

Abstract
Throughout history, natural products have played a dominant role in the treatment of human ailments. For example, the legendary discovery of penicillin transformed global existence. Presently, natural products comprise a large portion of current-day pharmaceutical agents, most notably in the area of cancer therapy. Examples include Taxol, vinblastine, and camptothecin. These structurally unique agents function by novel mechanisms of action; isolation from natural sources is the only plausible method that could have led to their discovery. In addition to terrestrial plants as sources for starting materials, the marine environment (e.g., ecteinascidin 743, halichondrin B, and dolastatins), microbes (e.g., bleomycin, doxorubicin, and staurosporin), and slime molds (e.g., epothilone B) have yielded remarkable cancer chemotherapeutic agents. Irrespective of these advances, cancer remains a leading cause of death worldwide. Undoubtedly, the prevention of human cancer is highly preferable to treatment. Cancer chemoprevention, the use of vaccines or pharmaceutical agents to inhibit, retard, or reverse the process of carcinogenesis, is another important approach for easing this formidable public health burden. Similar to cancer chemotherapeutic agents, natural products play an important role in this field. There are many examples, including dietary phytochemicals such as sulforaphane and phenethyl isothiocyanate (cruciferous vegetables) and resveratrol (grapes and grape products). Overall, natural product research is a powerful approach for discovering biologically active compounds with unique structures and mechanisms of action. Given the unfathomable diversity of nature, it is reasonable to suggest that chemical leads can be generated that are capable of interacting with most or possibly all therapeutic targets.
Key Words: Antitumor drugs, Chemoprevention, Drug discovery, Natural product drug sources
Introduction
Throughout history, natural products have played a dominant role in the treatment of human ailments. The associations of salicylates with the willow and quinine with cinchona are renowned examples; similarly, the legendary discovery of penicillin transformed global existence. In addition, traditional remedies, largely based on terrestrial plants, still dominate therapeutic practices throughout the world, and natural products comprise a large portion of current-day pharmaceutical agents, most notably in the areas of antibiotic and cancer therapies. For the treatment of cancer, early diagnosis and definitive tumor eradication through radiation therapy or surgical resection offer greatest hope. However, when dealing with malignant, metastatic disease, it is generally necessary to resort to chemotherapy. As described herein, many of the meaningful advances that have been realized for the treatment of cancer are directly correlated with the discovery of natural product drugs.
Irrespective of these advances, however, cancer remains a leading cause of death worldwide. In the United States, for example, 589,430 deaths and 1,658,370 new cases of cancer are projected for 2015 [1]. Given the morbidity and mortality associated with the disease, as well as the significant economic burden, there continues to be a critical need for more effective strategies. Undoubtedly, the prevention of human cancer is highly preferable to treatment. In this sense, the advent of vaccines for the prevention of hepatitis and consequential liver cancer is probably the greatest success, and the more recent development of vaccines for the prevention of cervical cancer offers promise. Cancer chemoprevention, the use of synthetic or natural agents to inhibit, retard, or reverse the process of carcinogenesis, is another important approach for easing this formidable public health burden [2].
In an ideal world, cancer chemoprevention would work as well as vaccines for the prevention of human ailments. Although this has yet to be accomplished, proof of principle has been established by seminal clinical trials conducted for the prevention of breast cancer with tamoxifen, and more recently with tamoxifen relatives such as raloxifene, and a separate class of aromatase inhibitors. Agents such as finasteride have shown promise for the prevention of prostate cancer. In terms of drugs under investigation, as is the case with cancer chemotherapeutic agents, natural products have played a critical role in cancer chemoprevention studies. An overview is presented herein.
Cancer Chemotherapy and the Role of Natural Products
Over 60% of the current anticancer drugs were derived in one way or another from natural sources [3]. Nature continues to be an abundant source of biologically active and diverse chemotypes, and while relatively few of the actual isolated natural products are developed into clinically effective drugs in their own right, these unique molecules often serve as models for the preparation of more efficacious analogues and prodrugs through the application of chemical methodology, such as total or combinatorial (parallel) synthesis, or the manipulation of biosynthetic pathways. In addition, improvements in formulation may result in more effective administration of the drug to patients, or conjugation of toxic natural molecules to monoclonal antibodies or polymeric carriers specifically targeting epitopes on tumors of interest can lead to the development of efficacious targeted therapies. The essential role played by natural products in the discovery and development of novel anticancer agents, and the importance of multidisciplinary collaboration in the optimization of novel molecular leads from natural product sources have been extensively reviewed [4,5,6,7,8,9].
The following sections briefly discuss some of the recent developments in this area, with discussions limited to new agents currently in some stage of advanced clinical development, or agents which have been approved for commercial use. It should be noted that effective cancer chemotherapy often involves the use of combinations of several agents (so-called combination chemotherapy), and these combination regimens can comprise agents derived from both natural and synthetic sources. Interested readers may gain information on recently completed and ongoing clinical trials of agents discussed below (e.g., vinblastine, VBL) by logging into www.clinicaltrials.gov and entering ‘cancer and name of agent (e.g., cancer and vinblastine)’ in the search box.
Cancer Chemotherapeutic Agents Derived from Terrestrial Plants
Historically, plants have been primary sources of natural product drug discovery, and in the anticancer area, plant-derived agents, such as VBL and vincristine (VCR), etoposide, paclitaxel (Taxol®), docetaxel, topotecan, and irinotecan, are among the most effective cancer chemotherapeutics currently available [8]. Nevertheless, many suffer from the liabilities of poor solubility in aqueous media and significant toxic side effects. Thus, there continues to be considerable research devoted to diminishing the impact of these factors, and numerous analogues and prodrugs of these agents have been synthesized, and methods devised for increasing aqueous solubility and targeting specific tumors.
Vinca Alkaloids
The first plant-derived agents to advance into clinical use were the vinca alkaloids, VBL (1; fig. 1) and VCR (2; fig. 1), isolated from the Madagascar periwinkle, Catharanthus roseus G. Don. (Apocynaceae). This plant was used by various cultures for the treatment of diabetes, and while under investigation as a source of potential oral hypoglycemic agents, it was noted that extracts of the plant reduced white blood cell counts and caused bone marrow depression in rats, and subsequently they were found to be active against lymphocytic leukemia in mice. This led to the isolation of VBL and VCR as the active agents, so their discovery may be indirectly attributed to the observation of an unrelated medicinal use of the source plant.
Fig. 1.

Vinca alkaloids, etoposide, taxanes, and CPTs.
The mechanism of action is to disrupt microtubules, causing the arrest of the cells at metaphase and leading to apoptotic cell death.
Semisynthetic Analogues
The effective semisynthetic analogues that have been developed include vinorelbine and vindesine, with the most recent being vinflunine, a second-generation bifluorinated analogue of vinorelbine.
These agents are primarily used in combination with other cancer chemotherapeutic drugs for the treatment of a variety of cancers, including leukemias, lymphomas, advanced testicular cancer, breast and lung cancers, and Kaposi's sarcoma. A comprehensive discussion of these agents is presented in the review by Roussi et al. [10].
Podophyllotoxins
The species Podophyllum peltatum L. (commonly known as the American mandrake or mayapple) and Podophyllumemodii Wallich from the Indian subcontinent have a long history of medicinal use, including the treatment of skin cancers and warts. The structure of the major active constituent, podophyllotoxin, first isolated in 1880, was only reported in the 1950s. Clinical trials of several closely related podophyllotoxin-like lignans, however, failed due to lack of efficacy and unacceptable toxicity. Extensive research led to the development of etoposide (3; fig. 1) and teniposide as clinically effective agents.
For mechanism of action, while podophyllotoxin reversibly binds to tubulin, etoposide and teniposide inhibit topoisomerase II, inducing topoisomerase II-mediated DNA cleavage.
For treatment, etoposide (3; fig. 1) and teniposide are used for lymphomas, and bronchial and testicular cancers. The history of the development of these agents and some related analogues under clinical investigation has been reviewed [11].
Taxanes
A more recent addition to the armamentarium of plant-derived chemotherapeutic agents are the taxanes, considered one of the most important classes of cancer chemotherapeutic drugs in clinical use. Currently, the two most clinically effective drugs of this class are paclitaxel (Taxol®: 4; fig. 1), originally isolated from the bark of the Pacific yew, Taxus brevifolia Nutt. (Taxaceae), and docetaxel (Taxotere®: 5; fig. 1), a semisynthetic analogue synthesized from DAB (10-deacetylbaccatin III) isolated from the leaves of the European yew, Taxus baccata. DAB has also been semisynthetically converted to paclitaxel, thereby providing a sustainable source of the drug. It is interesting to note that the leaves of T. baccata are used in the traditional Asiatic Indian (ayurvedic) medicine system, with one reported use being in the treatment of ‘cancer’ [12].
Regarding the mechanism of action, paclitaxel and other taxanes promote the polymerization of tubulin heterodimers to microtubules, suppressing dynamic changes in microtubules resulting in mitotic arrest.
Paclitaxel is used in the treatment of breast, ovarian, and non-small cell lung cancer (NSCLC), and has also shown efficacy against Kaposi's sarcoma, while docetaxel is primarily used in the treatment of breast cancer and NSCLC. A comprehensive review of the taxanes as well as ongoing research into the development of improved analogues and methods of delivery has been published by Kingston [13].
