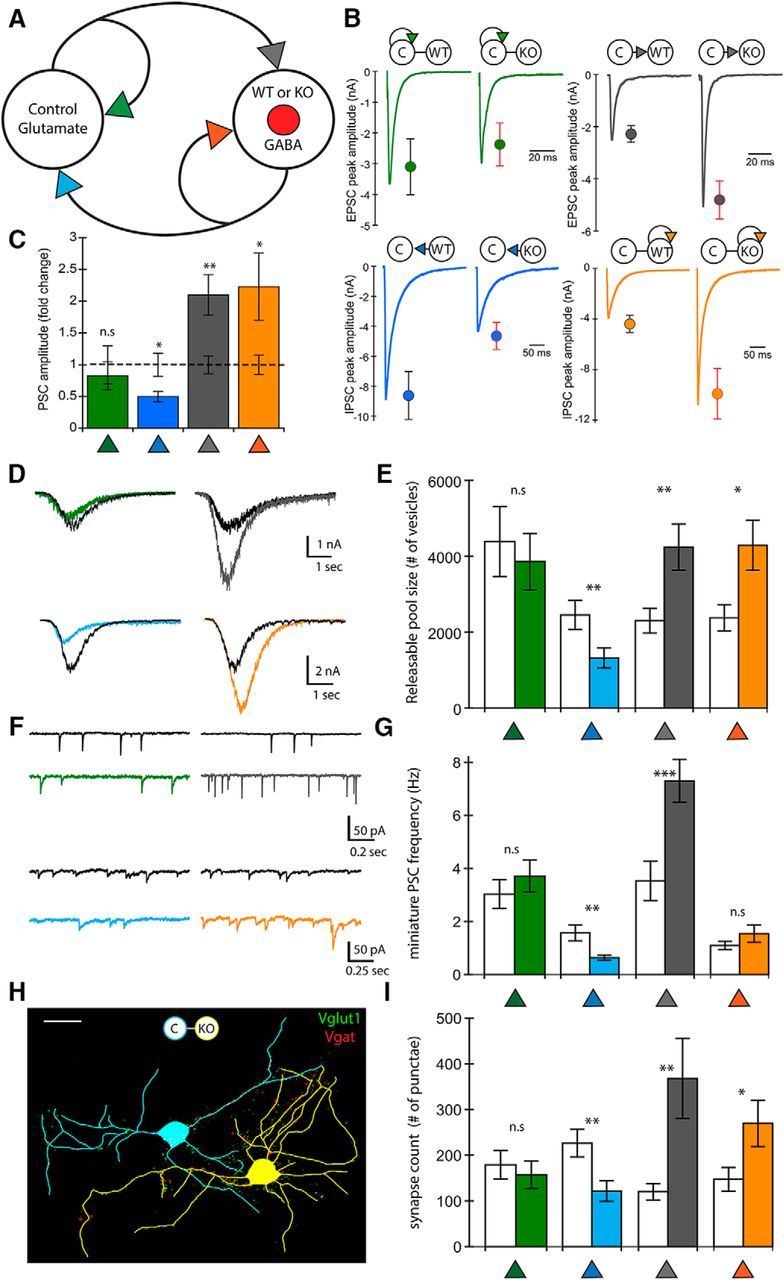
Loss of PTEN in E-I pairs leads to increased synaptic strength onto the postsynaptic neuron but decreased GABAergic output. A, Schematic of an E-I pair showing the four synaptic connections tested (color-coded arrowheads). B, Representative traces and mean ± SEM values for Control (left traces and circles) and Mutant (right traces and circles) pairs are shown for each of the four connections. Color code matches the connections shown in A. A summary of all values, numbers of neurons tested, and statistical results are in Table 1. C, Bar graph represents the normalized mean ± SEM PSC peak amplitude values for each of the four connections [Mutant pair relative to Control pair (dotted line)]. D, Representative traces indicate the current response to the 500 mm sucrose application used to assay the size of the readily releasable pool of synaptic vesicles at each of the four connections. The traces from Control pairs are in black, and those from Mutant pairs are colored to match the schematic in A. E, Bar graph represents the increase in the number of available synaptic vesicles in the two connections that showed increased synaptic strength but the decrease at the GABAergic output synapse of Pten-KO neurons. Control pair data (n = 21) are indicated by clear bars and Mutant pair data (n = 18) by colored bars to match the schematic in A. F, Representative traces indicate mEPSC and mIPSC activity. The traces from Control pairs are in black and those from Mutant pairs are colored to match the schematic in A. G, Bar graph represents the increase in the mEPSC frequency onto the Pten-KO GABAergic neuron and the decrease in the mIPSC frequency onto the Con neuron in Mutant pairs. Control pair data (n = 21) are indicated by clear bars and Mutant pair data (n = 18) by colored bars to match the schematic in A. H, Representative tracing of a Con glutamate (blue) and Pten-KO GABA (yellow) E-I Mutant pair showing VGLUT1 (green punctae) and VGAT (red punctae) immunofluorescence. Scale bar, 25 μm. I, Bar graph represents the changes in the number of VGLUT1 or VGAT punctae at each of the four connections. Control pair data (n = 15) are indicated by clear bars and Mutant pair data (n = 17) by colored bars to match the schematic in A. *p < 0.05 (Mann-Whitney test). **p < 0.01 (Mann-Whitney test).
