

Abstract
Activation of somatic μ-opioid receptors (MORs) in hypothalamic proopiomelanocortin (POMC) neurons leads to the activation of G-protein-coupled inward rectifier potassium (GIRK) channels and hyperpolarization, but in response to continued signaling MORs undergo acute desensitization resulting in robust reduction in the peak GIRK current after minutes of agonist exposure. We hypothesized that the attenuation of the GIRK current would lead to a recovery of neuronal excitability whereby desensitization of the receptor would lead to a new steady state of POMC neuron activity reflecting the sustained GIRK current observed after the initial decline from peak with continued agonist exposure. However, electrophysiologic recordings and GCaMP6f Ca2+ imaging in POMC neurons in mouse brain slices indicate that maximal inhibition of cellular activity by these measures can be maintained after the GIRK current declines. Blockade of the GIRK current by Ba2+ or Tertiapin-Q did not disrupt the sustained inhibition of Ca2+ transients in the continued presence of agonist, indicating the activation of an effector other than GIRK channels. Use of an irreversible MOR antagonist and Furchgott analysis revealed a low receptor reserve for the activation of GIRK channels but a >90% receptor reserve for the inhibition of Ca2+ events. Altogether, the data show that somatodendritic MORs in POMC neurons inhibit neuronal activity through at least two effectors with distinct levels of receptor reserve and that differentially reflect receptor desensitization. Thus, in POMC cells, the decline in the GIRK current during prolonged MOR agonist exposure does not reflect an increase in cellular activity as expected.
SIGNIFICANCE STATEMENT Desensitization of the μ-opioid receptor (MOR) is thought to underlie the development of cellular tolerance to opiate therapy. The present studies focused on MOR desensitization in hypothalamic proopiomelanocortin (POMC) neurons as these neurons produce the endogenous opioid β-endorphin and are heavily regulated by opioids. Prolonged activation of somatic MORs in POMC neurons robustly inhibited action potential firing and Ca2+ activity despite desensitization of the MOR and reduced activation of a potassium current over the same time course. The data show that somatic MORs in POMC neurons couple to multiple effectors that have differential sensitivity to desensitization of the receptor. Thus, in these cells, the cellular consequence of MOR desensitization cannot be defined by the activity of a single effector system.
Keywords: action potential, calcium imaging, electrophysiology, GIRK, hypothalamus, receptor reserve
Introduction
Opioid receptors are G-protein-coupled receptors (GPCRs) that couple to inhibitory G-proteins (Gi and Go) that signal through a range of secondary effectors including the inhibition of voltage-gated Ca2+ channels (VGCCs), the inhibition of adenylyl cyclase (AC), and the activation of potassium channels (Williams et al., 2001). In response to a maximal concentration of agonist, opioid receptor signaling, as measured by the activation or inhibition of effectors, reaches a peak effect. Continued signaling often causes a reduction in the peak agonist effect over the course of minutes, at which point a new steady-state level of signaling is achieved. This reduction in peak agonist effect is referred to as acute desensitization and is thought to be an early indication of the long-term homeostatic changes that result in the development of tolerance to opiates (Martini and Whistler, 2007; Christie, 2008). Thus, continued signaling of opioid receptors can proceed in the following three phases: first reaching a peak, then undergoing desensitization, and finally reaching a fully desensitized state. Although the mechanism of opioid receptor desensitization is not completely understood, agonist binding to the receptor initiates a stereotypical chain of events, starting with the activation of intracellular signaling cascades, that results in phosphorylation of the receptor, binding of β-arrestin, and eventual internalization of the receptor (Reiter and Lefkowitz, 2006; Williams et al., 2013).
Opioid receptors are widely expressed throughout the brain and have been heavily studied in circuits conferring nociception/pain and reward (Quirion et al., 1983; Walwyn et al., 2010). In addition, neurons that produce endogenous opioids often express opioid receptors and are regulated by opioid-sensitive inputs. For example, proopiomelanocortin (POMC) neurons of the arcuate nucleus (ARC) of the hypothalamus release, among other peptides, the endogenous opioid β-endorphin (O'Donohue and Dorsa, 1982) and are regulated by opioids (Slugg et al., 2000; Hentges et al., 2009). The POMC neurons express μ-opioid receptors (MORs) in the somatodendritic compartment, where they couple to G-protein-coupled inwardly rectifying K+ (GIRK) channels (Loose and Kelly, 1990; Pennock and Hentges, 2011). The activation of GIRK channels initiates an outward flux of K+ that hyperpolarizes the resting membrane potential (RMP), inhibiting action potential (AP) firing in POMC neurons (Kelly et al., 1990). Acute desensitization of the MOR results in a ∼50% decrease from the peak magnitude of the GIRK current after 5 min of agonist exposure (Pennock et al., 2012), which in turn results in a relative increase in the resting membrane potential from peak hyperpolarization (Kelly et al., 1990; Harris and Williams, 1991). Therefore, we hypothesized that acute desensitization of the MOR would result in a recovery of POMC neuron activity during the desensitization phase (first 5 min of agonist exposure) before reaching a new steady state of activity following complete desensitization (>5 min of agonist exposure) that would be closer to resting activity. To assess cellular activity, AP firing rate and Ca2+ influx were examined.
Contrary to the hypothesis, robust, persistent inhibition of POMC neuron AP firing and Ca2+ activity was observed throughout a 10 min application of opioid agonist. This persistent inhibition occurs despite the decline in the GIRK channel current and apparent desensitization of the MOR. The data provided indicate that in POMC neurons, MORs couple to a secondary effector pathway for which there is a high receptor reserve, rendering this effector pathway insensitive to desensitization of the MOR. Thus, for POMC neurons, the desensitization observed with the GIRK current as the readout does not correlate with a return to basal activity, as was expected. The data highlight the shortcoming of reliance on the readout from one effector to infer signaling at other effectors or overall changes in cellular activity. Further, divergent actions and desensitization at different effectors could help to explain seemingly disparate results in some studies comparing receptor desensitization and opioid tolerance (Ingram et al., 1998; Martini and Whistler, 2007; Allouche et al., 2014).
Materials and Methods
Animals.
Transgenic mice expressing discosoma red (DsRed) under the control of the Pomc gene promoter (Hentges et al., 2009) were originally obtained from Dr. Malcolm Low (University of Michigan, Ann Arbor, MI), as were mice expressing enhanced green fluorescent protein in POMC neurons [C57BL/6J-Tg(Pomc-EGFP)1Low/J; stock #009593, The Jackson Laboratory]. Mice where Cre recombinase is driven by the Pomc promoter were previously described (Xu et al., 2005) and were originally obtained from Dr. Gregory Barsh (Stanford University, Stanford, CA). All lines of mice were backcrossed onto the C57BL/6 background for 11 generations. Mice were housed at controlled temperatures (22–24°C) with a constant 12 h light/dark cycle. Standard rodent chow and tap water were provided ad libitum. Standard PCR genotyping was performed to identify transgenic mice. All animal procedures were approved by the Colorado State University Institutional Animal Care and Use Committee and met the United States Public Health Services guidelines.
Brain slice preparation.
Sagittal brain slices (240 μm) containing the arcuate nucleus were prepared as previously described (Pennock and Hentges, 2011). Briefly, mice were deeply anesthetized with isoflurane, and brains were rapidly removed and placed into an ice-cold artificial CSF (aCSF) solution containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11 glucose. All aCSF solutions were saturated with a 95% O2/5% CO2 mixture. Slices were prepared using a VT 1200S vibratome (Leica). Brain slices containing the arcuate nucleus were then transferred into warm (37°C) aCSF containing MK-801 (15 mm; Sigma-Aldrich) to block NMDA glutamate receptors. Slices were allowed to rest for at least 60 min before transfer to the recording chamber. Brain slices were prepared from both male and female mice between 8 and 14 weeks of age and were distributed evenly over treatment conditions.