The many structural analogues of Taxol® synthesized and new formulations developed have been recently reviewed by a number of authors [14,15]. Most of the analogues and new formulations have been engineered in attempts to overcome the clinical limitations of paclitaxel and docetaxel, including poor solubility, allergic reactions, dose-limiting toxicities such as myelosuppression or peripheral sensory neuropathy, and the development of drug resistance due to P-glycoprotein-mediated efflux. The analogue cabazitaxel (Jevtana®: 6; fig. 1) has been approved for the treatment of metastatic prostate cancer (in combination with prednisone and/or prednisolone) [16,17], and is in clinical trials, either as a single agent or in combination with other agents, for the treatment of a range of cancers including prostate, bladder, brain (glioblastoma multiforme), head-and-neck, ovarian, stomach and urinary tract cancers, and NSCLC www.clinicaltrials.gov). Other structural analogues in various stages of clinical development against a variety of cancers [14,15] include Taxoprexin or 7-docosahexaenoic acid (DHA)-paclitaxel (Protarga), a prodrug of paclitaxel covalently bound to the naturally occurring ω-3 fatty acid DHA which enables delivery directly to tumor tissue; larotaxel (XRP9881); ortataxel [18]; tesetaxel (DJ-927) [19]; TPI-287 [20], and paclitaxel poliglumex (PPX, CT-2103, previous trade name Xytotax®, now known as Opaxio®), an α-poly-L-glutamic acid conjugate of paclitaxel [21,22]. PPX is a potent radiation sensitizer, possibly enhancing radiation for glioblastoma [23], and in 2012 the US Food and Drug Administration (FDA) designated orphan drug status to PPX for the treatment of glioblastoma multiforme.
Several new formulations and nanoparticle preparations of paclitaxel have been either approved or are undergoing clinical trials [14,24], and promising developments are being reported. Of the approved agents, Abraxane® (nab-paclitaxel, ABI-007) is an albumin-stabilized nanoparticle formulation of paclitaxel which solubilizes paclitaxel without the use of the emulsifying agent Cremophor (polyethoxylated castor oil), thereby enabling larger doses of paclitaxel to be administered while avoiding the toxic effects associated with Cremophor. It has been approved for the treatment of refractory breast cancer [25], NSCLC [26], and pancreatic cancer as a less toxic, though less effective, alternative to the 4-drug regimen (leucovorin, 5-FU, irinotecan, and oxaliplatin) [27,28]. Other new formulations include LEP-ETU, a stable lyophilized liposome-based paclitaxel formulation which has shown bioequivalence with paclitaxel formulated with Cremophor [29]; EndoTAG-1, which is formed by encapsulation of paclitaxel in positively charged lipid-based complexes [granted orphan drug status for the treatment of pancreatic cancer by the European Medical Association (EMA) and FDA in 2006 and 2009, respectively] and has shown efficacy in the treatment of advanced triple-negative breast cancer [30] and head-and-neck squamous cell carcinoma [31]; Genexol-PM, a Cremophor EL-free polymeric micelle formulation of paclitaxel, which has shown activity against gemcitabine-sensitive and -resistant pancreatic ductal adenocarcinoma cell lines [32], and Tocosol® (S-8184), a tocopherol-based Cremophor-free formulation of paclitaxel, which reportedly has a potentially higher bioavailability of unbound paclitaxel compared to Cremophor EL-formulated paclitaxel in human subjects due to direct release at the target site [33], while the relative exposure of unbound paclitaxel at the site of toxicity was reported to be twice as large for Tocosol compared to other paclitaxel formulations [34].
Camptothecins
Camptothecins (CPTs; 7; fig. 1) were isolated from the Chinese ornamental tree Camptotheca acuminata Decne (Nyssaceae).
The mechanism of action is through binding to the topoisomerase I-DNA binary complex resulting in a stable ternary complex, thereby preventing DNA religation and causing DNA damage, which results in apoptosis. Clinical trials of the water-soluble sodium salt in the 1970s, however, were terminated due to severe bladder toxicity.
Comprehensive reviews of CPT and its analogues have been published [35]. Extensive research led to the development of semisynthetic derivatives, topotecan (8; fig. 1), irinotecan (9; fig. 1), and belotecan (CKD-602: 10; fig. 1), which had been approved for clinical use.
To avoid the problems related to oral bioavailability, unfavorable metabolism, toxicity, and drug resistance associated with currently marketed CPTs, many structural analogues of CPT have been designed and developed [35]. Cositecan (Karenitecin; BNP1350: 11; fig. 1) and silatecan (AR-67) are lipophilic silicon-containing CPTs modified in the 7-position. Further lipophilic analogues are gimatecan, another 7-substituted CPT derivative [36], and diflomotecan (BN80915), a 10,11-difluoro- homoCPT [37]. Water-soluble analogues currently in clinical development are the hydrochloride salts of elemotecan, lurtotecan, and namitecan, as well as DRF-1042 being developed by Dr. Reddy's Research Foundation.
CPT and its derivative, SN-38 (7-ethyl-10-hydroxy CPT: 12; fig. 1), are both poorly soluble in water and pharmaceutically approved solvents. This has led to extensive research into the development of macromolecular prodrugs and nanomedicine formulations of these compounds in efforts to improve the preferential delivery of these agents to cancer cells and tissues. This has resulted in the development of products exhibiting improved efficacy and reduced side effects. Among the products developed from CPT are: CRLX101, a conjugate with a polymeric nanoparticle comprised of cyclodextrin-poly(ethylene glycol) copolymer, which has an apparent solubility increase of >1,000-fold as compared to CPT [38], and has demonstrated encouraging safety, pharmacokinetic, and efficacy results in an early clinical trial in patients with advanced solid malignancies [39], and XMT-1001 (MER-1001 or PHF-CPT), a novel, water-soluble macromolecular prodrug of CPT (molecular weight: 70 kDa), in which CPT is conjugated to a hydrophilic, biodegradable polyacetal polymer, poly(1-hydroxymethylethylene hydroxymethylformal), also known as PHF or Fleximer®, and is slowly released [40]. The recent advances with SN-38 (7-ethyl-10-hydroxy CPT: 12; fig. 1) have been summarized in a recent review [41]. Among the promising products developed are DTS-108, a soluble prodrug in which SN-38 is covalently linked to a 20- amino acid peptide through a cross-linker that releases SN-38 by esterase bond cleavage; IMMU-130 (labetuzumab-SN-38; hMN14-SN-38), an antibody-drug conjugate (ADC) containing labetuzumab, a reduced anti-CEACAM5 humanized monoclonal antibody, conjugated to SN-38 at the C-20 hydroxyl group through a maleimidocaproyl-Phe-Lys-p-aminobenzyl-CO linker, and IMMU-132 (hRS7-SN-38), another ADC comprising SN-38 and the humanized monoclonal antibody hRS7 linked at the C-20 hydroxyl group in the same manner as outlined above for IMMU-130. In November 2013, the FDA designated IMMU-132 orphan drug status for the treatment of small cell lung cancer, and in May 2014, orphan drug status was designated for the treatment of pancreatic cancer.
Combretastatins
The combretastatins are a family of stilbenes originally isolated from the root bark Combretum caffrum, also known as the Cape bushwillow in southern Africa.
They act as vascular disrupting agents, selectively targeting the endothelial cells lining the tumor vasculature. They also disrupt the tubulin cytoskeleton and remodel the actin cytoskeleton, inducing a significant change in the three-dimensional shape of immature endothelial cells, thereby stopping blood flow through the capillary and starving the tumor of nutrients, causing tumor cell death. This mechanism of action differentiates the combretastatins from angiogenesis inhibitors that are designed to work by preventing the growth of new blood vessels. The discovery and development of these agents is presented in a comprehensive review by Pinney et al. [42].
Combretastatin Prodrugs
Combretastatin A4 phosphate (CA4P; fosbretabulin; Zybrestat: 14; fig. 2) is a phosphate prodrug of CA4 (13; fig. 2). In 2003, CA4P was granted orphan drug designation by the FDA for the treatment of anaplastic thyroid cancer, medullary thyroid cancer, and stage IV papillary or follicular thyroid cancer. Later trials in the treatment of patients with advanced anaplastic thyroid carcinoma, using CA4P as a single agent [43] or in combination with paclitaxel/carboplatin [44], reported no objective responses, though the treatments were reported to be well tolerated, and one third of the patients in the single-agent CA4P trial survived more than 6 months. In 2006, CA4P was granted orphan drug designation by the FDA for the treatment of ovarian cancer, and treatment of patients with platinum-resistant ovarian cancer with a combination of CA4P, carboplatin, and paclitaxel reportedly produced a higher response rate in this patient population compared to chemotherapy given without CA4P [45]. Numerous analogues of CA4 have been synthesized and their biological activities reviewed [46].
Fig. 2.

Combretastatins, HHT, and ingenol mebutate.
Combretastatin A1 diphosphate (CA1P; OXI-4503: 16; fig. 2) is a phosphate prodrug of CA1 (15; fig. 2), and it has shown promising efficacy in the treatment of patients with relapsed and refractory acute myelogenous leukemia (AML) and myelodysplastic syndromes [47]. In 2012, orphan drug designation was assigned by the FDA for the treatment of AML.
Homoharringtonine
The isolation and structure of homoharringtonine (HHT; omacetaxine mepesuccinate; Synribo: 17; fig. 2) was first stated in 1970 from the Chinese tree, Cephalotaxus harringtonia var. drupacea (Sieb and Zucc.; Cephalotaxaceae). The bark of Cephalotaxus species has a long history of use in traditional medicine in China for the treatment of various indications, and in 1983 Chinese investigators reported significant cytotoxicity of the total alkaloid fraction of Cephalotaxus fortunei Hook F.
