

Abstract
Ventilator-induced lung injury (VILI) results from mechanical disruption of blood-gas barrier and consequent edema and releases of inflammatory mediators. A transpulmonary pressure (PL) of 17 cmH2O increases baby lung volume to its anatomical limit, predisposing to VILI. Viscoelastic property of lung makes pulmonary mechanics time dependent so that stress (PL) increases with respiratory rate. Alveolar inhomogeneity in acute respiratory distress syndrome acts as a stress riser, multiplying global stress at regional level experienced by baby lung. Limitation of stress (PL) rather than strain (tidal volume [VT]) is the safe strategy of mechanical ventilation to prevent VILI. Driving pressure is the noninvasive surrogate of lung strain, but its relations to PL is dependent on the chest wall compliance. Determinants of lung stress (VT, driving pressure, positive end-expiratory pressure, and inspiratory flow) can be quantified in terms of mechanical power, and a safe threshold can be determined, which can be used in decision-making between safe mechanical ventilation and extracorporeal lung support.
Keywords: Baby lung, driving pressure, strain, stress, transpulmonary pressure, ventilator-induced lung injury
INTRODUCTION
“The lungs of one man may bear, without injury, as great a force as those of another man can exert; which by the bellows cannot always be determined.”
John fothergill said the above statement while describing a case of cardiac arrest revived by mouth to mouth ventilation, in the era of bellow resuscitation.[1] In 1745, Fothergill suggested limitation of delivered pressure in artificial respiration, to prevent iatrogenic lung injury.
Ventilator-induced lung injury (VILI) is the collateral damage suffered by the bystander healthy baby lung, caught between the cross fire between mechanical ventilator and diseased lung. The effect of this collateral damage can only be attenuated, not eliminated in acute respiratory distress syndrome (ARDS).
The term VILI was initially used for macroscopic injury related to high airway pressure, leading to pneumothorax. Over the years, gradual understanding of pathophysiology of this grievous complication of mechanical ventilation revealed the hidden aspect of increased capillary permeability, alveolar edema, and local as well as distant organ inflammatory injury. As we acknowledge today, VILI can be difficult to differentiate from ARDS, diseases for which mechanical ventilations may have been started.
In 1940, Macklin described that high airway pressure in mechanical ventilation causes tear of the alveolar membrane and pneumothorax, proposing barotrauma as cause of VILI.[2] Three decades later, Mead et al. devised the mathematical model of VILI and further explained the concept of barotrauma and stress risers.[3]
However, the pathophysiology of VILI was still alluding, as high airway pressure was not the prerequisite to cause lung injury. Negative airway pressure created by iron lung produced alveolar edema and pneumothorax while on the other end, very high airway pressure did not cause lung injury, as demonstrated in musicians who play blowing instruments such as trumpet and flute. These musicians can develop airway pressure as high as 150 cmH2O without sustaining barotrauma.[4]
Later in 1988, Dreyfuss et al. demonstrated that restricting the lung volume by chest strapping, prevented pneumothorax despite high airway pressure. They proposed that it is not pressure, but overdistension of lung that causes VILI, suggesting term volutrauma.[5]
In 1974, Webb and Tierney showed that positive end-expiratory pressure (PEEP) can attenuate the effect of barotrauma.[6] In experimental studies on rats ventilated with high-peak inspiratory pressure, application of PEEP prevented pulmonary edema. They ascribed it to preservation of surfactant activity by PEEP. This protective effect of PEEP attracted little attention and clinical application for many years to come.[7]
After several years, further studies confirmed that PEEP reduces VILI, and it was attributed to reduction of cyclic opening and closing of alveoli (atelectotrauma).
In 1997, Tremblay and Slutsky added the biological aspect to pathophysiology of VILI. They suggested local inflammation caused by volutrauma and atelectrauma, leading to increased capillary permeability and release of inflammatory biomarker culminating in distant organ injury and coined term biotrauma.[8]
At microscopic level, histopathology of normal and ARDS lung brought more clarity to the understanding of VILI. Alveolar epithelium and capillary endothelium are separated by a thin basement membrane, constituting blood-gas barrier.[9] Basement membrane consist of elastin, collagen, and proteoglycans. Elastin is like stretchable spring and contributes to elastic property of lung. Collagen is inextensible and act-like safety limit at total lung capacity (TLC). Proteoglycans stabilize the network of elastin and collagen and provide viscoelastic behavior to lung parenchyma.[10] Blood-gas barrier has a thickness of 50–100 nm but can withstand a transpulmonary pressure (PL) up to 35 cmH2O, due to the tensile strength of collagen fibers.[9]
VILI is the result of mechanical disruption of the blood-gas barrier, at PL which increases the alveolar volume, beyond safety limit provided by inextensible collagen fibers. Resulting increases permeability, edema, and release of inflammatory mediators further deranges the pulmonary mechanics, aggravating the adverse interaction between mechanical ventilator and lung.