Electrophysiology.
Brain slices placed in the recording chamber were continuously perfused with warm (37°C) aCSF saturated with 95%O2/5%CO2 at a consistent flow rate (1.5 ml/min). Whole-cell voltage-clamp recordings were made with an internal recording solution containing the following (in mm): KCl 57.5, K-methyl sulfate 57.5, NaCl 20, MgCl2 1.5, HEPES 5, EGTA 0.1, ATP 2, GTP 0.5, and phosphocreatine 10, pH 7.3. Recording pipettes had a tip resistance of 1.5–2.2 MΩ when filled with internal solution. POMC cells were identified by EGFP or DsRed fluorescence. After obtaining a seal of 1 GΩ, negative pressure was applied to rupture the cell and enter whole-cell mode. Cells were held at −60 mV, no series resistance compensation was applied, and liquid junction potentials were not corrected. Recordings were excluded from analysis if access resistance changed significantly (>15%) during the recording or if access resistance increased >18 MΩ. When measuring GIRK currents, 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μm) and bicuculline methiodide (BMI; 10 μm) were used to block synaptic AMPA and GABAA currents. GIRK currents were measured at a holding potential of −60 mV as part of a continuous chart recording or extracted from 100 ms voltage steps between −110 and −50 mV. For loose-patch recordings, the recording electrode contained aCSF and a loose patch was established by applying negative pressure until the membrane resistance reached between 10 and 25 MΩ. Action currents were measured continuously in voltage-clamp mode with a command potential of 0 mV. Action potentials were inferred from extracellular loose-patch recordings by using template-based event detection to identify action currents using Axograph X (version 1.6.1) software.
Viral gene transfer in vivo.
Pomc-Cre transgenic mice (8–10 weeks of age) were anesthetized with isoflurane and placed in a stereotaxic frame (David Kopf Instruments) with motorized axis control (NeuroStar). A viral vector containing a double-floxed sequence for GCaMP6f (AAV9.CAG.Flex.GCaMP6f.WRPE.SV40); obtained from the Penn Vector Core (University of Pennsylvania School of Medicine, Philadelphia, PA; made available by Dr. Vivek Jayaraman, Dr. Rex A. Kerr, Dr. Douglas S. Kim, Dr. Loren L. Looger, and Dr. Karel Svoboda, GENIE Project, Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA) was injected into the ARC of Pomc-Cre mice. For each animal, an injection (200 nl, 2.1E12 GC/ml) was made into each side of the ARC using a Hamilton syringe at bregma: X, ±0.30 mm; Y, −1.35 mm; Z, −6.10 mm. After microinjection, the wound was sutured and animals were left to recover for a minimum of 7 d, but no longer than 21 d before experiments, to allow GCaMP6f expression.
GCaMP6f imaging.
Before imaging experiments, the overall health of GCaMP6f-expressing slices was evaluated by looking for nuclear exclusion of GCaMP6f fluorescence, and several fields of view were explored to find intrinsically active POMC neurons. GCaMP6f was excited using 470 nm light supplied by an LED/LEDD1B driver (Thorlabs) through a 40× water-immersion objective (40×/0.8 W; LUMPlanFL N, Olympus). Images were captured on an electron-multiplying charge-coupled device (Evolve 512, Photometrics) at ∼10 Hz with a 50 ms exposure time using MetaFluor software (Molecular Devices). Image stacks were imported into ImageJ software (Schindelin et al., 2015) where they underwent image stabilization (Dubbs et al., 2016) and background subtraction (50 nm rolling ball) before signal extraction from individual neurons using the Time Series Analyzer plugin (J. Balaji, Indian Institute of Science, Bangalore, India). Raw fluorescence intensities for individual POMC neurons were imported into Axograph X software where they were normalized to the baseline fluorescence intensity at the beginning of the recording (F/F0) and separated into 60 s epochs. Event detection was performed in Axograph X using a derivative threshold to detect upward deflections in the fluorescence intensity. Detected events were manually compared with the normalized fluorescence trace for each individual neuron to ensure that all clearly visible events were detected by the software. Reported measurements of event frequency for baseline or drug applications are the mean of at least three sequential time points (60 s epochs) for each individual neuron.
Drugs.
Stock solutions of DNQX (100 mm; catalog #D0540, Sigma-Aldrich), (+)-MK-801 (100 mm; catalog #M107, Sigma-Aldrich), and picrotoxin (400 mm) were prepared in DMSO and diluted in aCSF just before application to the slice. DMSO never exceeded 0.035% of the total volume of aCSF in working solutions. [Met5]-enkephalin (ME; catalog #M6638, Sigma-Aldrich), BMI (catalog #2503, Tocris Bioscience), nociceptin (catalog #0910, Tocris Bioscience), the cyclized peptide d-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP; catalog #1560, Tocris Bioscience), tetrodotoxin (catalog #1069, Tocris Bioscience), β-chlornaltrexamine (β-CNA; catalog #O001, Sigma-Aldrich), and Tertiapin-Q (TertQ; catalog #STT-170, Alomone Labs) were prepared as stock solutions in distilled water. All drugs were diluted in aCSF to achieve the working concentrations. β-CNA was divided into aliquots and then stored at −80°C. For each experiment performed using β-CNA, an individual aliquot was thawed, diluted, and perfused into the slice within 5 min to minimize degradation of the compound before use. Dose–response curves were made for ME to determine the maximal and EC50 concentrations used in the present studies.
Estimation of MOR receptor reserve.
Dose–response curves representing the inhibition of GCaMP6f event frequency by postsynaptic MORs on POMC neurons with and without treatment with an irreversible antagonist (β-CNA) were fitted using GraphPad Prism software (GraphPad). Values for the predicted concentration of ME necessary to reduce GCaMP6f event frequency by given values between 20% and 70% were obtained from these curves. Concentrations that were predicted to achieve an equivalent inhibition of GCaMP6f event frequency in the absence of β-CNA ([ME]control) and in the presence of β-CNA ([ME]β-CNA) were plotted against one another as a double-reciprocal plot. The plot was fitted according to method of Furchgott (1966) using the following equation:
 |
where q is equal to the fraction of MORs still functional after β-CNA treatment and KA is the dissociation constant of ME at postsynaptic MORs on POMC neurons. Estimated values for q and KA were obtained using GrapPad Prism (GraphPad).
Experimental design and statistical analyses.
All data are presented as the mean ± SEM, unless otherwise specified. For electrophysiological recordings, only one recording was made per slice, and no more than two slices per animal. For Ca2+ imaging recordings, the number of neurons, brain slices, and animals are reported in the Results section and the figure legend for each experiment. Male and female mice were used for all experiments. The results were initially analyzed for sex differences, but, since no sex differences were detected, data from both sexes were pooled for all experiments. Comparisons between two groups were evaluated using paired and unpaired two-tailed Student's t tests. Dose–response curves (best fit) were constructed and analyzed using GraphPad Prism software (GraphPad). Each dose had data from 13 to 135 individual neurons included in the mean. The extra sum of squares (ESS) F test was performed to determine whether there was a significant shift in the dose–response curve compared with the control condition based on EC50 values. One-way ANOVA followed by Dunnett's multiple-comparison post hoc test was used for comparisons between multiple time points. For all experiments, p < 0.05 was considered to be significant.