A racemic mixture of HHT and harringtonine has been used in China for the treatment of AML and chronic myelogenous leukemia (CML), and purified HHT has shown efficacy against various leukemias, including some resistant to standard treatment, with complete hematologic remission being reported in the treatment of patients with late chronic phase CML. The development of HHT and related compounds has been comprehensively reviewed by Itokawa et al. [48]. HHT (Synribo) is thought to act as a broad-spectrum protein tyrosine kinase inhibitor and was approved in 2012 by the FDA for the treatment of adult patients with CML or accelerated phase CML [49]. The therapeutic response of patients with CML in myeloid blast crisis (CML-MBC) is generally very poor, but a combination of HHT and cytarabine has been reported to be an effective treatment for CML-MBC [50].
Ingenol Mebutate (Ingenol-3-Angelate)
The common Australian plant Euphorbia peplus (Euphorbiaceae) is widely used as a home remedy for the treatment of various skin conditions, and clinical studies with crude E. peplus sap in the 1970s provided compelling evidence of its efficacy. The active agent was identified as the hydrophobic diterpene ester, ingenol-3-angelate (PEP005; ingenol-3-mebutate; Picato: 18; fig. 2), and it was shown to act through activation of protein kinase C (PKC). Several reports of clinical trials for the treatment of actinic keratosis have been published [51,52,53], and in 2012 it was approved as a topical gel formulation (Picato) for this indication by the FDA and the EMA.
Cancer Chemotherapeutic Agents Derived from Marine Organisms
Despite covering roughly 70% of the planet, less than 5% of the deep sea has been pharmacologically explored in any way, and less than 0.01% of the deep-sea floor has been sampled in detail [54]. A significant number of marine-derived antitumor agents showing potent growth inhibition of human tumor cells in vitro and, in a number of cases, in in vivo murine models and in humans have been isolated, but although many agents have entered clinical trials in cancer, to date only four have been approved for use in humans [55]. These agents are cytarabine [AraC (cytosine arabinoside): 19; fig. 3], Yondelis® (ET743; trabectedin: 20; fig. 3), eribulin (Halaven®: 22; fig. 3), a synthetic derivative based on the structure of halichondrin B (21; fig. 3), and the ADC Adcetris® (brentuximab vedotin) containing the dolastatin 10 derivative, monomethyl auristatin E (MMAE or vedotin) as a warhead, which is discussed later. The status of these agents has recently been reviewed [55].
Fig. 3.

Cytarabine, trabectedin, halichondrin B, and eribulin.
Cytarabine and Nucleoside Analogues
It can be argued that the discovery by Bergmann and Burke [56] in the early 1950s of the arabinose-containing bioactive nucleosides, spongothymidine and spongouridine, from the Caribbean sponge Tethya crypta sparked the exploration of the marine environment as a source of novel bioactive compounds that could serve as leads to potential drugs. This discovery led to the identification and development of analogues such as cytarabine (AraC: 19; fig. 3) as a potent antileukemic agent, and the antiviral agent, AraA (adenine arabinoside) [55].
Various nucleoside analogues showing significant anticancer activity have been developed. Clofarabine is a second-generation purine nucleoside analogue which has shown a broad range of clinical activity and great potency in damaging the DNA of leukemia cells. In 2004, it received FDA approval for the treatment of pediatric patients with acute lymphoblastic leukemia, and there have been further extensive studies in the treatment of these patients at different ages [57]. In addition, its role in the treatment of adult patients with AML (also called acute myeloid leukemia, acute myeloblastic leukemia, acute granulocytic leukemia, and acute nonlymphocytic leukemia) and the myelodysplastic syndrome has been reviewed [58,59]. Sapacitabine (CYC-682), an orally bioavailable nucleoside analogue prodrug, shows a unique mechanism of action, causing single-strand breaks after incorporation into DNA, which are converted into double-strand breaks when cells enter a second S-phase. It has been shown to be active in the treatment of elderly patients with AML [60,61]. Orphan drug designation for the treatment of AML and for the treatment of myelodysplastic syndromes was assigned by the FDA in 2010.
Trabectedin (ET743; Yondelis®)
Although the isolation and structural elucidation of ecteinascidin derivatives were first reported in 1986, articles on the identification of trabectedin (ET743: 20; fig. 3), the most active of the ecteinascidins, only appeared in 1990.
It was approved for use in Europe for the treatment of advanced soft-tissue sarcoma and has been granted orphan drug status for the treatment of soft-tissue sarcoma and ovarian cancer by the FDA and the EMA. It was the first ‘unmodified’ marine-derived natural product to be approved for the treatment of cancer, and it is undergoing clinical trials for the treatment of breast, prostate, and pediatric sarcomas. For further details, readers are referred to a comprehensive review by Cuevas et al. [62]. The structurally related agents, Zalypsis (PM-10450) and lurbinectedin (PM-01183), have progressed into clinical trials, and their discovery and development have been discussed [55].
Halichondrin B and Eribulin
The isolation and structural elucidation of the complex natural product, halichondrin B (21; fig. 3), along with several other halichondrin derivatives, were first reported from the marine sponge Halichondria okadai in 1986, and closely followed by reports of the isolation of the same series of compounds from different sponges collected in different areas, ranging from the Central Pacific to the Indian Ocean to waters off New Zealand [55]. It was shown to act as a tubulin-destabilizing agent and was approved for preclinical development by the US National Cancer Institute in early 1992. The procurement of sufficient supplies of source raw material proved to be challenging, but large-scale collections and in-sea aquaculture of the sponge Lissodendoryx sp. proceeded off the coast of New Zealand in collaboration with local scientists, and sufficient quantities of halichondrin B were isolated for preclinical studies.
Meanwhile, several years prior to its approval for preclinical development, the Kishi group at Harvard University had been exploring the total synthesis of halichondrin B, and, in 1992, they reported that they had synthesized halichondrin B and norhalichondrin B. Working in collaboration with scientists at the then Eisai Research Institute, the biological activity was determined to reside predominately in the ring portion of the molecule, and close to 200 derivatives of the truncated natural product were made and evaluated. Head-to-head comparisons of pure halichondrin B and the best synthetic analogues, using in vitro time course assays and in vivo studies in mice with human xenografts, showed that the truncated halichondrin B analogue, now known as eribulin (22; fig. 3), with structural similarities to halichondrin B (21; fig. 3; in red, see online version) showed significantly more potent activity in the in vivo studies. This compound was chosen for advanced preclinical and then clinical studies, using materials made under cGMP conditions by total synthesis. Following extended clinical trials, eribulin (with the proprietary name of Halaven®) was approved for the treatment of refractory breast cancer by the FDA in 2010.
The discovery and development of this compound, including the progression from the synthesis of halichondrin B to the initial synthesis of eribulin, have been reviewed [63]. In addition, papers on the industrial methodologies that enabled the large-scale production of eribulin have been published [64,65,66,67]. To date, eribulin is by far the most complex drug ever produced by total synthesis, and it is a testament to all of the investigators in three countries and multiple organizations that cooperated to make it a success.
Cancer Chemotherapeutic Agents Derived from Bacteria and Fungi
Antitumor antibiotics are amongst the most important of the cancer chemotherapeutic agents. These include members of the actinomycin, ansamycin, anthracycline, bleomycin, epothilone, and staurosporin classes. Except for the epothilones, which are metabolites of the myxobacterium Sorangium cellulosum, metabolites of the other classes were isolated from various Streptomyces species. Comprehensive reviews of these agents have been published [8]. Some recent advances in the development of other microbial-derived anticancer agents are given in the following sections.
Rapamycins
The discovery of rapamycin (sirolimus: 23; fig. 4), a 31-membered macrocyclic antibiotic produced by the fermentation of a strain of Streptomyces hygroscopicus isolated from soil samples in Rapa Nui (Easter Island), was first reported in 1975. Initially reported to have antifungal activity, it was approved for use as an immunosuppressive agent (trade name, Rapamune) in 1999. While reported to have antitumor activity in 1984, only reports of the identification of TOR (‘target of rapamycin’) as the molecular target in yeast in 1991, followed by mTOR as the mammalian homologue in 1994, ultimately led to the development of a wide variety of anticancer and other pharmacologic agents.
Fig. 4.

Rapamycins, carfilzomib, and midostaurin.
Chemical modifications have yielded two clinically approved anticancer drugs. Everolimus (Afinitor®: 24; fig. 4) was initially approved as an immunosuppressive agent in 2004, but approval was granted for the treatment of kidney, brain, pancreatic, and breast cancers in 2009, 2010, 2011, and 2012, respectively. It is also currently in, or has recently completed, phase III trials for treating diffuse large B-cell lymphoma (NCT00790036), liver (NCT01035229), and stomach (NCT00879333) cancers. Temsirolimus (Torisel®; CCI-779: 25; fig. 4) was first approved as a treatment for renal carcinoma in 2007 and is currently in phase II trials for the treatment of various carcinomas. Another rapamycin derivative showing promise in the treatment of cancer is ridaforolimus (26; fig. 4), which has recently completed a phase III trial for the treatment of soft-tissue carcinoma and bone cancer. The remarkable application of rapamycin derivatives to the treatment of cancer and various other diseases has been covered in a recent review [68].
Carfilzomib (Kyprolis™)
Carfilzomib (Kyprolis™: 27; fig. 4), a synthetic analogue of epoxomicin, a peptide α′,β′-epoxyketone isolated from an actinomycete strain, is a proteasome inhibitor which binds through a covalent, selective, and stereospecific linkage to the chymotryptic subunit (20S) of the proteasome [69]. It was approved by the FDA in 2012 for the treatment of patients with relapsed and refractory multiple myeloma who had received prior treatment with bortezomib, thalidomide, or lenalidomide [70,71,72]. Phase II trials are ongoing for the treatment of a variety of other cancers.