The changes in glutamatergic and GABAergic synaptic strength input in E-I pairs are due to changes in synaptic connectivity
We assayed the number of synaptic vesicles available for release at each connection in the E-I pairs by combining 500 mm sucrose with 3 mm KYN to block glutamate receptors, or 20 μm BIC to block GABA receptors (Fig. 5D). Consistent with the alterations in synaptic strength observed in the E-I pairs, the number of available vesicles was increased in Mutant pairs at the two synapses onto the Pten-KO (Fig. 5D,E, gray and orange bars), whereas the number of vesicles available at the output of the Pten-KO neuron onto its partner (Fig. 5D,E, blue symbols) was significantly decreased. Next, we measured the miniature current frequencies for each connection. The mEPSC (Fig. 5F,G, gray symbols) frequency onto the GABAergic Pten-KO neurons was increased compared with those onto Pten-WT neurons, although we did not detect an increase in mIPSC frequency in the Pten-KO neurons (Fig. 5F,G, orange symbols). The mEPSC frequency of the glutamatergic self-synapses was also unchanged (Fig. 5F,G, green symbols), and the mIPSC frequency at the Pten-KO output synapses was significantly lower in Mutant pairs (Fig. 5F,G, blue symbols).
To determine whether these changes in synaptic strength and vesicle number are due to changes in synapse number, we visualized glutamatergic and GABAergic synapses with immunostaining against VGLUT1, VGAT, and MAP2 (Fig. 5H). There was a significant increase in the number of glutamatergic and GABAergic synapses onto the GABAergic Pten-KO neurons (Fig. 5I, gray and orange bars) and a significant decrease in the number of GABAergic synapses onto glutamatergic neurons paired with Pten-KO neurons (Fig. 5I, blue bar). Thus, postsynaptic loss of PTEN in hippocampal inhibitory neurons is sufficient to increase glutamatergic and GABAergic synaptic connectivity onto these neurons but decreases the number of synapses that they make onto glutamatergic neurons.
Loss of PTEN in hippocampal slice cultures increases functional connectivity and synchrony
Previous studies have shown that increased synaptic connectivity predicts an increase in synchronous network activity and functional connectivity (Lillis et al., 2015; Okun et al., 2015). To test whether this is true for loss of PTEN, we prepared hippocampal slice cultures from P4-P5 Ptenwt/wt and Ptenloxp/loxp pups and injected two AAVs: one expressing GCaMP6m, a fluorescent calcium indicator that is widely used to image neural activity, and one expressing the mCherry-Cre fusion protein (Fig. 6A,B). This preparation resulted in Control slices that, analogous to Control neuron pairs, contained a mixture of non–cre-expressing (Con) and cre-expressing (Pten-WT) neurons, and Mutant slices that, analogous to Mutant neuron pairs, contained a mixture of non–cre-expressing neurons (Con) and cre-expressing (Pten-KO) neurons (Fig. 6B). After 7–10 d, we imaged spontaneous fluorescence changes of neurons in the pyramidal cell layer of the slice cultures with two-photon microscopy and tested whether loss of PTEN in this preparation leads to increases in neuronal activity, synchronous activity, and functional connectivity.