Results
Sustained inhibition of firing and Ca2+ events during agonist application that causes reduced GIRK channel activation
To examine the effect of postsynaptic opioid receptor activation and desensitization on POMC excitability, several approaches were used including whole-cell voltage-clamp recordings of GIRK channel activation, extracellular loose-patch recordings of AP firing and fluorescence recordings of the genetically encoded Ca2+ indicator GCaMP6f (Chen et al., 2013) in POMC neurons in mouse brain slices containing the hypothalamus. Consistent with previous reports (Pennock et al., 2012), the application of the MOR agonist ME (10 μm) caused an outward current that robustly desensitized during a 10 min exposure (Fig. 1A). The magnitude of the GIRK current in Figure 1A reached a peak of 41 pA (Fig. 1A, top dashed line) above baseline (Fig. 1A, bottom dashed line) and reached a steady state of 23 pA (Fig. 1A, bottom dashed line) after 10 min of ME exposure, a 44% reduction. The remaining steady-state GIRK current was completely reversed by the MOR selective antagonist CTAP (500 nm). Overall, the magnitude of the steady-state current was 53% of the peak current (Fig. 1B; n = 6 neurons, 4 animals), indicating a 47% reduction of the GIRK current due to MOR desensitization. Based on this result, we hypothesized that the ∼50% decrease in the GIRK current over a 10 min ME application would lead to a recovery of AP firing over the course of the drug application.
Figure 1.

Sustained inhibition of AP firing and Ca2+ activity by MOR activation. A, Voltage-clamp recording from a POMC neuron showing attenuation of the GIRK current elicited by ME (10 μm) during a 10 min application. The residual GIRK current is eliminated the MOR selective antagonist CTAP (500 nm). Dotted lines are overlaid to highlight the baseline current (bottom dashed line), the peak GIRK current (top dashed line), and the steady-state GIRK current (middle dashed line) following desensitization. B, Summary data showing the attenuation of the GIRK current during prolonged ME exposure (n = 6 neurons, 4 animals). C, Extracellular loose-patch recording of a Pomc-Cre neuron expressing GCaMP6f during a 10 min ME (10 μm) application with subsequent reversal by CTAP (500 nm). Top trace, Action currents recorded in loose patch, with the bottom plot showing AP frequency over time. D, Summary data of the sustained inhibition of AP frequency by ME (10 μm; n = 6 neurons, 6 animals) over a 10 min application. E, Optical recording of the same neuron as in C. Top trace is the F/F0 normalized GCaMP6f fluorescence intensity with measured Ca2+ event frequency plotted below. In this cell, ME (10 μm) completely eliminated both AP firing and resolvable Ca2+ events. F, Summary data for the inhibition of Ca2+ events by ME (n = 29 neurons, 5 slices, 3 animals).
To assess AP firing, extracellular loose-patch recordings were made from POMC neurons. In these recordings, AP firing was persistently inhibited during the full 10 min of ME (10 μm) exposure. This is illustrated in a sample trace (Fig. 1C, minutes 10–20) and in the compiled data (Fig. 1D, minutes 7–16) where the maximum inhibition is 97 ± 3% (minute 7), and this inhibition does not significantly change during the 10 min ME application (F(1.5,7.7) = 0.8, p = 0.46, ANOVA for minutes 7–16; n = 6 neurons, 6 animals). Inhibition of AP firing was fully reversed by CTAP (500 nm).
We initially chose the loose-patch extracellular recording approach to avoid disrupting the intracellular milieu but later focused on GCaMP6f fluorescence imaging to increase the number of cells analyzed and to avoid recording artifacts induced by the positioning of the recording pipette on the cell body. GCaMP6f was expressed specifically in arcuate nucleus POMC neurons by stereotaxic injection of an adeno-associated virus containing a double-floxed inverse orientation GCaMP6f coding sequence into a mouse line expressing cre-recombinase driven by the Pomc promoter (Pomc-Cre). Figure 1E shows an F/F0 value normalized GCaMP6f fluorescence trace from the same cell as Figure 1C above a plot of GCaMP6f events in 1 min bins. Similar to the firing data, there is full inhibition of resolvable Ca2+ events for the entire 10 min application of ME (10 μm), and the inhibition is fully reversed by CTAP. This paired recording demonstrates the generally good correlation between the rate of GCaMP6f events and the AP firing rate. However, the relationship between APs and GCaMP6f events is not 1:1, indicating that it was only possible to resolve Ca2+ events due to multiple temporally grouped APs. Overall, GCaMP6f events were almost completely inhibited by 10 μm ME (97 ± 1%, 8.2 ± 0.2 events/min at baseline [minutes 1–4], 0.2 ± 0.0 events/min in 10 μm ME [minutes 6–15]; n = 29 neurons, 5 slices, 3 animals; Fig. 1F) for a full 10 min period. Here, there is no indication of a decrease in the inhibition of Ca2+ events, as expected based on the decline in the GIRK current during the same treatment paradigm (Fig. 1A). Altogether, these data suggest that the acute desensitization of postsynaptic MORs as detected by the decline in GIRK channel conductance does not drastically affect their ability to inhibit tonic firing or Ca2+ transients in POMC neurons.
Activation of GIRK current is not necessary for the inhibition of Ca2+ events
The decline in the GIRK channel current during prolonged agonist exposure generally results from MOR desensitization (Williams, 2014; Pennock et al., 2012), and, thus, it was surprising to see sustained inhibition of firing and Ca2+ activity during the desensitization observed for GIRK channel activation. To determine whether the sustained inhibition is independent of the hyperpolarization caused by GIRK channel activation, the magnitude of the GIRK current was diminished by preapplying barium chloride (Ba2+), a nonselective blocker of inward-rectifier K+ channels (IRKs; Standen and Stanfield, 1978; Dascal et al., 1993). Perfusion of Ba2+ (100 μm) caused a dramatic increase in POMC Ca2+ events, likely due to the blockade of IRKs, but GCaMP6f events were still robustly inhibited for the duration of the ME (10 μm) application (Fig. 2A). The mean inhibition of GCaMP6f events by ME (10 μm) in the presence of Ba2+ (100 μm) was 89 ± 2% (8.3 ± 0.7 events/min in 100 μm Ba2+, 1.0 ± 0.3 events/min following ME; n = 22 neurons, 3 slices, 3 animals) with no clear evidence of MOR desensitization, yielding a recovery of Ca2+ events (Fig. 2B; time points from minute 11 to minute 20 were not significantly different than at minute 10; F(2.8,22) = 1.859, p = 0.17, ANOVA).
Figure 2.