Midostaurin
Midostaurin (PKC-412: 28; fig. 4)is a semisynthetic derivative of staurosporine, an indolocarbazole alkaloid isolated from Streptomyces staurosporeus[73].
Midostaurin, a PKC and Flt3 (FLK2/STK1) inhibitor, has completed a phase IIB clinical trial in patients with AML and high-risk myelodysplastic syndrome with either wild-type or mutated Flt3 [74], and is in phase III clinical development for the oral treatment of AML. Further clinical trials for the treatment of patients with AML using midostaurin, either as single agent or in combination with agents such as azacitidine, bortezomib, cytarabine, daunorubicin, decitabine, and all-trans-retinoic acid, are ongoing, and phase III clinical trials are being conducted for the treatment of aggressive systemic mastocytosis or mast cell leukemia. Orphan drug designation for the treatment of AML was assigned by the EMA in 2004 and the FDA in 2009, as well asfor the treatment of mastocytosis by the EMA and FDA in 2010.
Targeted Agents
As mentioned earlier, cytotoxic natural products frequently suffer from liabilities such as limited solubility in aqueous solvents and considerable toxicity, often resulting in narrow therapeutic indices. This has led to the failure in clinical trials of a number of natural products, such as the plant-derived agent maytansine, but recently several of these agents have shown potential as ‘warheads’ which can be attached to monoclonal antibodies specifically targeting epitopes on the tumor of interest. Such agents are called ADCs, and the promise of this approach to cancer therapy has been the subject of several reviews [75,76,77,78,79,80], as well as a recent book chapter [81].
Maytansinoids
Maytansine (29; fig. 5), a novel macrocyclic compound, was isolated in extremely low yield in the early 1960s from the Ethiopian plant, Maytenus serrata (Hochst. ex A. Rich.) Wilczek [82], and was shown to exhibit very potent in vitro antitumor activity. Thus, further development was pursued, but despite the promising activity observed in preclinical animal testing, insignificant efficacy was observed in human clinical trials, and studies were terminated in the early 1980s. Due to its structural similarity to the ‘ansa’ antibiotics, such as the rifamycins, natural product chemists had wondered if maytansine was of microbial origin. In 1977, the isolation of the closely related ansamitocins from the bacterium Actinosynnema pretiosum[82] strengthened this speculation, and a report by members of the Leistner group, who identified a very closely related Actinosynnema sp. in the microbial root system of plants producing maytansine, coupled to the complete absence of a required AHBA synthase gene in plant cell cultures of the nominal host plant, provided further circumstantial evidence for the bacterial source of the maytansinoids [83]. The ansamitocins provided a ready and sustainable source of maytansinoids, and the derivatives DM1 (30; fig. 5) and DM4 (31; fig. 5) have been prepared from appropriate ansamitocins.
Fig. 5.

Targeted agents: maytansanoids and brentuximab vedotin. mAb = Monoclonal antibody.
Conjugation with Monoclonal Antibodies
DM1 and DM4 have been conjugated through either thioether or disulfide linkages with various monoclonal antibodies targeting a variety of cancers [82,84]. Linkage of DM1 to the approved her2neu-targeted antibody, trastuzumab, gives T-DM1 or adotrastuzumab emtansine (Kadcyla®), which showed significant efficacy in the treatment of patients with advanced or metastatic HER2-positive breast cancer who had failed at least two treatments with the currently approved drugs, trastuzumab and the tyrosine kinase inhibitor lapatinib [85,86,87]. Based on these results, Kadcyla® was approved by the FDA in 2013 as a new therapy for patients with HER2-positive, late-stage (metastatic) breast cancer. A review by Teicher and Chari [88], together with the article by Koehn [81], gives the clinical status of these and several other ADCs. Thus a bioactive natural product macrocycle that had failed clinical trials in the early 1980s has now become a potent and active treatment for specific breast cancers by using it as a warhead.
Brentuximab Vedotin (Adcetris®)
Brentuximab vedotin (Adcetris®: 32; fig. 5)is formed by conjugation of monomethyl auristatin E (vedotin), a ‘warhead’ derived from dolastatin 10, a secondary metabolite originally isolated from the marine mollusk, Dolabella auricularia, but later isolated from a Symploca species of cyanophyte which was shown to be in the diet of the mollusk [89].
It was approved for treatment of CD30-positive lymphoproliferative disorders such as Hodgkin's lymphoma by the FDA and the EMA in 2011 and 2012, respectively. Details of the use of this ADC agent against a variety of lymphomas have been discussed in the review by Newman and Cragg [55]; this review also discusses the clinical development of over 20 other ADCs prepared by conjugation of vedotin or closely related analogues with antibodies targeting epitopes found in various cancers, including breast, gastrointestinal, pancreatic, prostate, ovarian and renal cancers, leukemias, melanomas and NSCLC.
Cancer Chemoprevention
For the treatment of cancer, early diagnosis and definitive tumor eradication through radiation therapy or surgical resection offer greatest hope. In these cases, 5-year survival rates for many types of cancer (e.g., breast, prostate, and colon cancers, and melanomas) typically exceed 90%. However, when dealing with malignant, metastatic disease, 5-year survival rates of less than 5% are not atypical (e.g., lung, pancreas, and stomach cancers) and rarely exceed 40%. Novel approaches are currently being explored, such as exploitation of chimeric antigen receptors [90], but mainstay treatment regimens continue to involve chemotherapy.
Another strategy takes the genesis of tumors into account. The process of carcinogenesis is not fully understood, but it is clear that numerous subcellular alterations occur prior to the overt detection of a tumor. These changes have been classified into stages, including initiation, promotion, and progression. Obviously this is a continuum and the entire process is extraordinarily complex. Nonetheless, rather than responding to the appearance of a tumor in a reactive manner, the approach of cancer chemoprevention attempts to delay, block, or reverse the process of carcinogenesis, thus negating the necessity of treatment in a proactive manner.
Cancer chemoprevention can be implemented with complete success in model systems, especially involving rodents [91]. Use of genetically altered animals or treatment with chemical carcinogens readily leads to tumors in select sites such as the lung, breast, prostate, colon, and bladder. Pre- or posttreatment regimens with chemopreventive agents have been devised to completely or significantly prevent tumorigenesis in the absence of toxicity. Indeed, from a conceptual point of view, cancer chemoprevention is very appealing.
Agents Currently in Clinical Use
As proof of concept, tamoxifen (33; fig. 6) and related compounds, such as raloxifene (34; fig. 6), are well-established agents that are currently in use for the prevention of cancer in high-risk breast cancer patients [92]. The key mechanism appears to involve estrogen receptor antagonism. Side effects are known, some of which are serious (such as endometrial cancer), and so it is important to consider risk-benefit ratios [93]. More recently, aromatase inhibitors capable of inhibiting the conversion of androgens to estrogens have entered the market (e.g., letrozole: 35; fig. 6) for the prevention of breast cancer. For males, clinical trials have indicated that prostate cancer risk can be reduced by administration of finasteride (36; fig. 6) [94], an inhibitor of steroid 5α-reductase.
Fig. 6.

Structures of tamoxifen (33), raloxifene (34), letrozole (35), finasteride (36), aspirin (37), and celecoxib (38),
More generally, aspirin (37; fig. 6) is considered as useful for the reduction of colon cancer due to anti-inflammatory activity [95,96]. Another anti-inflammatory agent, celecoxib (38; fig. 6), an inhibitor of cyclooxygenase 2, can effectively inhibit the formation of familial adenomatous polyposis (a precursor of colon cancer), but routine use is precluded due to adverse cardiovascular events that have been observed with other drugs functioning in a similar manner.
Discovery and Evaluation of New Cancer Chemopreventive Agents
From a historical perspective, the concept of cancer chemoprevention appears to have evolved through epidemiological observations. For example, cancer incidence rates appear to be lower in population groups with diets rich in fruits and vegetables [97] or the Mediterranean diet [98], although this point remains moot [99]. In part, this has led to the isolation and study of many dominant components of dietary materials, such as β-carotene (39; fig. 7) from carrots, lycopene (40; fig. 7) from tomatoes, indoles (41; fig. 7) from cruciferous vegetables, curcumin (42; fig. 7) from turmeric, catechins (43; fig. 7) from green tea, and anthocyanins (44; fig. 7) from blueberries. Some clinical trials have been performed with resounding negative results [100] and the entire concept of cancer chemoprevention has been disclaimed [101]. Nonetheless, nearly 300 clinical trials are currently ongoing www.clinicaltrials.gov, most of which are investigating fairly generic agents or structural derivatives of known agents believed to have some activity.
Fig. 7.

Structures of β-carotene (39), lycopene (40), indole-3-carbinol (41), curcumin (42), catechins (43), and anthocyanins (44).
Natural Product-Derived Agents Currently in Clinical Trials
As noted above, cancer chemoprevention has been associated with aspirin, and 13 clinical trials are currently underway with this substance. Other examples of natural product-derived agents in clinical trials include metformin (45; fig. 8; 7 trials), Polyphenon E (46; fig. 8; 9 trials), retinoids (47; fig. 8; 20 trials), soy isoflavones (48; fig. 8; 16 trials), vitamin C (49; fig. 8; 10 trials), vitamin D (50; fig. 8; 20 trials), and vitamin E (51; fig. 8; 16 trials). In addition, curcumin has not done well in clinical trials (ostensibly due to problems with absorption and metabolism), but chemical derivatives (compound 52; fig. 9, and compound 53; fig. 9) are currently being investigated (12 trials).
Fig. 8.