As in the two-neuron cultures (Fig. 1E), Pten-KO neurons showed an increase in cell body size compared with those of Pten-WT and Con neurons, providing evidence of mTOR pathway activation in the infected hippocampal slices (Fig. 6C). To assess the ability of the GCaMP6m calcium indicator to accurately report action potentials, we first performed cell-attached recordings of individual neurons in the slices and confirmed that increases in GCaMP6m fluorescence were due to action potential firing (Fig. 6D). Next, we monitored GCaMP6m fluorescence changes in groups of 20–60 neurons from Control (mean: 50.9 ± 4.5 neurons, n = 11 slices, 6 mice) and Mutant slices (mean: 47.8 ± 3.5 neurons, n = 12 slices, 6 mice). The number of active cells per slice was not significantly different between Control and Mutant slices (Control: 39.5 ± 4.6 neurons vs Mutant 43.0 ± 3.7 neurons, p = 0.56). To test for changes in neuronal activity, we recorded the frequency and amplitude of spontaneous calcium events. The mean frequency of cellular events was not significantly different between Control and Mutant slices (Control: 1.18 ± 0.2 events/min vs Mutant: 1.34 ± 0.3 events/min, p = 0.98) (Fig. 6E,F). However, the mean amplitude of the calcium events was greater in neurons from Mutant slices than in neurons from Control slices (Control: 0.32 ± 0.07 ΔF/F vs Mutant: 0.65 ± 0.10 ΔF/F, p = 0.019) (Fig. 6G,H). This increased amplitude could be due to an increase in the amount of calcium influx per action potential or to an increase in the number of action potentials. To distinguish between these possibilities, we measured the mean amplitude of the calcium events accompanying individual action potentials in neurons from each slice group using loose-patch recordings, and found that they were not significantly different between Pten-WT and Pten-KO neurons (Pten-WT: 0.042 ± 0.002 ΔF/F vs Pten-KO: 0.040 ± 0.002 ΔF/F, p = 0.82), indicating that calcium influx due to action potentials is not altered by PTEN loss. Together, these data suggest that neurons in Mutant slices are not more likely to be active than neurons in Control slices; however, when Mutant slice neurons are active, they fire more action potentials than neurons in Control slices.
To examine the occurrence of synchronous activity, we searched for network events, which we defined as periods in which a significant number of neurons were simultaneously active based on the total number of calcium activations of all active neurons within a sliding 1 s window (Fig. 7A,B). The mean frequency of network events was not significantly different in Control slices (1.20 ± 0.23 events/min) and Mutant slices (1.43 ± 0.25 events/min, p = 0.61) (Fig. 7C, left). However, the number of neurons that participated in network events was greater in Mutant slices than in Control slices. This was true both when expressed as the absolute number of neurons participating in the network event (Control: 15.2 ± 2.8 neurons vs Mutant: 26.2 ± 2.8 neurons, p = 0.015), as well as the fraction of the active neurons that participated in the events (Control: 0.37 ± 0.04 neurons vs Mutant: 0.62 ± 0.06 neurons, p = 0.002) (Fig. 7C, middle), indicating an increased recruitment of neurons into network events in Mutant slices. As another measure of network synchronization and functional connectivity, we calculated the fraction of active cells in each image that were significantly correlated (p < 0.01 vs scrambled data). The fraction of significantly correlated neurons was greater in Mutant slices (0.74 ± 0.05) than in Control slices (0.53 ± 0.04) (Fig. 7C, right), indicating that the increased synaptic connectivity, which we observed in the two-neuron culture system, leads to an increase in functional connectivity and synchrony.
Figure 7.

Loss of PTEN increases synchrony in hippocampal slice cultures. A, Representative fluorescence traces for 20 simultaneously recorded neurons from a Control (left) and a Mutant (right) slice. Red boxes represent network events. B, Event histograms corresponding to the traces in A that illustrate the percentage of total active neurons that had event onsets within each 2 s time bin. C, Dot plots show the frequency of network events (left), the fraction of active neurons that participated in network events (middle), and the fraction of cells displaying a significant correlation (right) in Control (black) and Mutant (red) slices. **p < 0.01 (Mann-Whitney test).
The changes in functional connectivity and synchrony involve the entire network, not just Pten-KO neurons
Finally, to determine whether the increases in neuron recruitment and functional connectivity that we observed in the Mutant slices are due to selective increases in Pten-KO neurons, or whether the changes are distributed throughout the network, we compared the neuronal activity of cre-expressing (Pten-WT or Pten-KO) to non–cre-expressing (Con) neurons in Control and Mutant slices (Fig. 8). In Control slices, there were no significant differences between Con and Pten-WT neurons in any of the parameters measured (Fig. 8B--E, left columns), verifying that the cre-expressing viruses had no effect on neuronal activity. Moreover, in Mutant slices, the mean frequency of cellular events was not different between Pten-KO and Con neurons (Con: 1.36 ± 0.28 events/min vs Pten-KO: 1.38 ± 0.26 events/min, p = 1; Fig. 8B), indicating that the Con neurons are behaving similarly to the Pten-KO neurons in the Mutant slices. Surprisingly, the mean event amplitude was significantly increased in the Con neurons compared with that of the Pten-KO neurons (Con: 0.68 ± 0.12 ΔF/F vs Pten-KO: 0.57 ± 0.09 ΔF/F, p = 0.01; Fig. 8C), indicating that Con neurons fired more action potentials than Pten-KO neurons in the Mutant slices. Together, these data suggest that Con neurons participate in the increased neural activity of the Mutant slices, and that the increase in event amplitude seen in Mutant slices versus Control slices is not solely due to increases in Pten-KO neurons.
