

Abstract
Outward current conducted by human ether-à-go-go–related gene type 1 (hERG1) channels is a major determinant of action potential repolarization in the human ventricle. Ginsenoside 20(S)-Rg3 [Rg3; (2S,3R,4S,5S,6R)-2-[(2R,3R,4S,5S,6R)-4,5-dihydroxy-2-[[(3S,5R,8R,9R,10R,12R,13R,14R,17S)-12-hydroxy-17-[(2S)-2-hydroxy-6-methylhept-5-en-2-yl]-4,4,8,10,14-pentamethyl-2,3,5,6,7,9,11,12,13,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-yl]oxy]-6-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol], an alkaloid isolated from the root of Panax ginseng, slows the rate of hERG1 deactivation, induces channels to open at more negative potentials than normal, and increases current magnitude. The onset of Rg3 action is extremely fast, suggesting that it binds to an extracellular accessible site on the channel to alter its gating. Here we used a scanning mutagenesis approach to identify residues in the extracellular loops and transmembrane segments of hERG1 that might interact with Rg3. Single or multiple residues of hERG1 were mutated to Ala or Cys and the resulting mutant channels were heterologously expressed in Xenopus oocytes. The effects of Rg3 on the voltage dependence of activation and the deactivation rate of mutant channel currents were characterized using the two-microelectrode voltage clamp technique. Mutation to Ala of specific residues in the S1 (Tyr420), S2 (Leu452, Phe463), and S4 (Ile521, Lys525) segments partially inhibited the effects of Rg3 on hERG1. The double mutant Y420A/L452A nearly eliminated the effects of Rg3 on voltage-dependent channel gating but did not prevent the increase in current magnitude. These findings together with molecular modeling suggest that Rg3 alters the gating of hERG1 channels by interacting with and stabilizing the voltage sensor domain in an activated state.
Introduction
Repolarization of the cardiac action potential is mediated in part by voltage-dependent, K+-selective human ether-à-go-go–related gene type 1 (hERG1) channels. Pharmacological block of these channels can prolong the action potential duration in ventricular myocytes and increase the risk of cardiac arrhythmia, in essence mimicking the phenotype of congenital long QT syndrome (LQTS). This serious cardiac side effect was found to be caused by so many common medications that preclinical screening of compounds for hERG1 channel activity was adopted as a routine assay during early stages of the drug discovery process. Extensive screening of compound libraries serendipitously resulted in the discovery of several compounds that activate rather than inhibit hERG1 channels (Kang et al., 2005; Zhou et al., 2005; Gerlach et al., 2010). The hERG1 activators discovered to date alter channel gating by one or more molecular mechanisms, including an increase in single-channel open probability, inhibition of inactivation, slowing of deactivation, and a shift in the voltage dependence of activation to more negative potentials (Sanguinetti, 2014).
In an attempt to relate mechanisms of action of hERG1 activators to channel structure, we have used a site-directed mutagenesis approach to identify residues that contribute to the putative binding site of these compounds. These studies suggest that RPR-260243 [RPR; (3R,4R)-4-[3-(6-methoxy-quinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5 trifluorophenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid] (Wu et al., 2015), PD-118057 [2-({4-[2-(3,4-dichlorophenyl)ethyl]phenyl}amino)benzoic acid], and ICA-105574 [3-nitro-N-(4-phenoxyphenyl)benzamide] (Wu et al., 2014) bind with 4-fold symmetry to specific residues in the pore domain (S5–S6 region) to alter gating of the channel.
Rg3 [(2S,3R,4S,5S,6R)-2-[(2R,3R,4S,5S,6R)-4,5-dihydroxy-2-[[(3S,5R,8R,9R,10R,12R,13R,14R,17S)-12-hydroxy-17-[(2S)-2-hydroxy-6-methylhept-5-en-2-yl]-4,4,8,10,14-pentamethyl-2,3,5,6,7,9,11,12,13,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-yl]oxy]-6-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol] is a large (785 Da) steroid glycoside isolated from the root of Panax ginseng that alters hERG1 gating by slowing the rate of channel closure (deactivation) and causing a leftward shift in the voltage dependence of channel activation (Choi et al., 2011a; Wu et al., 2016). The onset of these effects is extremely fast, suggesting that this large compound exerts its actions by binding to an extracellular accessible site. A previously published study reported that S631C hERG1 channels were resistant to the effects of Rg3, leading to the suggestion that Ser631 is a major structural determinant of Rg3 interaction with the channel (Choi et al., 2011a). Here, we re-examine the importance of Ser631 in mediating the effects of Rg3 on channel gating and search for other potential interacting residues using a thorough mutagenesis scan of residues in the hERG1 subunit that could be accessible by a large molecule from the extracellular space.
Materials and Methods
Molecular Biology.