Increased transmural pressure across the blood-gas barrier produces small breaks and ultimately disruption, leading to hemorrhage, inflammation, and edema formation, a process called stress failure. Transmural pressure can rise either due to increases in alveolar pressure (airway pressure) or capillary pressure (pulmonary vascular pressure). Stress failure can occur under physiological as well as pathological conditions. Pulmonary hemorrhage encountered in galloping thoroughbred racehorses, and human athletes are an example of physiological stress failure. Pathological stress failure can happen with high alveolar pressure as in VILI and high capillary pressure as in congestive heart failure.
PL across the blood-gas barrier is the real culprit in VILI. If PL increases alveolar volume beyond it anatomical limit, mechanical disruption of blood gas membrane ensues, culminating in inflammation, edema formation, and distant organ injury.
Continued relearning of pulmonary mechanics and VILI, led to the concept of stress-strain, a theory borrowed from physical mechanics. Stress is defined as equal and opposite force developed in a material when exposed to external force while strain is change in the length or volume from baseline, brought about in this process. Stress-strain relationship abolishes the difference between the barotrauma and volutrauma and may be considered a conceptual evolution of barotrauma and volutrauma, respectively.
Relationship between stress and strain is determined by the physical properties of materials, for example solid, viscous (fluid) or viscoelastic.
In elastic materials, stress is directly proportional to strain, governed by a proportionality constant called Young's modulus (Y),[6]
Stress = Y × strain
In the context of pulmonary mechanics, stress is PL and strain is the ratio of tidal volume (VT) and functional residual capacity (FRC),[10]
PL = K × (VT/FRC)
Where K is specific lung elastance, which is equivalent to a PL, which inflates lung volume equal to FRC.
In humans, specific lung elastane (K) has been calculated in clinical studies, to be nearly 13.5 cmH2O.[11] It remains normal in baby lung in ARDS, supporting the concept that baby lung is not stiff, but small healthy lung.[12]
The ratio between TLC and FRC is nearly 1.3, considering FRC and TLC of 35 ml and 80 ml/kg body weight, respectively, in human being.[13] Therefore, a PL of 17 cmH2O will inflate lung from FRC to TLC, a limit beyond which collagen fibers will rupture.
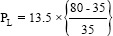
PL = 13.5 × 1.3 = 17 cmH2O
As baby lung has normal-specific lung elastance, PL of 17 cmH2O is the stress, which inflates baby lung to a strain of 1.3, predisposing to VILI, irrespective of its volume.
However, viscoelastic property of lung and alveolar heterogeneity in ARDS further complicate the stress-strain relationship and consequent safety limit for the development of VILI.
The viscoelastic lung
The equation of stress-stain is determined by the physical property of material, i.e., solid, viscous, or viscoelastic. Lung behaves more like a viscoelastic material, with elastin and proteoglycans contributing to elasticity and viscosity, respectively.[14]
In viscoelastic lung, stress is proportional to strain as well as strain rate.[15]
Stress = Y × strain + ή × strain rate
Where Y is Young's modulus, proportionality constant of elastic material and ή is viscosity constant, proportionality constant of viscous material.
In other words, PL is proportional to VT as well as inspiratory flow (rate of change in VT − VT/Ti).
PL = K × VT/FRC + ή × (VT/Ti)
As inspiratory time, inspiratory flow and respiratory rate are closely related to each other, PL increases with respiratory rate, for a given VT.
Mechanical ventilation imposes lung to a PEEP-related continuous strain (static strain-VPEEP) and VT related cyclic strain (dynamic strain-VT).[16] Lung behaves more like viscous material during dynamic strain and elastic material during static strain.[17] In viscoelastic lung during dynamic strain, stress is proportional to strain and strain rate while during static strain, it shows phenomenon of stress relaxation. That is, stress is higher during dynamic strain than static strain.
Thus, viscoelasticity makes pulmonary mechanics time dependent, so that, stress (PL) is variable with respiratory rate, for the same strain (VT). In other words, for a given strain, a safe stress at lower respiratory rate may become unsafe at higher respiratory rate. At the same time, for the same end inspiratory lung volume, combination of low VT (dynamic strain) and high PEEP (static strain) will generated lesser stress and lung injury than a combination of high VT (dynamic strain) and low PEEP (static strain).
Therefore, in ARDS, stress is governed by VT, PEEP and respiratory rate.
Alveolar heterogeneity
In a healthy lung, PL is equally distributed, so that, each alveolar unit and its skeletal fibers bear similar stress and strain. However, in heterogeneous ARDS lung, some of the alveoli remains collapsed throughout the respiratory cycle. In technical term, these collapsed alveoli undergo stress but not strain. Consider two neighboring alveoli, one open and other collapsed, sharing one wall (fiber skeleton). In response to applied PL, the open alveoli will experience much greater individual stress and strain at the expense of collapsed alveoli.