Figure 8.
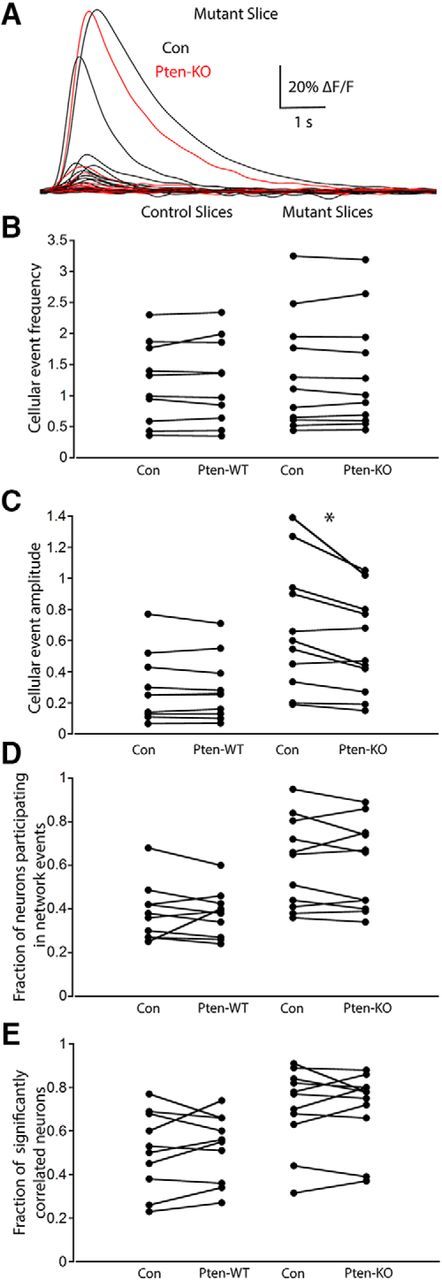
The changes observed in Mutant slices cannot be accounted for by changes in the activity of Pten-KO neurons alone. A, Fluorescence traces from Con (black traces) and Pten-KO (red traces) neurons during an identified network event in a Mutant slice. B–E, Dot plots showing the measurements of Con and Pten-WT neurons in Control slices (left columns) and Con and Pten-KO neurons in Mutant slices (right columns), for the following: B, the mean frequency of spontaneous events per cell (events per minute); C, the mean amplitude of the spontaneous events (ΔF/F); D, the fraction of the active neurons that participated in network events; and E, the fraction of significantly correlated active neurons. All comparisons were made with the Wilcoxon matched-pairs signed ranks test. *p < 0.05 (Wilcoxon matched-pairs signed ranks test).
We next asked whether the increase in the number of neurons that participated in the network events that we observed in Mutant slices is likewise due to increased participation by both Con and Pten-KO neurons. Indeed, the fraction of Con neurons that participated in the network events was not significantly different from the fraction of Pten-KO neurons that participated in network events in the Mutant slices (Con: 0.61 ± 0.06 fraction of neurons vs Pten-KO: 0.59 ± 0.06 fraction of neurons, p = 0.53; Fig. 8D). Similarly, there was no significant difference between the fraction of Con and Pten-KO neurons that displayed correlated activity in the Mutant slices (Con: 0.75 ± 0.05 fraction of neurons vs Pten-KO: 0.71 ± 0.04 fraction of neurons, p = 0.56). Thus, these data demonstrated that the changes that we observed in Mutant slices were manifest throughout the network and not confined to the Pten-KO neurons.
Discussion
Here, we performed a comprehensive analysis of the effects of PTEN loss on the four synaptic motifs formed by the two major classes of neurons in the hippocampus. Loss of PTEN caused postsynaptic, presynaptic, and transsynaptic changes. The postsynaptic effect was independent of the connection type, as it always increased connectivity onto the Pten-KO neurons, but the presynaptic and transsynaptic effects were only evident at specific connections. We then deleted Pten in an ex vivo model of the developing hippocampus in a mosaic manner and found that it increased the number of action potentials during periods of activity, and the number of neurons that participated in network activity. Importantly, these alterations occurred across the network in Con and Pten-KO neurons. Together, our data indicate that PTEN loss causes changes at multiple connection motifs that rewire the network and combine to induce the network pathology.