Structures of metformin (45), Polyphenon E (46), retinoids (47), soy isoflavones (48), vitamin C (49), vitamin D3 (50), and vitamin E (51).
Fig. 9.

Structures of demethoxycurcumin (52) and bis-demethoxycurcumin (53).
New Natural Product Cancer Chemopreventive Agents
A traditional approach that led to the discovery of many well-known natural product cancer chemotherapeutic agents such as Taxol and CPT is bioassay-guided isolation. Following a similar approach for chemoprevention, a number of assays can be employed, such as investigating effects on epigenetics [102] or signaling pathways including Keap1-Nrf2 [103], and the Keap1-Nrf2-ARE signaling pathway has proven to be very important for cell defense and survival [104,105,106]. We have tended to focus on broad-based assays reflective of the initiation, promotion, and progression stages of carcinogenesis [107]. In addition to terrestrial plants [108], marine microorganisms have been utilized as starting materials for the isolation and identification of unique cancer chemopreventive agents [109]. Using this strategy, agents have been discovered or produced that fall into the categories of dietary natural products, nondietary natural products, semisynthetic derivatives, and known natural products with hitherto unknown mechanisms of action (i.e., repurposing).
A resounding example of the latter is the quassinoid bruceantin (54; fig. 10) and the close structural relative brusatol (55; fig. 10). Bruceantin was originally discovered as a cancer chemotherapeutic agent and dropped due to poor efficacy in advanced-stage cancer patients [110,111]. It was ‘rediscovered’ as a potent inducer of cell differentiation and subsequently found to inhibit tumor growth at low doses in the absence of toxicity [112,113,114]. More recently, additional mechanisms have been described [115]. This is a good example of how a chemopreventive agent may function at the interface of carcinogenesis/tumorigenesis. Like tamoxifen, an agent that can mediate an antitumor effect with an acceptably low level of toxicity, it can be considered for use in the arena of chemoprevention.
Fig. 10.
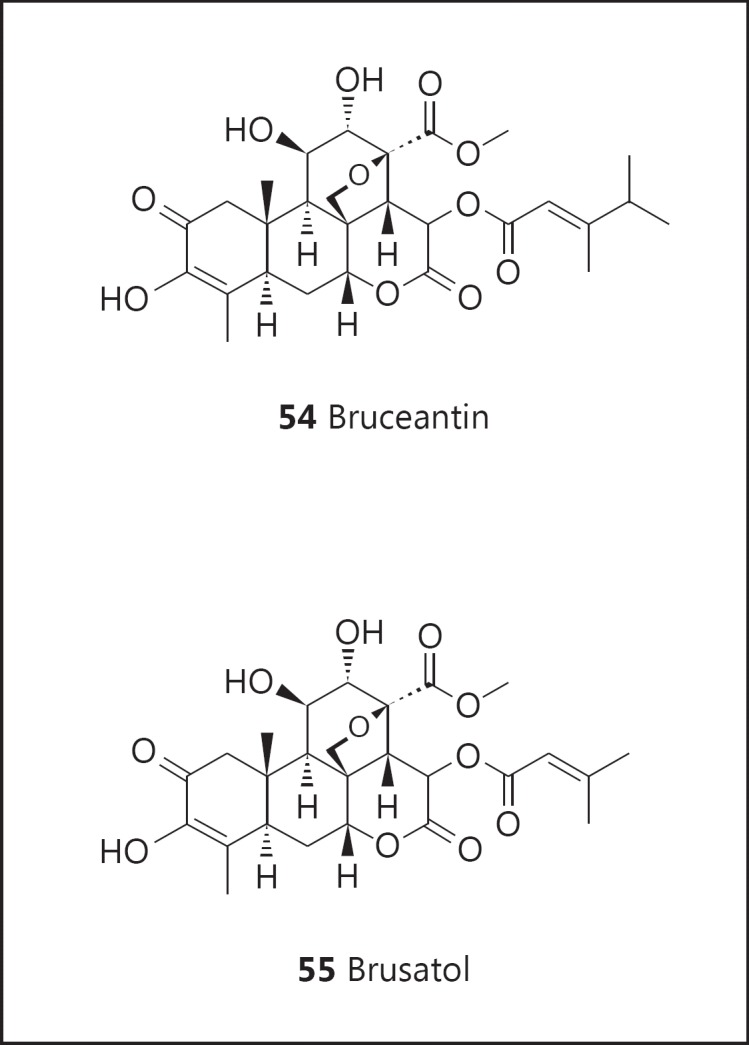
Structures of bruceantin (54) and brusatol (55).
Resveratrol
One of the most notable discoveries resulting from this program of natural product cancer chemopreventive agent discovery is resveratrol (56; fig. 11). In the context of chemoprevention, resveratrol was isolated from a nonedible legume following bioassay-guided fractionation with inhibition of cyclooxygenase as the objective. Following this discovery, the compound was characterized as an inhibitor of skin carcinogenesis functioning by a pleiotropic mechanism of action [116]. Due to the presence of resveratrol in grapes and grape products such as wine, the work attracted a great deal of attention. During the ensuing years, approximately 15,800 papers have appeared in the scientific literature, of which approximately 6,700 are related to cancer. Well beyond cancer chemoprevention, amelioration of a plethora of disease states has been investigated, most notably cardiovascular, diabetes, and age-related diseases [117]. Currently, 97 ongoing trials are listed on www.clinicaltrials.gov.
Fig. 11.
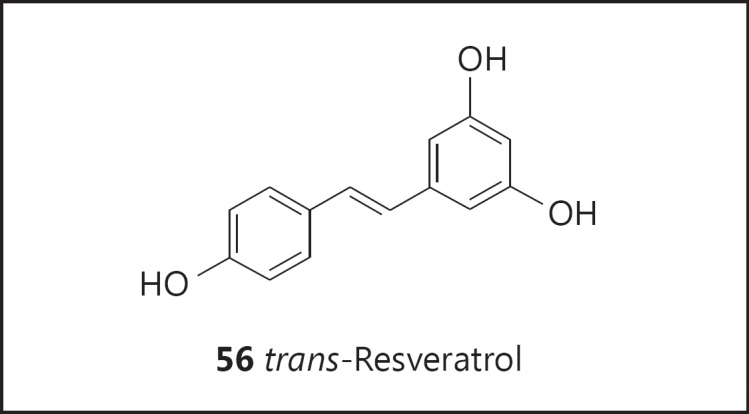
Structure of resveratrol (56).
Concluding Remarks and Future Prospects
It is clear that there continue to be multiple opportunities for the development of novel analogues, prodrugs, and methods of administration of agents from well-established drug classes, such as CPTs, nucleosides, taxanes, and vinca alkaloids, and that in many instances these can lead to the development of products having superior clinical efficacy and decreased toxicity. The conjugation of potent cytotoxic natural products to monoclonal antibodies specifically targeting epitopes on tumors of interest offers another promising approach to developing effective chemotherapeutic anticancer agents. In addition, new lead compounds are being discovered from natural sources, and these are providing new avenues for the development of novel and effective chemotherapeutic agents.
The scope and potential of natural product drug discovery is being greatly expanded by the investigation of plant endophytic fungi, which have proven to be a good source of bioactive metabolites, including antitumor agents [118,119]. In fact, it is significant that major anticancer agents, such as Taxol, CPT, podophyllotoxin, and the vinca alkaloids, have been isolated from endophytic fungi. Symbiotic microbes found growing in association with host macroorganisms have also been a rich source of marine-derived bioactive metabolites [120], as well as bioactive metabolites from insects [121,122]. Further exciting opportunities for the discovery of multiple novel microbial metabolites are emerging in the rapidly growing area of research into the power of microbial genomics through genome mining [123] and metagenomic exploration [124,125,126]. In addition, the exploration of extreme environments, including marine sediments [127], thermophiles from deep-sea vents and hot springs [128], psychrophiles from arctic and antarctic regions and alpine lakes [129], and toxic lakes and dump sites [130], as well as from the venoms of centipedes, cone snails, snakes, spiders, and other venomous creatures [131], is providing a range of novel therapeutic agents.
Relative to the development of cancer chemopreventive agents, the development of cancer chemotherapeutic agents is more straightforward. For example, much higher levels of toxicity can be tolerated under life-threatening conditions, the phases of clinical trials are clear, and therapeutic endpoints are well defined. There is often a ‘gold standard’ threshold that must be exceeded and extension of life for relatively short periods is considered a success. In the case of cancer chemoprevention, acceptable biomarkers are rare and massive trials are required to prove efficacy. Little or no toxic side effects are tolerated with the possible exception of individuals at high risk for the development or recurrence of cancer. As a result, it is difficult to make definitive health claims, and consequently investment is relatively low and progress is relatively slow. In one perspective, the entire effort has been declared a failure [101]. However, to be frank, thinking along these lines and avoiding difficult but important challenges is obtuse. Certainly, some lifestyle and environmental risk factors are well-known and appropriate modifications or precautions should be recommended or implemented. But in many cases, the etiology of malignancy is simply unknown. In such cases, the availability of an efficacious chemopreventive agent is imperative. Thus, failure to continue attempts providing such agents would be irresponsible.
It is clear that effective drug discovery and development will require close multidisciplinary collaboration embracing natural product lead discovery, as described above, coupled with optimization through the application of combinatorial and medicinal chemistry, total synthesis, and combinatorial biochemistry, all combined with good biology. Natural product research is a powerful approach for discovering biologically active compounds with unique structures and mechanisms of action. Given the unfathomable diversity of nature, it is reasonable to suggest that chemical leads can be generated that are capable of interacting with most or possibly all therapeutic targets. With the advent of high-throughput screening, a large number of potential starting materials can be readily evaluated, so informed selections can be made for unearthing prototype ligands worthy of further development as therapeutic agents.