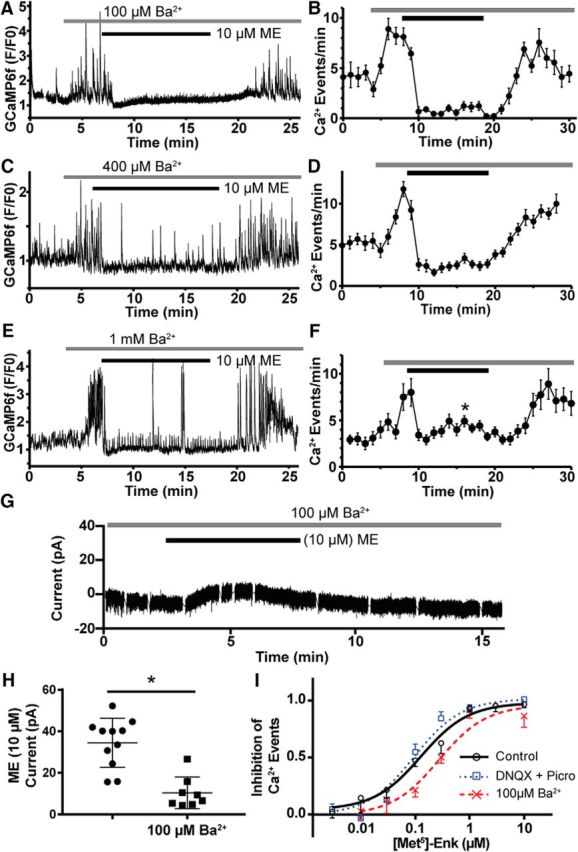
Blockade of GIRK current by barium fails to prevent the inhibition of Ca2+ events by MORs or uncover receptor desensitization. A, GCaMP6f fluorescence recording (F/F0) where Ba2+ (100 μm) was applied before ME (10 μm) showing a complete cessation of resolvable Ca2+ events. B, Summary plot of the inhibition of GCaMP6f events by ME (10 μm) in the presence of 100 μm Ba2+ (n = 22 neurons, 3 slices, 3 animals). C, Experiment as performed in A, but with [Ba2+] increased to 400 μm. D, Summary plot of inhibition of GCaMP6f events by ME (10 μm) in the presence of 400 μm Ba2+ (n = 18 neurons, 3 slices, 3 animals). E, Experiment as performed in A, but with [Ba2+] increased to 1 mm. F, Summary plot of inhibition of GCaMP6f events by ME (10 μm) in the presence of 1 mm Ba2+ (n = 12 neurons, 2 slices, 2 animals). Only the time point at minute 16 is significantly different than that at minute 10. G, Whole-cell voltage-clamp recording of the GIRK current elicited by ME (10 μm) in the presence of Ba2+ (100 μm) illustrating the blunted GIRK current compared with Figure 1A. H, Summary of the magnitude of GIRK currents from POMC neurons elicited by ME (10 μm) alone (n = 11 neurons, 7 animals, black circles), which is significantly reduced in the presence of 100 μm Ba2+ (n = 8 neurons, 6 animals, black squares). I, Dose–response curves for the inhibition of GCaMP6f events by ME. Under control conditions (solid black line) the EC50 value was 130 nm, and blockade of AMPA and GABAA receptors (dotted blue line, DNQX and picrotoxin, respectively) did not significantly alter the EC50 value (90 nm). In the presence of Ba2+ (100 μm), there was a significant twofold rightward shift of the EC50 value (270 nm).
The experiment illustrated in Figure 2, A and B, was repeated in the presence of higher concentrations of Ba2+ (Fig. 2C,D, 400 μm, E,F, 1 mm), which more completely block GIRK channels (Lancaster et al., 2000). In the presence of 400 μm Ba2, ME (10 μm) inhibited GCaMP6f events by 76 ± 16% (9.8 ± 3.1 events/min baseline, 2.5 ± 2.0 events/min following 10 μm ME; n = 18 neurons, 3 slices, 3 animals). Here, there was still no evidence for a recovery of Ca2+ events (time points from minute 11 to minute 20 were not significantly different than at minute 10; F(4.4,75) = 1.8, p = 0.1248, ANOVA). Even with the addition of 1 mm Ba2+, ME (10 μm) still inhibited the events by 55 ± 18% (8.8 ± 3.9 events/min baseline, 3.7 ± 1.5 events/min following 10 μm ME; n = 12 neurons, 2 slices, 2 animals). Only the time point at minute 16 is significantly different than at minute 10 (F(3.8,41) = 11.3, p < 0.0001, ANOVA; minute 10 vs 16, p = 0.04, Dunnett's multiple-comparison test). Thus, even under conditions where the GIRK current is minimized and overall activity is increased due to the blockade of IRKs, ME (10 μm) is still able to potently inhibit POMC Ca2+ transients with no clear attenuation of inhibition following MOR desensitization. These data suggest that maximal activation of postsynaptic MORs has a profound ability to silence POMC signaling, even in the absence of GIRK current. To ensure that Ba2+ was indeed reducing the GIRK current elicited by ME, recordings were made in POMC neurons after the addition of Ba2+. A sample recording is shown in Figure 2G, which is scaled the same as Figure 1A to highlight the inhibition of the GIRK current. Here, the application of 100 μm Ba2+ caused a small inward current to develop, presumably due to the blockade of IRKs, while ME (10 μm) induced a small outward current that failed to reach the same sharp peak as the control recording (Fig. 1A). Overall, the magnitude of the GIRK current elicited by ME was 34 ± 4 pA (n = 11, 7 animals), and preapplication of 100 μm Ba2+ reduced the magnitude of the ME-induced current to 10 ± 3 pA (t(17) = 5.04, p = 0.0001, unpaired t test; n = 8, 6 animals), a 70% reduction (Fig. 2H) indicating significant blockade of the GIRK current even at the lowest concentration of Ba2+ used.
To better understand the relationship between MOR activation and inhibition of POMC neuron Ca2+ events at submaximal concentrations of agonist, dose–response curves were built. Under baseline conditions, the inhibition of GCaMP6f events by ME has an EC50 value of 130 nm (95% CI, 84–195 nm; Fig. 2I). Blockade of GABAA and AMPA receptors (DNQX, 10 μm; picrotoxin, 100 μm) before ME application to occlude the potential effects of ME on presynaptic terminals did not significantly shift the EC50 value (95 nm; 95% CI, 70–129 nm; F(1,805) = 1.75, p = 0.19, ESS F test; Fig. 2I). Application of Ba2+ (100 μm) to block GIRK currents before activation of MORs by ME caused a twofold rightward shift in the EC50 value (270 nm; 95% CI, 184–394 nm; F(1,749) = 9.3, p = 0.002, ESS F test) with no decrease in the maximal inhibition. The significance of the modest shift in the dose–response curve is unclear since 100 μm Ba2+ also causes a significant increase in the frequency of Ca2+ events from 5.1 ± 0.2 events/min (n = 235 neurons) to 8.2 ± 0.3 events/min (t(502) = 9.44, p < 0.0001, unpaired t test; n = 269 neurons), which could be partially responsible for the rightward shift in the dose–response curve. Although these data provide strong evidence that the inhibition of POMC Ca2+ events is distinct from the activation of postsynaptic GIRK channels by ME, it is possible that the residual GIRK current in the presence of 100 μm Ba2+ blockade (Fig. 2H) is sufficient to potently inhibit Ca2+ events. Blockade of GIRK channels could be improved by using a higher [Ba2+], but this increasingly blocks IRKs, which depolarizes the RMP and further increases Ca2+ activity. To avoid this potential confound, we used a more selective GIRK blocker, TertQ. We made whole-cell voltage-clamp recordings from POMC neurons where a brief 90 s pulse of ME (10 μm) was applied to assess the amplitude of the GIRK current before the application of TertQ (500 nm). After the perfusion of TertQ, a 3 min pulse of ME (10 μm) was used to assess the amplitude of the remaining GIRK current (Fig. 3A). Overall, the mean GIRK current elicited by ME was 41 ± 9 pA, and this was reduced to 2 ± 1 pA following the application of TertQ (Fig. 3B; n = 8 neurons, 5 animals). Last, we recorded Ca2+ events from POMC neurons using a protocol similar to whole-cell voltage-clamp recordings where a brief pulse of ME was applied before the perfusion of TertQ (Fig. 3C). In the presence of TertQ (500 nm), ME (10 μm) inhibited Ca2+ events by 98% (6.2 events/min at baseline vs 0.1 events/min; n = 60 neurons, 9 slices, 3 animals), and TertQ did not increase overall POMC Ca2+ activity as Ba2+ had done (Fig. 3D). Thus, the inhibition of POMC Ca2+ events by ME does not require the activation of a GIRK current. Since MORs are known to couple to a wide variety of effector pathways (Williams et al., 2013), it is possible that an alternate effector pathway contributes to sustained inhibition of POMC neuron activity.