Loss of PTEN increases connectivity via postsynaptic mechanisms, independent of the connection type
In all four connection types studied, loss of PTEN in the postsynaptic neuron was sufficient to cause an increase in synaptic strength onto that neuron. Three distinct assays supported the conclusion that this was due to an increase in synapse number, and the effect persisted whether the input was GABAergic or glutamatergic. Although these changes were shown in vitro, hyperactivation of PI3K-mTOR signaling also potentially affects these connections in vivo. Whether it does or not may depend on many factors particular to each circuit, including anatomical constraints, homeostatic plasticity, activity-dependent plasticity, and molecular heterogeneity. Previous studies in acute slices generally support the conclusion that postsynaptic PTEN loss increases excitatory-excitatory synaptic strength via increases in synapse formation (Xiong et al., 2012; Williams et al., 2015). Our model confirms these results but further argues that this increase in synapse number occurs at GABAergic connections, both those onto glutamatergic neurons and those between GABAergic neurons. Thus, the mechanism involved in the increased synapse formation is likely to involve signaling molecules that are not unique to GABAergic or glutamatergic synapse formation.
The consistent increase in mPSC frequency when the postsynapse was PTEN-deficient supports our conclusion that the increased synaptic strength is due to an increase in synapse number. Previously, we reported that loss of PTEN in single-neuron cultures did not increase mPSC frequency but did increase synapse number and RRP size, which we interpreted as PTEN loss causing a decrease in the spontaneous release rate via a presynaptic process (Weston et al., 2012). There are at least two possibilities for this discrepancy. First, it is probable that PTEN loss exerts two effects: a postsynaptic increase in synapse formation that leads to an increase in mPSC frequency and a presynaptic decrease in spontaneous release rate. In single-neuron cultures, where both the presynapse and postsynapse are devoid of PTEN, these effects cancel each other out and produce no change. In two-neuron cultures, where many of the synapses onto a postsynaptic PTEN-KO are made by a Con presynaptic neuron, mEPSC frequency is increased. However, in two-neuron culture recordings where the presynapse is PTEN-KO and the postsynapse is Con (Figs. 3G; 5G), mPSC frequency is depressed. Second, it is possible that the lack of mPSC frequency increase due to PTEN loss in single-neuron cultures is due to the lack of heterogeneous connectivity in this preparation, similar to findings in synaptotagmin I-knock-out neurons, and that this prevented us from detecting this increase in our previous study (Liu et al., 2009; Wierda and Sørensen, 2014).
Loss of PTEN in E-I and I-E pairs causes unique changes in synaptic strength and connectivity
In addition to the general effects of postsynaptic PTEN loss, we found two additional connection-specific effects. The first was a transsynaptic effect, where PTEN loss in the glutamatergic neuron caused an increase in the connection that the partner GABAergic neuron made with itself (Fig. 3). GABAergic neurons in the cortex (Tamás et al., 1997; Bacci et al., 2003), cerebellum (Pouzat and Marty, 1998), and amygdala use autaptic connections to regulate their firing; thus, loss of PTEN in glutamatergic neurons in these areas could alter the activity of inhibitory neurons. In more intact circuits, this transsynaptic effect may not be limited to autaptic connections, and it may serve to increase global GABAergic interconnectivity. This effect, in combination with the strengthening of I-I connections due to PTEN loss in GABAergic neurons themselves (Fig. 4), could lead to disinhibition and contribute to the hypersynchronization that we observed in the slice experiments (Fig. 7).
The second unique effect of PTEN loss was a presynaptic effect in which Pten deletion in the GABAergic neuron decreased its output onto the glutamatergic partner (Fig. 5). Heterozygous Pten deletion in parvalbumin-expressing interneurons results in loss of GABAergic connectivity onto pyramidal neurons (Baohan et al., 2016), suggesting that even a modest reduction in PTEN activity may reduce inhibitory output. However, more widespread homozygous Pten deletion in interneurons using the Nkx2.1 promoter increases spontaneous IPSC frequency onto layer 2/3 pyramidal neurons (Vogt et al., 2015). Our results support the interpretation that the initial effect of Pten deletion in inhibitory neurons is a reduction in output connectivity, which may contribute to the increase in action potential firing that we observed in the Mutant slices (Fig. 6G,H).