This was demonstrated in mathematical model by Mead et al., in 1970.[3] They calculated that a tenfold reduction in collapsed alveolar size will increase the stress on neighboring normal alveoli by 4.57 times of generated PL. In other words, a PL of 30 cmH2O will be multiplied to 132 cmH2O (30 × 4.57) of stress to normal alveoli, neighboring the collapsed alveoli.
Alveolar heterogeneity acts as stress multiplier in the sense that a safe PL can generate dangerous stress at alveolar level, which would be experienced by baby lung predisposing to VILI.
Thus, in ARDS, viscoelasticity and alveolar heterogeneity make lung stress variable, according to applied PEEP and respiratory rate well as disease severity. The critical PL is determined by the severity of ARDS (baby lung and stress risers), VT, PEEP, and respiratory rate.
A safe strategy of mechanical ventilation is limitation of stress below critical level and attenuation of stress risers. Stress can be limited by a combination of low dynamic strain, high static strain, and low respiratory rate. Optimal PEEP and prone position exert their protective effect by reducing dynamic strain at the cost of static strain, as well as attenuation of stress risers.
As baby lung volume in ARDS is variable with severity, VT as per predicted body weight is inaccurate surrogate of lung strain, as similar VT may produce different stain and stress depending on baby lung volume. Therefore, targeting VT as independent variable in mechanical ventilation is not a safe strategy. Rather, limiting stress by making PL as primary variable and VT as dependent variable is the strategy to prevent VILI.
In recently published study, it was shown that keeping driving pressure (plateau pressure above PEEP) lower than 14 cmH2O predicted improved survival.[18]
Driving pressure seems to be promising strategy of safe mechanical ventilation. However, it becomes unpredictable surrogate of PL in patients with low chest compliance such as morbid obesity, raised intraabdominal pressure, and chest deformity.
Stress, strain, inspiratory flow, and respiratory rate are intricately related to each other and collectively contribute to the pathogenesis of VILI, in a predisposed inhomogeneous ARDS lung. Cressoni et al. conceptualized this in terms of mechanical power delivered to the respiratory system.[19] They quantified VT, PL and respiratory rate as mechanical power in joules per minute, in experimental studies on piglets. The animals were ventilated with similar VT and PL but varying respiratory rates, and a mechanical power threshold was identified. It was observed that healthy piglets developed extensive alveolar edema when delivered mechanical power was >12.1 J/min.
Gattinoni et al. calculated mechanical power by mechanical power equation, derived from equation of motion and compared it with measured mechanical power from P to V loop, in healthy controls and ARDS patients.[20] They found that both were equivalent to each other and the mechanical power increases with VT, PEEP, driving pressure, inspiratory flow, and respiratory rate.
Failure of high-frequency oscillatory ventilation may be attributed to delivered mechanical power. Although very low VT (delta volume) seems to be innocuous in terms of VILI, its combination with high frequency may have produced mechanical power exceeding the threshold for VILI.
More clinical studies are needed to identify safe threshold of mechanical power. It is quite possible that in many patients with severe ARDS, prevention of mechanical ventilation is the only strategy to prevent VILI. Mechanical power equation can be incorporated into ventilator software, which may help in decision-making between safe mechanical ventilation and extracorporeal lung support.