Figure 3.

Blockade of the ME-induced GIRK current by the GIRK selective blocker Tertiapin-Q fails to prevent the inhibition of Ca2+ events by ME. A, Whole-cell voltage-clamp recording from a POMC neuron. A brief (90 s) pulse of ME (10 μm) was used to elicit a GIRK current just before the addition of the GIRK selective blocker Tertiapin-Q (500 nm). Following blockade with Tertiapin-Q, a 3 min pulse of ME was used to determine the amplitude of the residual GIRK current. B, Dot plot showing the amplitude of the GIRK current elicited by ME (10 μm) before and after the addition of Tertiapin-Q (500 nm; n = 8 cells, 5 animals). C, Slice average (mean ± SEM) plotting the frequency of GCaMP6f events in response to ME (10 μm) before and after the addition of Tertiapin-Q (500 nm) to block GIRK channels. D, Summary data for the inhibition of Ca2+ events by ME (10 μm) in the presence of 500 nm Tertiapin-Q (n = 60 neurons, 9 slices, 3 animals).
MOR desensitization does not limit the inhibition of Ca2+ events by a maximal concentration of agonist
Postsynaptic GPCRs that couple to GIRK channels in POMC neurons show acute desensitization only minutes after peak GIRK current is achieved (Pennock et al., 2012; Fig. 1A), and this desensitization is likely receptor delimited (Williams et al., 2013). However, we saw no clear evidence of acute desensitization of the inhibition of Ca2+ events by ME under any conditions examined. To determine whether the inhibition of Ca2+ events by ME may be maintained even after desensitization of the MOR, we used a more sensitive measure of acute desensitization where the response to a low concentration of agonist is compared before and after the application of a saturating concentration of agonist.
In the presence of 100 μm Ba2+ to reduce the contribution of the GIRK current, a near-EC50 concentration of ME (300 nm) was applied to clearly, but incompletely, inhibit POMC Ca2+ events. After a brief washout, a maximal concentration of ME (10 μm) was applied, which almost completely inhibited POMC Ca2+ events for a full 10 min to fully desensitize the MORs. Following a full washout of ME (10 μm) and a return to baseline activity, a second 300 nm concentration of ME was applied and a less robust inhibition of POMC Ca2+ events, compared with the first application of 300 nm ME, occurred (Fig. 4A,B). By comparing the magnitude of Ca2+ event inhibition by the first application of 300 nm ME to the second application of 300 nm ME, the percentage decrease in inhibition, presumably due to desensitization of the receptor, was calculated. The mean inhibition of Ca2+ events by the first application of 300 nm ME was 55 ± 5% (n = 30 neurons, 6 slices, 4 animals) as expected for a near-EC50 concentration. The mean inhibition of Ca2+ events by the second application of 300 nm ME was 29 ± 5% (t(29) = 6.04, p < 0.0001, paired t test), which is a 47% reduction in inhibition compared with the first application (Fig. 4C,D). Thus, the inhibition of Ca2+ events by the activation of MORs is subject to the same receptor-delimited desensitization as the activation of the GIRK current, although this is not apparent when a maximal concentration of agonist is applied.
Figure 4.

Desensitization of the MOR reduces the inhibition of Ca2+ events by an EC50 concentration of ME. A, Normalized (F/F0) fluorescent GCaMP6f recording illustrating the protocol used to detect MOR desensitization. In the presence of Ba2+ (100 μm), to minimize the GIRK current, a near-EC50 concentration of ME (300 nm) was briefly applied then washed out. Then a 10 min ME (10 μm) application was used to induce MOR desensitization. After washout, a second application of 300 nm ME was used to assess the level of desensitization. B, Slice average (mean ± SEM) for the experiment in A. C, Plot of the percentage inhibition of Ca2+ events before (open circles) and after MOR desensitization (open squares). Each point is an individual neuron, and the horizontal bars are the mean ± SEM. The mean inhibition of Ca2+ events for the first application of 300 nm ME was significantly greater than the mean inhibition for the second application of 300 nm ME (black asterisk; n = 30, neurons, 6 slices, 4 animals). D, Same data as in C paired to show the downward trend between the first and second applications of 300 nm ME.
Receptor reserve differs for GIRK channel activation and Ca2+ inhibition
The inability to observe any attenuation in the magnitude of inhibition of Ca2+ events by a maximal concentration of ME is not due to an artificial ceiling effect, as shown by the increased baseline activity in the Ba2+ experiments in Figure 2A–F, or due to a lack of desensitization of the receptor for the inhibition of Ca2+ events, as shown in Figure 4A–D. An alternative possibility is that there is a high level of receptor reserve for the inhibition of Ca2+ events, such that MOR desensitization is insufficient to attenuate a maximal ME response. To test this hypothesis, a proportion of MORs was inactivated with the irreversible antagonist β-CNA as previously described (Chavkin and Goldstein, 1984). To ensure the efficacy of β-CNA to remove a portion of MORs, the effect of β-CNA on the magnitude of the GIRK current elicited by ME (10 μm) was determined by perfusing β-CNA (50 nm) for 2 min, before agonist exposure, a concentration and perfusion time previously used to study receptor reserve in presynaptic MORs (Pennock et al., 2012). A representative trace from a voltage-clamp recording held at −60 mV, where the slice had been perfused with 50 nm β-CNA for 2 min just before the application of DNQX and the GABAA receptor blocker BMI to eliminate EPSCs and IPSCs followed by ME, is shown in Figure 5A. ME (10 μm) typically causes a sharp upward deflection in the current trace due to rapid activation of GIRK channels (Figs. 1A, 3A). Here, only a slow gradual upward deflection of the current trace after the application of ME (10 μm) is observed, indicating successful inactivation of a proportion of MORs. After washout of the ME, nociception (200 nm) was applied to elicit a typical GIRK current through the nociceptin receptor (Pennock et al., 2012). Under control conditions, the mean amplitude of the GIRK current elicited by ME was 34 ± 4 pA (Fig. 5B; n = 11 neurons, 7 animals; same as in Fig. 2H), while the mean amplitude following β-CNA perfusion was 8.3 ± 2.7 pA (t(17.5) = 5.9, p < 0.0001, unpaired t test; n = 9 neurons, 6 animals), a reduction of ∼75% (Fig. 5B). The mean GIRK current elicited by nociceptin (200 nm) following β-CNA perfusion was 31.4 ± 7.0 pA (n = 5 neurons, 4 animals; Fig. 5B), indicating that the lack of GIRK current in response to 10 μm ME was not due to a lack of functional GIRK channels.
Figure 5.

Blockade of the ME-induced GIRK current by the irreversible antagonist β-CNA. A, Whole-cell voltage-clamp recording from a POMC neuron treated for 2 min with β-CNA (50 nm) before a 2 min application of ME (10 μm) used to assess GIRK current amplitude. After washout of ME, nociceptin (200 nm) was used to activate the GIRK current through the nociceptin receptor. B, Plot of the amplitude of the GIRK current from individual neurons. Horizontal bars are the mean ± SEM. The mean amplitude of the GIRK current induced by ME (10 μm, open circles; n = 11 neurons, 7 animals) was significantly reduced by β-CNA pretreatment (50 nm, open squares; n = 9 neurons, 6 animals). The mean amplitude of the GIRK current elicited by nociceptin (200 nm, open triangles; n = 5 neurons, 4 animals) after β-CNA (50 nm) pretreatment was similar to the current elicited by ME in the absence of β-CNA.