Loss of PTEN in hippocampal slice cultures increases synchrony
In the hippocampal slice culture model, Pten deletion increased the amplitude, but not frequency, of the calcium events. Because we determined that Pten-KO neurons did not show a greater increase in fluorescence per action potential, we interpret this increase as the neurons in Pten-KO slices firing more action potentials when active, although they were not more likely to enter into a period of activity. This change could be due to increased afferent excitation, weakened local inhibition, or even changes in membrane excitability. Previous studies have not generally found increases in membrane excitability due to PTEN loss (Garcia-Junco-Clemente et al., 2013; Weston et al., 2014; Williams et al., 2015; Cupolillo et al., 2016), suggesting that the increased firing is due to synaptic changes. Of the synaptic changes that we found, the increase in E-E pair connectivity and the decrease in GABAergic output in E-I pairs could both contribute to the increase. A previous study found increased evoked population spike numbers in the dentate gyrus circuit containing Pten-knock-out neurons (LaSarge et al., 2016), which is in agreement with our finding of increased spikes in individual neurons.
We also found that the network bursts that occur in the developing hippocampus recruited a greater proportion of neurons in Mutant slices and increased the number of significantly correlated neurons during spontaneous activity (Fig. 7). Previous studies showed that synchronous network activity in the hippocampus evolves in parallel to increases in glutamatergic and GABAergic synaptic connectivity (Allene et al., 2012; Lillis et al., 2015), and that the activity could be blocked by pharmacological inhibition of PI3K and mTOR (Berdichevsky et al., 2013). Our results extend these findings by showing that PTEN loss increased synaptic connectivity in the two-neuron culture in the absence of network synchrony, supporting the idea that the enhanced synaptic connectivity downstream of mTOR hyperactivation drives abnormal network synchronization. Moreover, the fact that the synaptic and network changes appear within 1 week after Pten deletion suggests that these are early events following mTOR hyperactivation. Increases in neuronal synchrony have also been reported in mouse models of Fragile X and Rett syndromes (Gonçalves et al., 2013; Lu et al., 2016), both of which also have high incidences of epilepsy and autism (Gatto and Broadie, 2010), and elevated synchrony is associated with epilepsy and autism in humans and animal models (Uhlhaas and Singer, 2006; Tyler et al., 2012; Luongo et al., 2016).
On one end, the effects of PTEN loss on cell signaling, cellular and subcellular neuroanatomy, and synaptic physiology have been well studied (Backman et al., 2001; Kwon et al., 2001; Jaworski et al., 2005; Fraser et al., 2008). On the other end, the effects of PTEN loss on behavior (Kwon et al., 2006; Lugo et al., 2014; Cupolillo et al., 2016) and the development of epilepsy (Ljungberg et al., 2009; Pun et al., 2012; Nguyen et al., 2015) have also been characterized. There is relatively little information, however, on how these changes at the cellular level manifest themselves in the activity of local circuits, and how these circuit changes predispose the brain to abnormal behavior and epilepsy. Our two-neuron culture preparation allowed us to test the effects of PTEN loss on the most basic circuit elements, and we found widespread changes. It is likely that some of these changes are masked or compensated for in vivo, and we do not know how each change may relate to the increase in synchronous activity and functional connectivity. Future studies may link specific changes in synaptic function to altered network function in Pten and other models of neurological disease, or find that the relationships are nonlinear. Either way, understanding the relationship between the two should lead to a better grasp of how gene variants lead to their neurological phenotypes.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant K99/R00 NS087095 as well as a COBRE Neuroscience Award P30 103498. We thank Dr. Andreas Tolias and Dr. Dimitry Yatsenko for their assistance with experimental design and analysis of calcium imaging experiments; Dr. Amy Shore for critical reading and editing of the manuscript; Dr. Vivek Jayaraman, Dr. Douglas S. Kim, Dr. Loren L. Looger, Dr. Karel Svoboda, and the GENIE Project, Janelia Research Campus, Howard Hughes Medical Institute for sharing the GCaMP6 construct.
The authors declare no competing financial interests.