Summary
VILI is the result of adverse interaction between mechanical ventilator and deranged mechanics in ARDS lung
Stress (PL) of 17 cmH2O increases baby lung volume to safety limit of collagen fibers, irrespective of its volume
Viscoelasticity makes lung mechanics time dependent, so that, for a given strain (VT), stress (PL) increases with respiratory rate and for a given end inspiratory volume, dynamic stress is more harmful than static strain
The protective effect of PEEP and prone position is the result of limiting dynamic strain (VT) at the cost of static strain and attenuation of stress risers
VT according to predicted body weight is inaccurate surrogate of lung strain, as baby lung volume is variable with severity of ARDS
Limiting stress by targeting PL as independent variable, and VT as derived variable in mechanical ventilation, is the safe strategy to prevent VILI
The cumulative effect of VT, PEEP, driving pressure, and respirator rate can be measured and calculated, as mechanical power delivered by the ventilator, and a limiting threshold can be determined to prevent VILI
Safety of low-tidal ventilation can be assessed by estimating baby lung volume by driving pressure and mechanical power threshold, which can become rationale indication for extracorporeal lung support.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Fothergill J. Observation on a case published in the last volume of the medical essays, and c. of recovering a man dead in appearance, by distending the lungs with air. Philos Trans R Soc Lond. 1745;43:275–81. [Google Scholar]
- 2.Macklin MT, Macklin CC. Malignant interstitial emphysema of lungs and mediastinum as an important occult complication in many respiratory diseases and other conditions: An interpretation of the clinical literature in the light of laboratory experiment. Medicine. 1944;23:281–358. [Google Scholar]
- 3.Mead J, Takishima T, Leith D. Stress distribution in lungs: A model of pulmonary elasticity. J Appl Physiol. 1970;28:596–608. doi: 10.1152/jappl.1970.28.5.596. [DOI] [PubMed] [Google Scholar]
- 4.Bouhuys A. Physiology and musical instruments. Nature. 1969;221:1199–204. doi: 10.1038/2211199a0. [DOI] [PubMed] [Google Scholar]
- 5.Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis. 1988;137:1159–64. doi: 10.1164/ajrccm/137.5.1159. [DOI] [PubMed] [Google Scholar]
- 6.Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974;110:556–65. doi: 10.1164/arrd.1974.110.5.556. [DOI] [PubMed] [Google Scholar]
- 7.Tierney DF. Ventilator-induced lung injury occurs in rats, but does it occur in humans? Am J Respir Crit Care Med. 2003;168:1414–5. doi: 10.1164/rccm.2310003. [DOI] [PubMed] [Google Scholar]
- 8.Tremblay LN, Slutsky AS. Ventilator-induced injury: From barotrauma to biotrauma. Proc Assoc Am Physicians. 1998;110:482–8. [PubMed] [Google Scholar]
- 9.West JB. Thoughts on the pulmonary blood-gas barrier. Am J Physiol Lung Cell Mol Physiol. 2003;285:L501–13. doi: 10.1152/ajplung.00117.2003. [DOI] [PubMed] [Google Scholar]
- 10.Gattinoni L, Carlesso E, Caironi P. Stress and strain within the lung. Curr Opin Crit Care. 2012;18:42–7. doi: 10.1097/MCC.0b013e32834f17d9. [DOI] [PubMed] [Google Scholar]
- 11.Blankman P, Hasan D, Bikker IG, Gommers D. Lung stress and strain calculation in mechanically ventilated patients in Intensive Care Unit. Acta Anaesthesiol Scand. 2016;60:69–78. doi: 10.1111/aas.12589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiumello D, Carlesso E, Cadringher P, Caironi P, Valenza F, Polli F, et al. Lung stress and strain during mechanical ventilation for acute respiratory distress syndrome. Am J Respir Crit Care Med. 2008;178:346–55. doi: 10.1164/rccm.200710-1589OC. [DOI] [PubMed] [Google Scholar]
- 13.Gibson C, Roberts F. Anaesthesia data. In: Allman K, editor. Oxford Handbook of Anesthesia. 4th ed. Oxford: Oxford University Press; 2016. [Google Scholar]
- 14.Protti A, Votta E, Gattinoni L. Which is the most important strain in the pathogenesis of ventilator-induced lung injury: Dynamic or static? Curr Opin Crit Care. 2014;20:33–8. doi: 10.1097/MCC.0000000000000047. [DOI] [PubMed] [Google Scholar]
- 15.Nihat O, editor. Fundamentals of Biomechanics: Equilibrium, Motion and Deformation. 3rd ed. New York: Springer; 2012. Mechanical properties of biological tissues; pp. 221–36. [Google Scholar]
- 16.Protti A, Andreis DT, Monti M, Santini A, Sparacino CC, Langer T, et al. Lung stress and strain during mechanical ventilation: Any difference between statics and dynamics? Crit Care Med. 2013;41:1046–55. doi: 10.1097/CCM.0b013e31827417a6. [DOI] [PubMed] [Google Scholar]
- 17.Suki B, Stamenović D, Hubmayr R. Lung parenchymal mechanics. Compr Physiol. 2011;1:1317–51. doi: 10.1002/cphy.c100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amato MB, Meade MO, Slutsky AS, Brochard L, Costa EL, Schoenfeld DA, et al. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med. 2015;372:747–55. doi: 10.1056/NEJMsa1410639. [DOI] [PubMed] [Google Scholar]
- 19.Cressoni M, Gotti M, Chiurazzi C, Massari D, Algieri I, Amini M, et al. Mechanical power and development of ventilator-induced lung injury. Anesthesiology. 2016;124:1100–8. doi: 10.1097/ALN.0000000000001056. [DOI] [PubMed] [Google Scholar]
- 20.Gattinoni L, Tonetti T, Cressoni M, Cadringher P, Herrmann P, Moerer O, et al. Ventilator-related causes of lung injury: The mechanical power. Intensive Care Med. 2016;42:1567–75. doi: 10.1007/s00134-016-4505-2. [DOI] [PubMed] [Google Scholar]