Gordon M. Cragg retired from the Natural Products Branch, National Cancer Institute, Frederick, Md., USA.
References
- 1.National Cancer Institute Surveillance, Epidemiology, and End Results Program. SEER. http://seer.cancer.gov/statfacts/html/all.html (accessed April 30, 2015).
- 2.Hong WK, Sporn MB. Recent advances in chemoprevention of cancer. Science. 1997;278:1073–1077. doi: 10.1126/science.278.5340.1073. [DOI] [PubMed] [Google Scholar]
- 3.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cragg GM, Newman DJ. Nature: a vital source of leads for anticancer drug development. Phytochem Rev. 2009;8:313–331. [Google Scholar]
- 5.Cragg GM, Grothaus PG, Newman DJ. Impact of natural products on developing new anti-cancer agents. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 6.Grothaus PG, Cragg GM, Newman DJ. Plant natural products in anticancer drug discovery. Curr Org Chem. 2010;14:1781–1791. [Google Scholar]
- 7.Demain AL, Vaishnav P. Natural products for cancer chemotherapy. Microb Biotechnol. 2011;4:687–699. doi: 10.1111/j.1751-7915.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cragg GM, Kingston DGI, Newman DJ. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. [Google Scholar]
- 9.Basmadjian C, Zhao Q, Djehal A, et al. Cancer wars: natural products strike back. Front Chem. 2014;2:20. doi: 10.3389/fchem.2014.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roussi F, Guéritte F, Fahy J. The vinca alkaloids. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 177–198. [Google Scholar]
- 11.Lee KH, Xiao Z. Podophyllotoxins and analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 95–122. [Google Scholar]
- 12.Hartwell JL. Plants Used against Cancer. Lawrence: Quarterman; 1982. [Google Scholar]
- 13.Kingston DGI. Taxol and its analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 123–175. [Google Scholar]
- 14.Yared JA, Tkaczuk KH. Update on taxane development: new analogs and new formulations. Drug Des Devel Ther. 2012;6:371–384. doi: 10.2147/DDDT.S28997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muggia F, Kudlowitz D. Novel taxanes. Anticancer Drugs. 2014;25:593–598. doi: 10.1097/CAD.0000000000000053. [DOI] [PubMed] [Google Scholar]
- 16.Villanueva C, Bazan F, Kim S, et al. Cabazitaxel: a novel microtubule inhibitor. Drugs. 2011;71:1251–1258. doi: 10.2165/11591390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 17.Mita AC, Figlin R, Mita MM. Cabazitaxel: more than a new taxane for metastatic castrate-resistant prostate cancer? Clin Cancer Res. 2012;18:6574–6579. doi: 10.1158/1078-0432.CCR-12-1584. [DOI] [PubMed] [Google Scholar]
- 18.Beer M, Lenaz L, Amadori D. Phase II study of ortataxel in taxane-resistant breast cancer (abstract). J Clin Oncol. 2008;26((suppl)):1066. [Google Scholar]
- 19.Seidman AD, Schwartzberg LS, O'Shaughnessy J, et al. Activity of an oral taxane as first line treatment in metastatic breast cancer (abstract). J Clin Oncol. 2012;30((suppl)):1016. [Google Scholar]
- 20.Modiano MR, Plezia P, Basche M, et al. A phase I study of TPI 287, a novel taxane, administered every 21 days in patients (pts) with advanced cancer (abstract). J Clin Oncol. 2008;26((suppl)):13510. [Google Scholar]
- 21.Edelman MJ. Novel taxane formulations and microtubule-binding agents in non-small-cell lung cancer. Clin Lung Cancer. 2009;10:S30–S34. doi: 10.3816/CLC.2009.s.005. [DOI] [PubMed] [Google Scholar]
- 22.Galic VL, Wright JD, Lewin SN, et al. Paclitaxel poliglumex for ovarian cancer. Expert Opin Investig Drugs. 2011;20:813–821. doi: 10.1517/13543784.2011.576666. [DOI] [PubMed] [Google Scholar]
- 23.Jeyapalan S, Boxerman J, Donahue J, et al. Paclitaxel poliglumex, temozolomide, and radiation for newly diagnosed high-grade glioma: a Brown University Oncology Group study. Am J Clin Oncol. 2014;37:444–449. doi: 10.1097/COC.0b013e31827de92b. [DOI] [PubMed] [Google Scholar]
- 24.Koudelka S, Turánek J. Liposomal paclitaxel formulations. J Control Release. 2012;163:322–334. doi: 10.1016/j.jconrel.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Montero AJ, Adams B, Diaz-Montero CM, et al. Nab-paclitaxel in the treatment of metastatic breast cancer: a comprehensive review. Expert Rev Clin Pharmacol. 2011;4:329–334. doi: 10.1586/ecp.11.7. [DOI] [PubMed] [Google Scholar]
- 26.Gupta N, Hatoum H, Dy GK. First line treatment of advanced non-small-cell lung cancer - specific focus on albumin bound paclitaxel. Int J Nanomedicine. 2014;9:209–221. doi: 10.2147/IJN.S41770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thota R, Pauff JM, Berlin JD. Treatment of metastatic pancreatic adenocarcinoma: a review. Oncology (Williston Park) 2014;28:70–74. [PubMed] [Google Scholar]
- 29.Slingerland M, Guchelaar HJ, Rosing H, et al. Bioequivalence of liposome-entrapped paclitaxel easy-to-use (LEP-ETU) formulation and paclitaxel in polyethoxylated castor oil: a randomized, two-period crossover study in patients with advanced cancer. Clin Ther. 2013;35:1946–1954. doi: 10.1016/j.clinthera.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 30.Awada A, Bondarenko IN, Bonneterre J, et al. A randomized controlled phase II trial of a novel composition of paclitaxel embedded into neutral and cationic lipids targeting tumor endothelial cells in advanced triple-negative breast cancer (TNBC) Ann Oncol. 2014;25:824–831. doi: 10.1093/annonc/mdu025. [DOI] [PubMed] [Google Scholar]
- 31.Strieth S, Dunau C, Michaelis U, et al. Phase I/II clinical study on safety and antivascular effects of paclitaxel encapsulated in cationic liposomes for targeted therapy in advanced head and neck cancer. Head Neck. 2014;36:976–984. doi: 10.1002/hed.23397. [DOI] [PubMed] [Google Scholar]
- 32.Shea JE, Nam KH, Rapoport N, et al. Genexol inhibits primary tumour growth and metastases in gemcitabine-resistant pancreatic ductal adenocarcinoma. HPB (Oxford) 2011;13:153–157. doi: 10.1111/j.1477-2574.2010.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bulitta JB, Zhao P, Arnold RD, et al. Mechanistic population pharmacokinetics of total and unbound paclitaxel for a new nanodroplet formulation versus Taxol in cancer patients. Cancer Chemother Pharmacol. 2009;63:1049–1063. doi: 10.1007/s00280-008-0827-2. [DOI] [PubMed] [Google Scholar]
- 34.Bulitta JB, Zhao P, Arnold RD, et al. Multiple-pool cell lifespan models for neutropenia to assess the population pharmacodynamics of unbound paclitaxel from two formulations in cancer patients. Cancer Chemother Pharmacol. 2009;63:1035–1048. doi: 10.1007/s00280-008-0828-1. [DOI] [PubMed] [Google Scholar]
- 35.Rahier NJ, Thomas CJ, Hecht SM. Camptothecin and its analogs. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 5–25. [Google Scholar]
- 36.Pecorelli S, Ray-Coquard I, Tredan O, et al. Phase II of oral gimatecan in patients with recurrent epithelial ovarian, fallopian tube or peritoneal cancer, previously treated with platinum and taxanes. Ann Oncol. 2010;2:759–765. doi: 10.1093/annonc/mdp514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trocóniz IF, Cendrós JM, Soto E, et al. Population pharmacokinetic/pharmacodynamic modeling of drug-induced adverse effects of a novel homocamptothecin analog, elomotecan (BN80927), in a phase I dose finding study in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;70:239–250. doi: 10.1007/s00280-012-1906-y. [DOI] [PubMed] [Google Scholar]
- 38.Svenson S, Wolfgang M, Hwang J, et al. Preclinical to clinical development of the novel camptothecin nanopharmaceutical CRLX101. J Control Release. 2011;153:49–55. doi: 10.1016/j.jconrel.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Weiss GJ, Chao J, Neidhart JD, et al. First-in-human phase 1/2a trial of CRLX101, a cyclodextrin-containing polymer-camptothecin nanopharmaceutical in patients with advanced solid tumor malignancies. Invest New Drugs. 2013;31:986–1000. doi: 10.1007/s10637-012-9921-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walsh MD, Hanna SK, Sen J, et al. Pharmacokinetics and antitumor efficacy of XMT-1001, a novel, polymeric topoisomerase I inhibitor, in mice bearing HT-29 human colon carcinoma xenografts. Clin Cancer Res. 2012;18:2591–2602. doi: 10.1158/1078-0432.CCR-11-1554. [DOI] [PubMed] [Google Scholar]
- 41.Bala V, Rao S, Boyd BJ, et al. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172:48–56. doi: 10.1016/j.jconrel.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 42.Pinney KG, Pettit GR, Trawick ML, et al. The discovery and development of the combretastatins. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 27–64. [Google Scholar]