References
- Allene C, Picardo MA, Becq H, Miyoshi G, Fishell G, Cossart R (2012) Dynamic changes in interneuron morphophysiological properties mark the maturation of hippocampal network activity. J Neurosci 32:6688–6698. 10.1523/JNEUROSCI.0081-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR, Prince DA (2003) Functional autaptic neurotransmission in fast-spiking interneurons: a novel form of feedback inhibition in the neocortex. J Neurosci 23:859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW (2001) Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet 29:396–403. 10.1038/ng782 [DOI] [PubMed] [Google Scholar]
- Baohan A, Ikrar T, Tring E, Xu X, Trachtenberg JT (2016) Pten and EphB4 regulate the establishment of perisomatic inhibition in mouse visual cortex. Nat Commun 7:12829. 10.1038/ncomms12829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky Y, Dryer AM, Saponjian Y, Mahoney MM, Pimentel CA, Lucini CA, Usenovic M, Staley KJ (2013) PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post-traumatic epilepsy. J Neurosci 33:9056–9067. 10.1523/JNEUROSCI.3870-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgalossi A, Jung S, Man KN, Nair R, Jockusch WJ, Wojcik SM, Brose N, Rhee JS (2012) Analysis of neurotransmitter release mechanisms by photolysis of caged Ca(2) (+) in an autaptic neuron culture system. Nat Protoc 7:1351–1365. 10.1038/nprot.2012.074 [DOI] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C (2005) Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 42:318–321. 10.1136/jmg.2004.024646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casillas-Espinosa PM, Powell KL, O'Brien TJ (2012) Regulators of synaptic transmission: roles in the pathogenesis and treatment of epilepsy. Epilepsia 53 [Suppl 9]:41–58. [DOI] [PubMed] [Google Scholar]
- Chamberland S, Topolnik L (2012) Inhibitory control of hippocampal inhibitory neurons. Front Neurosci 6:165. 10.3389/fnins.2012.00165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Trimbuch T, Chao HT, Jordan JC, Herman MA, Rosenmund C (2014) Investigation of synapse formation and function in a glutamatergic-GABAergic two-neuron microcircuit. J Neurosci 34:855–868. 10.1523/JNEUROSCI.0229-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB. (2011) mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 17:734–742. 10.1016/j.molmed.2011.07.008 [DOI] [PubMed] [Google Scholar]
- Cupolillo D, Hoxha E, Faralli A, De Luca A, Rossi F, Tempia F, Carulli D (2016) Autistic-like traits and cerebellar dysfunction in Purkinje cell PTEN knock-out mice. Neuropsychopharmacology 41:1457–1466. 10.1038/npp.2015.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euro EPINOMICS-RESC Consortium (2014) De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 95:360–370. 10.1016/j.ajhg.2014.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ (2008) Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 151:476–488. 10.1016/j.neuroscience.2007.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Junco-Clemente P, Chow DK, Tring E, Lazaro MT, Trachtenberg JT, Golshani P (2013) Overexpression of calcium-activated potassium channels underlies cortical dysfunction in a model of PTEN-associated autism. Proc Natl Acad Sci U S A 110:18297–18302. 10.1073/pnas.1309207110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto CL, Broadie K (2010) Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front Synaptic Neurosci 2:4. 10.3389/fnsyn.2010.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves JT, Anstey JE, Golshani P, Portera-Cailliau C (2013) Circuit level defects in the developing neocortex of Fragile X mice. Nat Neurosci 16:903–909. 10.1038/nn.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman KN, Pal SK, Burrone J, Murthy VN (2006) Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat Neurosci 9:642–649. 10.1038/nn1677 [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M (2005) Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci 25:11300–11312. 10.1523/JNEUROSCI.2270-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Shen S, Cadwell CR, Berens P, Sinz F, Ecker AS, Patel S, Tolias AS (2015) Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350:aac9462. 10.1126/science.aac9462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ (2001) Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet 29:404–411. 10.1038/ng781 [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50:377–388. 10.1016/j.neuron.2006.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasarge CL, Danzer SC (2014) Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front Mol Neurosci 7:18. 10.3389/fnmol.2014.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaSarge CL, Pun RY, Muntifering MB, Danzer SC (2016) Disrupted hippocampal network physiology following PTEN deletion from newborn dentate granule cells. Neurobiol Dis 96:105–114. 10.1016/j.nbd.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG (2012) De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 44:941–945. 10.1038/ng.2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillis KP, Wang Z, Mail M, Zhao GQ, Berdichevsky Y, Bacskai B, Staley KJ (2015) Evolution of network synchronization during early epileptogenesis parallels synaptic circuit alterations. J Neurosci 35:9920–9934. 10.1523/JNEUROSCI.4007-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Dean C, Arthur CP, Dong M, Chapman ER (2009) Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci 29:7395–7403. 