As β-CNA was effectively removing a portion of MORs, the dose–response curves for ME-induced inhibition of Ca2+ events were built in the presence of β-CNA as a step toward determining receptor reserve. After β-CNA treatment, ME inhibited Ca2+ events over a range of concentrations (Fig. 6A shows the mean inhibition for all cells in a single slice exposed to 300 nm, 1 μm, and 10 μm ME). The dose–response curves for ME-induced inhibition of Ca2+ events under control conditions and following exposure to β-CNA (50 or 100 nm) show that 50 nm β-CNA for 2 min results in an ∼10-fold rightward shift of the EC50 value of inhibition of Ca2+ events (control: 95 nm; 95% CI, 70–129 nm; vs 50 nm β-CNA: 1260 nm; 95% CI, 860–1850 nm; F(1,687) = 234.4, p < 0.0001, ESS F test) but does not reduce the maximal inhibition of Ca2+ events (Fig. 6B). Increasing the concentration of β-CNA to 100 nm for 2 min only slightly shifts the EC50 value compared with 50 nm β-CNA (1410 nm; 95% CI, 790–2490 nm; F(1,731) = 0.07, p = 0.79, ESS F test) but causes a decrease in the maximal inhibition of Ca2+ events to 80% (Fig. 6B). The 10-fold rightward shift in the dose–response curve by 50 nm β-CNA, without a loss in maximal inhibition, suggests that a significant receptor reserve exists for the inhibition of Ca2+ events. To determine the proportion of MORs that were inactivated by 50 and 100 nm β-CNA, we constructed a double-reciprocal plot (Fig. 6C) of the agonist concentration needed to achieve a given inhibition of Ca2+ events before and after 50 and 100 nm β-CNA according to the method of Furchgott (1966, their Eq. 1). This analysis indicates that 50 nm β-CNA reduced the proportion of receptors by 91% (95% CI for q, 0.089–0.092), while 100 nm β-CNA reduced the proportion of receptors by 93% (95% CI for q, 0.065–0.071). This indicates that the MOR has a >90% reserve for the inhibition of Ca2+ events. The potent reduction in the magnitude of the GIRK current following 50 nm β-CNA treatment (Fig. 5A,B) suggests that the receptor reserve for the activation of the GIRK current is much lower than the reserve for the inhibition of Ca2+ events. The high level of receptor reserve for the inhibition of Ca2+ events is sufficient to explain why we fail to observe acute desensitization, since desensitization of 50% of MORs would be insufficient to attenuate the maximal inhibition of Ca2+ events by 10 μm ME.
Figure 6.
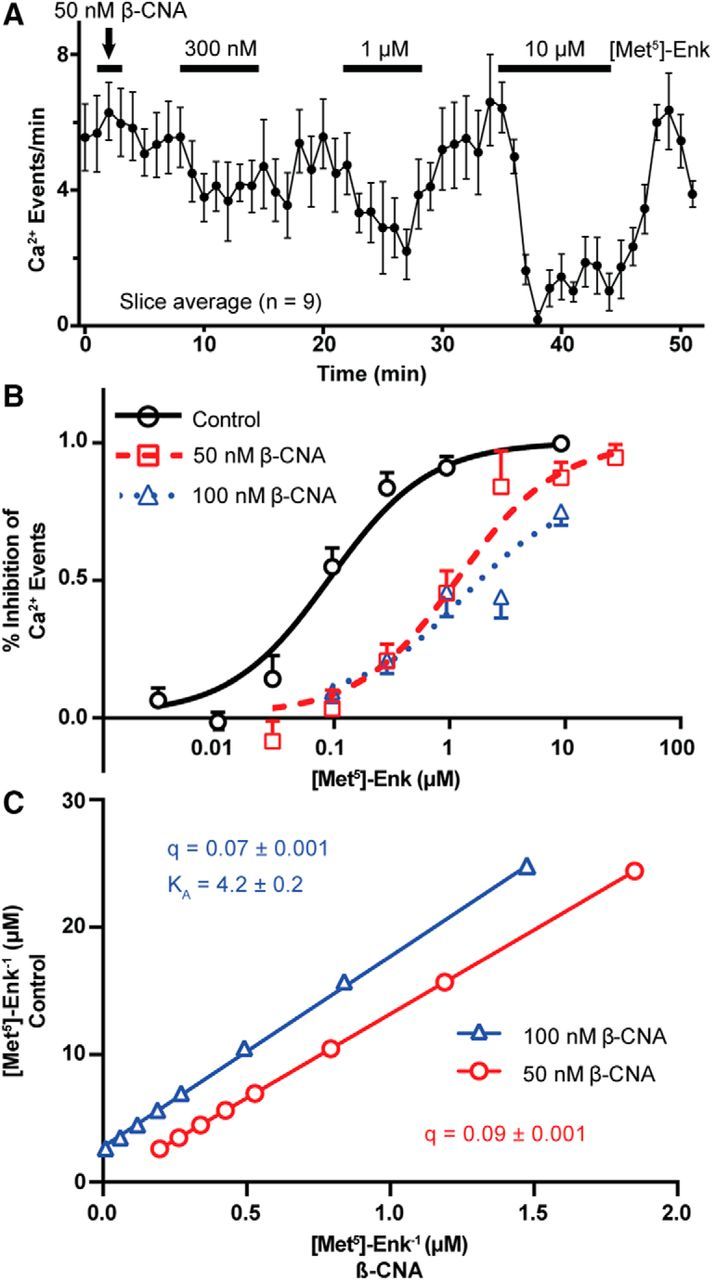
Determination of MOR reserve for the inhibition of Ca2+ events by ME. A, Slice average (mean ± SEM) plotting the frequency of GCaMP6f events in response to increasing concentrations of ME (300 nm, 1 μm, and 10 μm) following a 2 min β-CNA (50 nm) pretreatment. GABAA and AMPA receptor blockers (DNQX and picrotoxin, respectively) were included throughout the recording to occlude the potential effects of presynaptic MORs. B, Dose–response curves for the inhibition of Ca2+ events by ME under control conditions or after pretreatment with 50 or 100 nm β-CNA. Treatment with 50 nm β-CNA caused a significant rightward shift of the EC50 concentration (control, 95 nm; 50 nm β-CNA, 1260 nm), without a concomitant decrease in the maximal inhibition, suggesting the presence of a large receptor reserve. Treatment with 100 nm β-CNA did not significantly shift the EC50 concentration (1410 nm) compared with 50 nm, but there was a decrease in the maximal inhibition to 80%. C, Double reciprocal plot of the concentration of agonist required for equally effective points on the dose–response curve between control (y-axis) and β-CNA treatments (x-axis) as used for the Furchgott (1966) analysis, to calculate q, the proportion of receptors still functional following β-CNA treatment, from the slope of the linear fits.
Discussion
The present data demonstrate that a prolonged application of a high concentration of MOR agonist continues to maximally inhibit AP firing and Ca2+ transients despite the decline observed in the GIRK current during agonist exposure. This result was surprising as we expected cellular activity to return toward baseline as MORs desensitized and GIRK current declined, although observations of differential desensitization across effectors have been described for other GPCRs, including 5HT2c (Stout et al., 2002) and muscarinic receptors (Pappano and Mubagwa, 1991), and for MORs in heterologous systems (Nowoczyn et al., 2013). For MORs in POMC neurons, this differential desensitization observed at two effectors correlates with receptor reserve. A low receptor reserve for the activation of GIRK channels was found and can explain why this effector system is susceptible to desensitization of the receptor. The high receptor reserve (>90%) for the inhibition of Ca2+ activity renders this effector system relatively insensitive to desensitization of the receptor, especially in response to stimulation with a maximal concentration of agonist. Together, the data indicate that the desensitization observed at one effector pathway may not be reflected in other effectors, and, thus, the cellular consequence of MOR desensitization may not be easily inferred from studies of one effector alone.