- 43.Mooney CJ, Nagaiah G, Fu P, et al. A phase II trial of fosbretabulin in advanced anaplastic thyroid carcinoma and correlation of baseline serum-soluble intracellular adhesion molecule-1 with outcome. Thyroid. 2009;19:233–240. doi: 10.1089/thy.2008.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sosa JA, Elisei R, Jarzab B, et al. Randomized safety and efficacy study of fosbretabulin with paclitaxel/carboplatin against anaplastic thyroid carcinoma. Thyroid. 2014;24:232–240. doi: 10.1089/thy.2013.0078. [DOI] [PubMed] [Google Scholar]
- 45.Zweifel M, Jayson GC, Reed NS, et al. Phase II trial of combretastatin A4 phosphate, carboplatin, and paclitaxel in patients with platinum-resistant ovarian cancer. Ann Oncol. 2011;22:2036–2041. doi: 10.1093/annonc/mdq708. [DOI] [PubMed] [Google Scholar]
- 46.Marelli M, Conforti F, Statti GA, et al. Biological potential and structure-activity relationships of most recently developed vascular disrupting agents: an overview of new derivatives of natural combretastatin A-4. Curr Med Chem. 2011;18:3035–3081. doi: 10.2174/092986711796391642. [DOI] [PubMed] [Google Scholar]
- 47.Turner D, Gonzalez A, Pettiford L, et al. A phase I study of the vascular disrupting combretastatin, OXi4503, in patients with relapsed and refractory acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) (abstract) Blood. 2013;122:1463. [Google Scholar]
- 48.Itokawa H, Hitotsuyanagi Y, Lee KH. Homoharringtonine and related compounds. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 65–94. [Google Scholar]
- 49.Chen Y, Li S. Omacetaxine mepesuccinate in the treatment of intractable chronic myeloid leukemia. Onco Targets Ther. 2014;7:177–186. doi: 10.2147/OTT.S41786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Deng Z, Zhu J, et al. Homoharringtonine combined with cytarabine to treat chronic myelogenous leukemia in myeloid blast crisis and its impact on bone marrow CD34+CD7+ cells. Acta Haematol. 2014;132:172–176. doi: 10.1159/000356742. [DOI] [PubMed] [Google Scholar]
- 51.Siller G, Gebauer K, Welburn P, et al. PEP005 (ingenol mebutate) gel, a novel agent for the treatment of actinic keratosis: results of a randomized, double-blind, vehicle-controlled, multicentre, phase IIa study. Australas J Dermatol. 2009;50:16–22. doi: 10.1111/j.1440-0960.2008.00497.x. [DOI] [PubMed] [Google Scholar]
- 52.Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366:1010–1019. doi: 10.1056/NEJMoa1111170. [DOI] [PubMed] [Google Scholar]
- 53.Gras J. Ingenol mebutate: a new option for actinic keratosis treatment. Drugs Today (Barc) 2013;49:15–22. doi: 10.1358/dot.2013.49.1.1910723. [DOI] [PubMed] [Google Scholar]
- 54.Ramirez-Llodra E, Brandt A, Danovaro R, et al. Deep, diverse and definitely different: unique attributes of the world's largest ecosystem. Biogeosciences. 2010;7:2851–2899. [Google Scholar]
- 55.Newman DJ, Cragg GM. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar Drugs. 2014;12:255–278. doi: 10.3390/md12010255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bergmann W, Burke DC. Marine products. XXXIX. The nucleosides of sponges. III. Spongothymidine and spongouridine. J Org Chem. 1955;20:1501–1507. [Google Scholar]
- 57.McGregor BA, Brown AW, Osswald MB, et al. The use of higher dose clofarabine in adults with relapsed acute lymphoblastic leukemia. Am J Hematol. 2009;84:228–230. doi: 10.1002/ajh.21365. [DOI] [PubMed] [Google Scholar]
- 58.Tran H, Yang D. Clofarabine in the treatment of newly diagnosed acute myeloid leukemia in older adults. Ann Pharmacother. 2012;46:89–96. doi: 10.1345/aph.1Q295. [DOI] [PubMed] [Google Scholar]
- 59.Ghanem H, Kantarjian H, Ohanian M, et al. The role of clofarabine in acute myeloid leukemia. Leuk Lymphoma. 2013;54:688–698. doi: 10.3109/10428194.2012.726722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kantarjian H, Faderl S, Garcia-Manero G, et al. Oral sapacitabine for the treatment of acute myeloid leukaemia in elderly patients: a randomised phase 2 study. Lancet Oncol. 2012;13:1096–1104. doi: 10.1016/S1470-2045(12)70436-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lim MY, Jamieson K. Profile of sapacitabine: potential for the treatment of newly diagnosed acute myeloid leukemia in elderly patients. Clin Interv Aging. 2014;9:753–762. doi: 10.2147/CIA.S39558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cuevas C, Francesch A, Galmarini CM, et al. Ecteinascidin-743 (Yondelis®), Aplidin®, and Irvalec®. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 291–316. [Google Scholar]
- 63.Yu MJ, Kishi Y, Littlefield BA. Discovery of E7389, a fully synthetic macrocyclic ketone analog of halichondrin B. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 317–345. [Google Scholar]
- 64.Yu MJ, Zheng W, Seletsky BM. From micrograms to grams: scale-up synthesis of eribulin mesylate. Nat Prod Rep. 2013;30:1158–1164. doi: 10.1039/c3np70051h. [DOI] [PubMed] [Google Scholar]
- 65.Austad BC, Benayoud F, Calkins TL, et al. Process development of Halaven®: synthesis of the C14-C35 fragment via iterative Nozaki-Hiyama-Kishi reaction-Williamson ether cyclization. Synlett. 2013;24:327–332. [Google Scholar]
- 66.Austad BC, Calkins TL, Chase CE, et al. Commercial manufacture of Halaven®: chemoselective transformations en route to structurally complex macrocyclic ketones. Synlett. 2013;24:333–337. [Google Scholar]
- 67.Chase CE, Fang FG, Lewis BM, et al. Process development of Halaven®: synthesis of the C1-C13 fragment from d-(-)-gulono-1,4-lactone. Synlett. 2013;24:323–326. [Google Scholar]
- 68.Cragg GM, Grothaus PG, Newman DJ. New horizons for old drugs and drug leads. J Nat Prod. 2014;77:703–723. doi: 10.1021/np5000796. [DOI] [PubMed] [Google Scholar]
- 69.Kim KB, Crews CM. From epoxomicin to carfilzomib: chemistry, biology, and medical outcomes. Nat Prod Rep. 2013;30:600–604. doi: 10.1039/c3np20126k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Redic K. Carfilzomib: a novel agent for multiple myeloma. J Pharm Pharmacol. 2013;65:1095–1106. doi: 10.1111/jphp.12072. [DOI] [PubMed] [Google Scholar]
- 71.Steele JM. Carfilzomib: a new proteasome inhibitor for relapsed or refractory multiple myeloma. J Oncol Pharm Pract. 2013;19:348–354. doi: 10.1177/1078155212470388. [DOI] [PubMed] [Google Scholar]
- 72.Thompson JL. Carfilzomib: a second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Ann Pharmacother. 2013;47:56–62. doi: 10.1345/aph.1R561. [DOI] [PubMed] [Google Scholar]
- 73.Prudhomme M. Staurosporines and structurally related indocarbazoles as antitumor agents. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 647–670. [Google Scholar]
- 74.Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28:4339–4345. doi: 10.1200/JCO.2010.28.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13:235–244. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 76.Teicher BA. Antibody-drug conjugate targets. Curr Cancer Drug Targets. 2009;9:982–1004. doi: 10.2174/156800909790192365. [DOI] [PubMed] [Google Scholar]
- 77.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 78.Caravella J, Lugovskoy A. Design of next-generation protein therapeutics. Curr Opin Chem Biol. 2010;14:520–528. doi: 10.1016/j.cbpa.2010.06.175. [DOI] [PubMed] [Google Scholar]
- 79.Hughes B. Antibody-drug conjugates for cancer: poised to deliver? Nat Rev Drug Discov. 2010;9:665–667. doi: 10.1038/nrd3270. [DOI] [PubMed] [Google Scholar]
- 80.Bouchard H, Viskov C, Garcia-Echeverria C. Antibody-drug conjugates - a new wave of cancer drugs. Bioorg Med Chem Lett. 2014;24:5357–5363. doi: 10.1016/j.bmcl.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 81.Koehn FE. Natural product cytotoxins as payloads for antibody drug conjugates. In: Koehn FE, editor. Natural Products and Cancer Drug Discovery. New York: Humana/Springer; 2013. pp. 97–119. [Google Scholar]
- 82.Yu JW, Floss HG, Cragg GM, et al. Ansamitocin (maytansinoids) In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 407–427. [Google Scholar]
- 83.Wings S, Muller H, Berg G, et al. A study of the bacterial community in the root system of the maytansine containing plant Putterlickia verrucosa. Phytochemistry. 2013;91:158–164. doi: 10.1016/j.phytochem.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 84.Lambert JM. Antibody-maytansinoid conjugates: a new strategy for the treatment of cancer. Drugs Fut. 2010;35:471–480. [Google Scholar]
- 85.Kümler I, Ehlers Mortensen C, Nielsen DL. Trastuzumab emtansine, tumor-activated prodrug (TAP) immunoconjungate, oncolytic. Drugs Fut. 2011;36:825–834. [Google Scholar]
- 86.LoRusso PM, Weiss D, Guardino E, et al. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17:6437–6447. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 87.Barginear MF, John V, Budman DR. Trastuzumab-DM1: a clinical update of the novel antibody-drug conjugate for HER2-overexpressing breast cancer. Mol Med. 2013;18:1473–1479. doi: 10.2119/molmed.2012.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Teicher BA, Chari RV. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res. 2011;17:6389–6397. doi: 10.1158/1078-0432.CCR-11-1417. [DOI] [PubMed] [Google Scholar]