HERG1 isoform 1a (KCNH2, NCBI reference sequence: NM_000238) was cloned into an pSP64 oocyte expression vector. Single or multiple point mutations in KCNH2 were introduced into wild-type (WT) channels using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). To prepare cRNA for use in oocyte expression studies, pSP64 plasmids were linearized with EcoRI prior to in vitro transcription using the mMessage mMachine SP6 transcription kit (Thermo Fisher Scientific, Waltham, MA).
Isolation and Voltage Clamp of Oocytes.
Xenopus laevis oocytes were harvested as previously described (Garg et al., 2012) using methods approved by the University of Utah Institutional Animal Care and Use Committee. Isolated oocytes were incubated at 17°C in Barth’s solution and studied 2–7 days after injection with 1–40 ng KCNH2 cRNA encoding WT or mutant hERG1 channels. Whole cell currents were measured by using standard two-electrode voltage-clamp techniques (Stühmer, 1992). Agarose-cushion electrodes (Schreibmayer et al., 1994) with a tip resistance of 0.5–1.5 MΩ when backfilled with 3 M KCl were used to minimize leakage of the pipette solution into oocytes. Data acquisition was performed with a Dell personal computer, a Digidata 1322A data acquisition system, and a GeneClamp 500 amplifier controlled by pCLAMP 8.2 software (all from Molecular Devices, LLC, Sunnyvale, CA).
For most experiments, hERG1 channel currents were activated with 4-second voltage steps to a test potential (Vt) that was varied from −80 to +40 mV in 10-mV increments from a holding potential of −70, −80, or −90 mV. To compare current-voltage relationships before and after treatment of an oocyte with Rg3, the time-dependent outward current (Ipeak) was measured at the end of each test pulse, normalized to the largest value determined under control conditions, and plotted as a function of Vt. Tail currents (Itail) were measured immediately after a test pulse by stepping the membrane voltage to a return potential (Vret) of −70 mV. The pulse to −70 was usually 4 seconds in duration but was extended to 10 seconds for slowly deactivating mutant channels. A brief prepulse to −70 was applied before each test pulse and the magnitude of leak current at this potential was measured and subtracted from Itail to quantify the peak tail current (Itail-peak).
To estimate the fold change in Itail-peak induced by Rg3, a Vt of sufficient amplitude and duration (e.g., 1 or 4 seconds at +40 mV) to fully activate channels was applied, followed by a return pulse to −70 mV. The leak-subtracted Itail at −70 mV was fitted with a single or double exponential function and extrapolated to the moment of membrane repolarization to estimate Itail-peak. This method was especially important for fast deactivating mutant channels where Itail-peak could be severely underestimated due to insufficient time for channels to recover from inactivation. To measure the kinetics of Itail over a wide voltage range, Y420A/L452A hERG1 channels were activated with a 1-second pulse to 40 mV followed by stepping to a Vret that was varied in 10-mV increments from 20 to −130 mV.
Solutions.
Barth’s solution contained 88 mM NaCl, 1 mM KCl, 0.41 mMCaCl2, 0.33 mM Ca(NO3)2, 1 mM MgSO4, 2.4 mM NaHCO3, 10 mM HEPES, and 1 mM pyruvate plus gentamycin (50 mg/l), amikacin (100 mg/l), and ciprofloxacin (25 mg/l), pH adjusted to 7.4 with NaOH. For voltage-clamp experiments, oocytes were bathed in an extracellular saline solution that contained 96 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES, pH adjusted to 7.6 with NaOH. Ginsenoside 20(S)-Rg3 (Sigma-Aldrich, St. Louis, MO) was dissolved in dimethylsulfoxide to make a 1- or 10-mM stock solution that was stored at –20°C. Before each experiment, a stock solution was diluted with the extracellular saline solution to produce the desired final concentration of Rg3.
Data Analysis.
PCLAMP 8.2 software (Molecular Devices) was used to analyze the magnitude and kinetics of digitized currents. Origin 8.6 software (OriginLab Corp., Northampton, MA) was used to prepare graphs and figures. Deactivation of hERG1 channels was quantified by fitting the time-dependent decay of Itail to a mono- or biexponential function:
where τf and τs are the fast and slow time constants and As/(As + Af) is the relative amplitude of the slow component of deactivation. Deactivation of WT hERG1 channel Itail is biexponential, but becomes essentially monoexponential when slowed by Rg3 (Wu et al., 2016). To simplify the comparison of deactivation rates before and after treatment with Rg3, Itail at −70 mV was fitted with a single exponential function, and the fold change in the deactivation time constant (τdeact) was calculated for each mutant channel type.