Response to prolonged agonist exposure varies by effector
The decline in the peak effect of MOR agonists as indicated by effector readout (desensitization) by the full agonist ME is thought to be receptor delimited, phosphorylation dependent, and an early step in the development of the tolerance to the analgesic actions of opioids (Williams et al., 2013; Yousuf et al., 2015). Interestingly, MOR desensitization is usually not complete. The decline in the peak response elicited by a desensitizing concentration of agonist often reaches a steady state that is not all the way back to baseline, and this can be attributed to the fact that some receptors remain active, although with lower affinity for agonist, after desensitization (Williams, 2014). The fact that some MORs continue to signal during prolonged exposure to a saturating concentration of agonist is illustrated by the residual GIRK current that remains late in the agonist exposure (steady-state current after a decline from peak that does not reach baseline, Fig. 1A,B). We reasoned that the residual hyperpolarizing GIRK current after desensitization may contribute to the sustained inhibition of AP firing and Ca2+ transients, but blockade of the GIRK channels with Ba2+ or Tertiapin-Q did not alter the persistent inhibition of Ca2+ events during prolonged exposure to a maximal concentration of ME, indicating sustained signaling through a non-GIRK effector. The fact that high concentrations of ME likely cause desensitization of the receptor itself is indicated by the results of previous studies (Harris and Williams, 1991; Kovoor et al., 1998), by the ability to observe a nociceptin-induced current after the prolonged application of a saturating concentration of ME (Fig. 5), and by the reduced inhibition of Ca2+ events by an EC50 concentration of ME after the application of a maximal concentration of ME for >10 min (Fig. 4). Therefore, the MOR appears to undergo acute desensitization, but this does not noticeably reduce the sustained effectiveness of a maximal concentration of ME to inhibit Ca2+ influx or AP firing in POMC neurons.
Differential receptor reserve for the inhibition of Ca2+ transients and GIRK conductance
While the residual GIRK current is not necessary for the sustained inhibition of cellular activity after receptor desensitization, it may be that the same receptors that maintain the residual GIRK current after desensitization are also responsible for the profound and sustained inhibition of Ca2+ transients observed. The sustained inhibition of Ca2+ events compared with the inhibition of GIRK conductance can be accounted for by the finding of high receptor reserve (∼90%) for Ca2+ event inhibition, indicating that only a small fraction of the receptor population (∼10%) needs to be activated for maximal efficacy. On the other hand, there is very little receptor reserve for the inhibition of GIRK conductance since even the lower concentration of β-CNA (50 nm) nearly abolished the ME-induced GIRK current. In fact, β-CNA reduced the GIRK current so dramatically that it was not feasible to perform a Furchgott (1966) analysis for an accurate estimate of reserve. Regardless, it is clear that there is low receptor reserve for the activation of the GIRK conductance by ME and, thus, any loss of functional MOR during agonist exposure results in decreased GIRK current. MORs in many cell types are known to couple to diverse effectors and the potency of agonist can vary widely for individual effectors (Kelly, 2013; Nowoczyn et al., 2013), which may indicate different receptor reserves. A differential receptor reserve for multiple effectors activated by a single receptor type has been clearly described for adenosine A1 receptors (Srinivas et al., 1997) and suggested for other receptors (Brown and Goldstein, 1986; Meller et al., 1987; James et al., 1994), but the contribution to apparent desensitization has generally not been explored.
What is the nature of the non-GIRK effectors?
MORs couple to a number of G-protein-dependent and G-protein-independent effector systems (Williams et al., 2013). Among the G-protein-dependent effector systems, MORs have been shown to activate K+ channels, including voltage-gated K+ channels and GIRK channels, inhibit VGCC (typically N and PQ types) and inhibit AC (Williams et al., 2013). The robust inhibition of Ca2+ events points to a direct inhibition of Ca2+ influx through VGCCs, although it is possible that the inhibition of AC would also have an inhibitory effect on POMC Ca2+ activity and AP firing (Alreja and Aghajanian, 1995). The use of a Ca2+ indicator as a readout precluded detailed studies to determine whether VGCCs are a direct effector for MORs in POMC neurons as blocking these channels would necessarily alter the assay readout. Future studies to determine the nature of the effector that resists desensitization and sustains inhibited cellular activity are warranted, as is determining whether some MOR agonists preferentially or more effectively activate this effector compared with other effectors in POMC neurons. Such information would provide a better understanding of dynamic opioid actions in these cells and perhaps would be translatable to other cell types that also have diverse MOR–effector coupling that may be differentially altered during agonist treatment.
Correlation between MOR signaling or desensitization and POMC neuron activity
It has been previously shown that the dose response of inhibition of IPSCs by the MOR-selective agonist DAMGO ([D-Ala2, N-MePhe4, Glyol5]-enkephalin) for presynaptic MORs (EC50, 80 nm DAMGO) is left shifted relative to the dose response of the activation of GIRK channels by postsynaptic MORs (EC50, 350 nm DAMGO) in POMC neurons (Pennock and Hentges, 2011). This led to the hypothesis that low concentrations of opioid agonists would preferentially activate MORs on the primarily GABAergic presynaptic terminals, leading to an overall disinhibition of POMC activity. Instead, we found that ME inhibits POMC Ca2+ activity at all concentrations tested and that the removal of presynaptic MOR effects by blocking GABAA and AMPA receptors did not alter this relationship. This result could be explained by the low receptor occupancy required to robustly inhibit POMC Ca2+ activity (EC50, 130 nm ME). Thus, for a systemic application of an MOR agonist, postsynaptic inhibition would likely predominate. Disinhibition of POMC activity through the activation of presynaptic MORs and the subsequent inhibition of GABA release would require opioid agonist release to be restricted to presynaptic terminals. Previous work has also shown that MORs presynaptic to POMC neurons resist acute desensitization and that this resistance is not due to a high receptor reserve, or the specific effectors that presynaptic MORs couple to, but instead seems to be a property of presynaptic MORs themselves (Pennock and Hentges, 2016). This is in stark contrast to what we observe for postsynaptic MORs on POMC neurons where certain effectors are more sensitive to desensitization than others. Together, the results highlight the complexity surrounding MOR function and regulation as compartment, effector, and receptor reserve all contribute to the net consequence of acute and prolonged activation.
Conclusion
The hyperpolarizing current generated by the GIRK channels can be sufficient to produce the acute reduction in firing observed in POMC neurons in response to μ-agonists. However, the blockade of GIRK channels by Ba2+ or Tertiapin-Q does not diminish the ability of ME to inhibit neuronal activity as measured by Ca2+ imaging, leading to the conclusion that MORs signal through redundant inhibitory mechanisms, which are each independently capable of inhibiting neuronal activity.
Footnotes
This work was supported by the National Institute on Drug Abuse (Grant R01-DA-032562) and the National Institute on Diabetes and Digestive and Kidney Diseases (Grant R01-DK-078749) to S.T.H. We thank Ms. Connie King for excellent technical assistance with the animal colony and Dr. Andrew Rau for critical comments on early versions of the manuscript.
The authors declare no competing financial interests.