- 89.Flahive E, Srirangam J. The dolastatins: novel antitumor agents from Dolabella auricularia. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. ed 2. Boca Raton: CRC/Taylor & Francis; 2012. pp. 263–289. [Google Scholar]
- 90.Maude SL, Frey N, Pamela A, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Steele VE, Lubet RA, Moon RC. Preclinical animal models for the development of cancer chemopreventive drugs. In: Kelloff GJ, Hawk ET, Sigman CC, editors. Cancer Chemoprevention; vol 2: Strategies for Cancer Chemoprevention. Totowa: Humana; 2005. pp. 39–46. [Google Scholar]
- 92.Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Natl Cancer Inst. 2005;97:1652–1662. doi: 10.1093/jnci/dji372. [DOI] [PubMed] [Google Scholar]
- 93.Nichols HB, DeRoo LA, Scharf DR, et al. Risk-benefit profiles of women using tamoxifen for chemoprevention. J Natl Cancer Inst. 2015;107:354. doi: 10.1093/jnci/dju354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thompson IM, Jr, Goodman PJ, Tangen CM, et al. Long-term survival of participants in the prostate cancer prevention trial. N Engl J Med. 2013;369:603–610. doi: 10.1056/NEJMoa1215932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arber N, Spicak J, Rácz I, et al. Five-year analysis of the Prevention of Colorectal Sporadic Adenomatous Polyps trial. Am J Gastroenterol. 2011;106:1135–1146. doi: 10.1038/ajg.2011.116. [DOI] [PubMed] [Google Scholar]
- 96.Rothwell PM, Wilson M, Price JF, et al. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. doi: 10.1016/S0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- 97.Block G, Patterson B, Subar A. Fruit, vegetables, and cancer prevention: a review of the epidemiological evidence. Nutr Cancer. 1992;18:1–29. doi: 10.1080/01635589209514201. [DOI] [PubMed] [Google Scholar]
- 98.Pauwels EKJ. The protective effect of the Mediterranean diet: focus on cancer and cardiovascular risk. Med Princ Pract. 2011;20:103–111. doi: 10.1159/000321197. [DOI] [PubMed] [Google Scholar]
- 99.Key TJ. Fruit and vegetables and cancer risk. Br J Cancer. 2011;104:6–11. doi: 10.1038/sj.bjc.6606032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Albanes D, Heinonen OP, Taylor PR, et al. α-Tocopherol and β-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88:1560–1570. doi: 10.1093/jnci/88.21.1560. [DOI] [PubMed] [Google Scholar]
- 101.Potter JD. The failure of cancer chemoprevention. Carcinogenesis. 2014;35:974–982. doi: 10.1093/carcin/bgu063. [DOI] [PubMed] [Google Scholar]
- 102.Gerhauser C. Cancer chemoprevention and nutri-epigenetics: state of the art and future challenges. In: Pezzuto JM, Suh N, editors. Natural Products in Cancer Prevention and Therapy. Top Curr Chem. Vol. 329. Heidelberg: Springer; 2013. pp. 73–132. [DOI] [PubMed] [Google Scholar]
- 103.Kensler TW, Egner PA, Agyeman AS, et al. Keap1-Nrf2 signaling: a target for cancer prevention. In: Pezzuto JM, Suh N, editors. Natural Products in Cancer Prevention and Therapy. Top Curr Chem. Vol. 329. Heidelberg: Springer; 2013. pp. 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang DD. The Nrf2-Keap1-ARE signaling pathway: the regulation and dual function of Nrf2 in cancer. Antioxid Redox Signal. 2010;13:1623–1626. doi: 10.1089/ars.2010.3301. [DOI] [PubMed] [Google Scholar]
- 105.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88(pt B):93–100. doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 107.Pezzuto JM, Kosmeder JW, II, Park EJ, et al. Characterization of natural product chemopreventive agents. In: Kelloff GJ, Hawk ET, Sigman CC, editors. Cancer Chemoprevention; vol 2: Strategies for Cancer Chemoprevention. Totowa: Humana; 2005. pp. 3–37. [Google Scholar]
- 108.Cuendet M, Nievergelt A, Pezzuto JM. Molecular targets of botanicals used for chemoprevention. In: Abrams DI, Weil AT, editors. Integrative Oncology. ed 2. New York: Oxford University Press; 2014. pp. 52–84. [Google Scholar]
- 109.Park E-J, Murphy BT, Pezzuto JM. Marine organisms in cancer chemoprevention. In: Baker BJ, editor. Marine Biomedicine: From Beach to Bedside. Boca Rotan: CRC; 2016. pp. 279–339. [Google Scholar]
- 110.Wiseman CL, Yap HY, Bedikian AY, et al. Phase II trial of bruceantin in metastatic breast carcinoma. Am J Clin Oncol. 1982;5:389–391. doi: 10.1097/00000421-198208000-00007. [DOI] [PubMed] [Google Scholar]
- 111.Arsenau JC, Wolter JM, Kuperminc M, et al. A phase II study of bruceantin (NSC 165563) in advanced malignant melanoma. Investig New Drugs. 1983;1:239–242. doi: 10.1007/BF00208896. [DOI] [PubMed] [Google Scholar]
- 112.Mata-Greenwood E, Cuendet M, Sher D, et al. Brusatol-mediated induction of leukemic cell differentiation and G1 arrest is associated with down-regulation of c-myc. Leukemia. 2002;16:2275–2284. doi: 10.1038/sj.leu.2402696. [DOI] [PubMed] [Google Scholar]
- 113.Cuendet M, Pezzuto JM. Antitumor activity of bruceantin: an old drug with new promise. J Nat Prod. 2004;67:269–272. doi: 10.1021/np030304+. [DOI] [PubMed] [Google Scholar]
- 114.Cuendet M, Christov K, Lantvit DD, et al. Multiple myeloma regression mediated by bruceantin. Clin Cancer Res. 2004;10:1170–1179. doi: 10.1158/1078-0432.ccr-0362-3. [DOI] [PubMed] [Google Scholar]
- 115.Ren D, Nicole F, Villeneuve NF, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci USA. 2011;108:1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jang M, Cai L, Udeani GO, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 117.Park E-J, Pezzuto JM. The pharmacology of resveratrol in animals and humans. Biochim Biophys Acta. 2015;1852:1071–1113. doi: 10.1016/j.bbadis.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 118.Kharwa RN, Mishra A, Gond SK, et al. Anticancer compounds derived from fungal endophytes: their importance and future challenges. Nat Prod Rep. 2011;28:1208–1228. doi: 10.1039/c1np00008j. [DOI] [PubMed] [Google Scholar]
- 119.Chandra S. Endophytic fungi: novel sources of anticancer lead molecules. Appl Microbiol Biotechnol. 2012;95:47–59. doi: 10.1007/s00253-012-4128-7. [DOI] [PubMed] [Google Scholar]
- 120.Abdelmohsen UR, Bayer K, Hentschel U. Diversity, abundance and natural products of marine sponge-associated actinomycetes. Nat Prod Rep. 2014;31:381–399. doi: 10.1039/c3np70111e. [DOI] [PubMed] [Google Scholar]
- 121.Crawford JM, Clardy J. Bacterial symbionts and natural products. Chem Commun (Camb) 2011;47:7559–7566. doi: 10.1039/c1cc11574j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ramadhar TR, Beemelmanns C, Currie CR, et al. Bacterial symbionts in agricultural systems provide a strategic source for antibiotic discovery. J Antibiot (Tokyo) 2014;67:53–58. doi: 10.1038/ja.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bachmann BO, Van Lanen SG, Baltz RH. Microbial genome mining for accelerated natural products discovery: is a renaissance in the making? J Ind Microbiol Biotechnol. 2014;41:175–184. doi: 10.1007/s10295-013-1389-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brady SF, Simmons L, Kim JH, et al. Metagenomic approaches to natural products from free-living and symbiotic organisms. Nat Prod Rep. 2009;26:1488–1503. doi: 10.1039/b817078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Leis B, Angelov A, Liebl W. Screening and expression of genes from metagenomes. Adv Appl Microbiol. 2013;83:1–68. doi: 10.1016/B978-0-12-407678-5.00001-5. [DOI] [PubMed] [Google Scholar]
- 126.Charlop-Powers Z, Milshteyn A, Brady SF. Metagenomic small molecule discovery methods. Curr Opin Microbiol. 2014;19:70–75. doi: 10.1016/j.mib.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Potts BC, Lam KS. Generating a generation of proteasome inhibitors: from microbial fermentation to total synthesis of salinosporamide A (marizomib) and other salinosporamides. Mar Drugs. 2010;8:835–880. doi: 10.3390/md8040835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Andrianasolo EH, Haramaty L, McPhail KL, et al. Bathymodiolamides A and B, ceramide derivatives from a deep-sea hydrothermal vent invertebrate mussel, Bathymodiolus thermophiles. J Nat Prod. 2011;74:842–846. doi: 10.1021/np100601w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Margesin R, Feller G. Biotechnological applications of psychrophiles. Environ Technol. 2010;31:835–844. doi: 10.1080/09593331003663328. [DOI] [PubMed] [Google Scholar]
- 130.Stierle AA, Stierle DB, Girtsman T. Caspase-1 inhibitors from an extremophilic fungus that target specific leukemia cell lines. J Nat Prod. 2012;75:344–350. doi: 10.1021/np200414c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.King GF. Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin Biol Ther. 2011;11:1469–1484. doi: 10.1517/14712598.2011.621940. [DOI] [PubMed] [Google Scholar]