10.1523/JNEUROSCI.1341-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D'Arcangelo G (2009) Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech 2:389–398. 10.1242/dmm.002386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Ash RT, He L, Kee SE, Wang W, Yu D, Hao S, Meng X, Ure K, Ito-Ishida A, Tang B, Sun Y, Ji D, Tang J, Arenkiel BR, Smirnakis SM, Zoghbi HY (2016) Loss and gain of MeCP2 cause similar hippocampal circuit dysfunction that is rescued by deep brain stimulation in a Rett syndrome mouse model. Neuron 91:739–747. 10.1016/j.neuron.2016.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo JN, Smith GD, Arbuckle EP, White J, Holley AJ, Floruta CM, Ahmed N, Gomez MC, Okonkwo O (2014) Deletion of PTEN produces autism-like behavioral deficits and alterations in synaptic proteins. Front Mol Neurosci 7:27. 10.3389/fnmol.2014.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo FJ, Horn ME, Sohal VS (2016) Putative microcircuit-level substrates for attention are disrupted in mouse models of autism. Biol Psychiatry 79:667–675. 10.1016/j.biopsych.2015.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M (2004) Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A 58:167–176. 10.1002/cyto.a.20022 [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, Poduri A (2014) Megalencephaly and hemimegalencephaly: breakthroughs in molecular etiology. Am J Med Genet C Semin Med Genet 166C:156–172. 10.1002/ajmg.c.31401 [DOI] [PubMed] [Google Scholar]
- Nelson SB, Valakh V (2015) Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87:684–698. 10.1016/j.neuron.2015.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LH, Brewster AL, Clark ME, Regnier-Golanov A, Sunnen CN, Patil VV, D'Arcangelo G, Anderson AE (2015) mTOR inhibition suppresses established epilepsy in a mouse model of cortical dysplasia. Epilepsia 56:636–646. 10.1111/epi.12946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun M, Steinmetz N, Cossell L, Iacaruso MF, Ko H, Barthó P, Moore T, Hofer SB, Mrsic-Flogel TD, Carandini M, Harris KD (2015) Diverse coupling of neurons to populations in sensory cortex. Nature 521:511–515. 10.1038/nature14273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouzat C, Marty A (1998) Autaptic inhibitory currents recorded from interneurones in rat cerebellar slices. J Physiol 509:777–783. 10.1111/j.1469-7793.1998.777bm.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun RY, Rolle IJ, Lasarge CL, Hosford BE, Rosen JM, Uhl JD, Schmeltzer SN, Faulkner C, Bronson SL, Murphy BL, Richards DA, Holland KD, Danzer SC (2012) Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75:1022–1034. 10.1016/j.neuron.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson T. (2016) Neural circuit dysfunction in schizophrenia: insights from animal models. Neuroscience 321:42–65. 10.1016/j.neuroscience.2015.06.059 [DOI] [PubMed] [Google Scholar]
- Smedler E, Malmersjö S, Uhlen P (2014) Network analysis of time-lapse microscopy recordings. Front Neural Circuits 8:111. 10.3389/fncir.2014.00111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J (2017) Molecular neurobiology of mTOR. Neuroscience 341:112–153. 10.1016/j.neuroscience.2016.11.017 [DOI] [PubMed] [Google Scholar]
- Tamás G, Buhl EH, Somogyi P (1997) Massive autaptic self-innervation of GABAergic neurons in cat visual cortex. J Neurosci 17:6352–6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE, Unser M (1998) A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process 7:27–41. 10.1109/83.650848 [DOI] [PubMed] [Google Scholar]
- Tyler AL, Mahoney JM, Richard GR, Holmes GL, Lenck-Santini PP, Scott RC (2012) Functional network changes in hippocampal CA1 after status epilepticus predict spatial memory deficits in rats. J Neurosci 32:11365–11376. 10.1523/JNEUROSCI.1516-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W (2006) Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron 52:155–168. 10.1016/j.neuron.2006.09.020 [DOI] [PubMed] [Google Scholar]
- Vogt D, Cho KK, Lee AT, Sohal VS, Rubenstein JL (2015) The parvalbumin/somatostatin ratio is increased in Pten mutant mice and by human PTEN ASD alleles. Cell Rep 11:944–956. 10.1016/j.celrep.2015.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MC, Chen H, Swann JW (2012) Multiple roles for mammalian target of rapamycin signaling in both glutamatergic and GABAergic synaptic transmission. J Neurosci 32:11441–11452. 10.1523/JNEUROSCI.1283-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MC, Chen H, Swann JW (2014) Loss of mTOR repressors Tsc1 or Pten has divergent effects on excitatory and inhibitory synaptic transmission in single hippocampal neuron cultures. Front Mol Neurosci 7:1. 10.3389/fnmol.2014.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Sørensen JB (2014) Innervation by a GABAergic neuron depresses spontaneous release in glutamatergic neurons and unveils the clamping phenotype of synaptotagmin-1. J Neurosci 34:2100–2110. 10.1523/JNEUROSCI.3934-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, DeSpenza T Jr, Li M, Gulledge AT, Luikart BW (2015) Hyperactivity of newborn Pten knock-out neurons results from increased excitatory synaptic drive. J Neurosci 35:943–959. 10.1523/JNEUROSCI.3144-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q, Oviedo HV, Trotman LC, Zador AM (2012) PTEN regulation of local and long-range connections in mouse auditory cortex. J Neurosci 32:1643–1652. 10.1523/JNEUROSCI.4480-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4:a009886. 10.1101/cshperspect.a009886 [DOI] [PMC free article] [PubMed] [Google Scholar]