References
- Allouche S, Noble F, Marie N (2014) Opioid receptor desensitization: mechanisms and its link to tolerance. Front Pharmacol 5:280. 10.3389/fphar.2014.00280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alreja M, Aghajanian GK (1995) Use of the whole-cell patch-clamp method in studies on the role of cAMP in regulating the spontaneous firing of locus coeruleus neurons. J Neurosci Methods 59:67–75. 10.1016/0165-0270(94)00195-M [DOI] [PubMed] [Google Scholar]
- Brown JH, Goldstein D (1986) Differences in muscarinic receptor reserve for inhibition of adenylate cyclase and stimulation of phosphoinositide hydrolysis in chick heart cells. Mol Pharmacol 30:566–570. [PubMed] [Google Scholar]
- Chavkin C, Goldstein A (1984) Opioid receptor reserve in normal and morphine-tolerant guinea pig ileum myenteric plexus. Proc Natl Acad Sci U S A 81:7253–7257. 10.1073/pnas.81.22.7253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300. 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ. (2008) Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol 154:384–396. 10.1038/bjp.2008.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascal N, Lim NF, Schreibmayer W, Wang W, Davidson N, Lester HA (1993) Expression of an atrial G-protein-activated potassium channel in Xenopus oocytes. Proc Natl Acad Sci U S A 90:6596–6600. 10.1073/pnas.90.14.6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbs A, Guevara J, Yuste R (2016) moco: fast motion correction for calcium imaging. Front Neuroinform 10:6. 10.3389/fninf.2016.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF. (1966) The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. Adv Drug Res 3:21–55. [Google Scholar]
- Harris GC, Williams JT (1991) Transient homologous μ-opioid receptor desensitization in rat locus coeruleus neurons. J Neurosci 11:2574–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ (2009) Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci 29:13684–13690. 10.1523/JNEUROSCI.3770-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ (1998) Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci 18:10269–10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SR, Vaziri C, Walker TR, Milligan G, Downes CP (1994) The turkey erythrocyte beta-adrenergic receptor couples to both adenylate cyclase and phospholipase C via distinct G-protein alpha subunits. Biochem J 304:359–364. 10.1042/bj3040359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E. (2013) Efficacy and ligand bias at the mu-opioid receptor. Br J Pharmacol 169:1430–1446. 10.1111/bph.12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Loose MD, Ronnekleiv OK (1990) Opioids hyperpolarize beta-endorphin neurons via mu-receptor activation of a potassium conductance. Neuroendocrinology 52:268–275. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Celver JP, Wu A, Chavkin C (1998) Agonist induced homologous desensitization of mu-opioid receptors mediated by G protein-coupled receptor kinases is dependent on agonist efficacy. Mol Pharmacol 54:704–711. [PubMed] [Google Scholar]
- Lancaster MK, Dibb KM, Quinn CC, Leach R, Lee JK, Findlay JB, Boyett MR (2000) Residues and mechanisms for slow activation and Ba2+ block of the cardiac muscarinic K+ channel, Kir3.1/Kir3.4. J Biol Chem 275:35831–35839. 10.1074/jbc.M006565200 [DOI] [PubMed] [Google Scholar]
- Loose MD, Kelly MJ (1990) Opioids act at mu-receptors to hyperpolarize arcuate neurons via an inwardly rectifying potassium conductance. Brain Res 513:15–23. 10.1016/0006-8993(90)91084-T [DOI] [PubMed] [Google Scholar]
- Martini L, Whistler JL (2007) The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol 17:556–564. 10.1016/j.conb.2007.10.004 [DOI] [PubMed] [Google Scholar]
- Meller E, Bohmaker K, Namba Y, Friedhoff AJ, Goldstein M (1987) Relationship between receptor occupancy and response at striatal dopamine autoreceptors. Mol Pharmacol 31:592–598. [PubMed] [Google Scholar]
- Nowoczyn M, Marie N, Coulbault L, Hervault M, Davis A, Hanouz JL, Allouche S (2013) Remifentanil produces cross-desensitization and tolerance with morphine on the mu-opioid receptor. Neuropharmacology 73:368–379. 10.1016/j.neuropharm.2013.06.010 [DOI] [PubMed] [Google Scholar]
- O'Donohue TL, Dorsa DM (1982) The opiomelanotropinergic neuronal and endocrine systems. Peptides 3:353–395. 10.1016/0196-9781(82)90098-5 [DOI] [PubMed] [Google Scholar]
- Pappano AJ, Mubagwa K (1991) Muscarinic agonist-induced actions on potassium and calcium channels in atrial myocytes: differential desensitization. Eur Heart J 12 [Suppl F]:70–75. [DOI] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST (2011) Differential expression and sensitivity of presynaptic and postsynaptic opioid receptors regulating hypothalamic proopiomelanocortin neurons. J Neurosci 31:281–288. 10.1523/JNEUROSCI.4654-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST (2016) Desensitization-resistant and -sensitive GPCR-mediated inhibition of GABA release occurs by Ca2+-dependent and -independent mechanisms at a hypothalamic synapse. J Neurophysiol 115:2376–2388. 10.1152/jn.00535.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock RL, Dicken MS, Hentges ST (2012) Multiple inhibitory G-protein-coupled receptors resist acute desensitization in the presynaptic but not postsynaptic compartments of neurons. J Neurosci 32:10192–10200. 10.1523/JNEUROSCI.1227-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirion R, Zajac JM, Morgat JL, Roques BP (1983) Autoradiographic distribution of mu and delta opiate receptors in rat brain using highly selective ligands. Life Sci 33 [Suppl 1]:227–230. [DOI] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ (2006) GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab 17:159–165. 10.1016/j.tem.2006.03.008 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Rueden CT, Hiner MC, Eliceiri KW (2015) The ImageJ ecosystem: an open platform for biomedical image analysis. Mol Reprod Dev 82:518–529. 10.1002/mrd.22489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slugg RM, Hayward MD, Ronnekleiv OK, Low MJ, Kelly MJ (2000) Effect of the mu-opioid agonist DAMGO on medial basal hypothalamic neurons in beta-endorphin knockout mice. Neuroendocrinology 72:208–217. 10.1159/000054589 [DOI] [PubMed] [Google Scholar]
- Srinivas M, Shryock JC, Dennis DM, Baker SP, Belardinelli L (1997) Differential A1 adenosine receptor reserve for two actions of adenosine on guinea pig atrial myocytes. Mol Pharmacol 52:683–691. [DOI] [PubMed] [Google Scholar]
- Standen NB, Stanfield PR (1978) A potential- and time-dependent blockade of inward rectification in frog skeletal muscle fibres by barium and strontium ions. J Physiol 280:169–191. 10.1113/jphysiol.1978.sp012379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout BD, Clarke WP, Berg KA (2002) Rapid desensitization of the serotonin(2C) receptor system: effector pathway and agonist dependence. J Pharmacol Exp Ther 302:957–962. 10.1124/jpet.302.3.957 [DOI] [PubMed] [Google Scholar]
- Walwyn WM, Miotto KA, Evans CJ (2010) Opioid pharmaceuticals and addiction: the issues, and research directions seeking solutions. Drug Alcohol Depend 108:156–165. 10.1016/j.drugalcdep.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT. (2014) Desensitization of functional micro-opioid receptors increases agonist off-rate. Mol Pharmacol 86:52–61. 10.1124/mol.114.092098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O (2001) Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 81:299–343. [DOI] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ (2013) Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65:223–254. 10.1124/pr.112.005942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS (2005) PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest 115:951–958. 10.1172/JCI24301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousuf A, Miess E, Sianati S, Du YP, Schulz S, Christie MJ (2015) Role of phosphorylation sites in desensitization of micro-opioid receptor. Mol Pharmacol 88:825–835. 10.1124/mol.115.098244 [DOI] [PubMed] [Google Scholar]