The relative conductance–voltage (g/gmax–Vt) relationship for hERG1 was determined by plotting normalized peak tail currents (Itail-peak) versus Vt for each cell. Origin 8.6 software was used for fitting the data with a Boltzmann function:
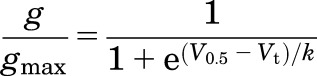 |
where V0.5 is the voltage required for half maximal Itail-peak activation and k is the slope factor for the relationship. V0.5 and k values were determined for each individual cell and the averaged data for all of the channels characterized in this study are presented in Supplemental Table 1. The fold change in the maximum values of Itail-peak induced by Rg3 treatment of WT and mutant channels is presented in Supplemental Table 2. Data are expressed as means ± S.D. (n is the number of oocytes).
Molecular Modeling.
A homology model of the voltage-sensing domain (VSD) in the activated state was built using the recently solved cryo-electron microscopy (EM) structure of the rat ether-a-go-go gene 1 (rEAG1) channel (Protein Data Bank identifier 5K7L; Whicher and MacKinnon, 2016) as a template and the Modeller9v14 program (http://www.mybiosoftware.com/) (Martí-Renom et al., 2000). Since this model is not fully consistent with data on charge pairings stabilizing the open state, an additional model (denoted “activated1”), with the alignment shifted +3 was built, consistent with experimental data for hERG1 (Zhang et al., 2005).
Coordinates of Rg3 were optimized with the Hartree–Fock 6-31G* basis set implemented in Gaussian 09 software (Gaussian, Inc., Wallingford, CT) (Frisch et al., 2009). Docking was performed using the Gold 4.0.1 program (Cambridge DataCentre, Cambridge, UK) (Jones et al., 1995). Coordinates of the geometric center calculated among residues Y420, L452, F463, I521, and K525 were taken as the binding site origin. The binding site radius was set to 20 Å. Then 150,000 operations of the GOLD genetic algorithm were used to dock the compound into the two different VSD models. The best snapshots (highest binding score) were subsequently relaxed by molecular dynamics (MD) simulations.
MD simulations were performed in Gromacs5.1.4 (http://www.gromacs.org/) and the amber99sb force field (Hornak et al., 2006). VSD domains were embedded in an equilibrated simulation box consisting of 118 1-palmitoyl-2-oleoylphosphatidylcholine lipids and the extended simple point charge model water model was used (Berendsen et al., 1987). Electrostatic interactions were calculated explicitly at a distance <1 nm and long-range electrostatic interactions were calculated at every step by particle-mesh Ewald summation (Darden et al., 1993). Lennard–Jones interactions were calculated with a cutoff of 1 nm. All bonds were constrained by using the LINCS (linear constraint solver) algorithm (Hess et al., 1997), allowing for an integration time step of 2 femtoseconds. The Nose–Hoover thermostat (Nose, 1984) was used for temperature coupling (τ = 0.1 picoseconds), and the Parrinello–Rahman barostat algorithm (Parrinello and Rahman, 1981) was used for pressure coupling. One-thousand conjugate gradient energy-minimization steps were performed, followed by 5 nanoseconds of equilibration, in which the protein atoms were restrained with a force constant of 1000 kJ × mol−1nm−2 to their initial position, and ions, lipids, and solvent were allowed to move freely. Each system was subjected to 2 × 50 nanoseconds of unrestrained MD, during which coordinates were saved every 20 picoseconds for analysis.
Results
The Onset of Rg3 is Rapid.
As reported previously (Choi et al., 2011a; Wu et al., 2016), the most obvious effect of Rg3 on hERG1 channels is a slowing in the rate of Itail deactivation and an altered voltage dependence of channel activation. Slowed deactivation is illustrated in Fig. 1A, where WT hERG1 channels were activated by 4-second pulses to a range of test voltages and Itail was measured at a fixed Vret of −70 mV. Rg3 increased the magnitude of Ipeak at Vt <0 mV, consistent with a negative shift in the voltage dependence of channel activation. This effect is confirmed in Fig. 1C, where Itail-peak is plotted as a function of Vt. The V0.5 for activation determined by fitting this relationship with a Boltzmann function was shifted from −31.5 mV under control conditions to −42.6 mV in the presence of 3 µM Rg3, a maximally effective concentration (Choi et al., 2011a; Wu et al., 2016). In addition, Rg3 increased the maximum amplitude of Itail-peak by 10% ± 4.5% (n = 5), an effect likely due to an increase in channel open probability because the alkaloid has no effect on the voltage dependence of inactivation (Wu et al., 2016). The kinetics of Rg3-induced changes in channel gating were probed by applying repetitive 1-second voltage steps, once every 5 seconds, to a Vt of 0 mV from a Vh of −90 mV before and after rapidly switching the perfusate from the control solution to a solution containing 3 µM Rg3 (Fig. 1D). After the addition of Rg3, current was increased midway through the first test pulse and Itail measured at −70 mV was dramatically slowed. Itail slowed further during the second pulse but was not further changed by continued pulsing. The magnitude of Itail was measured at 1 second after stepping to −70 mV to estimate changes in deactivation kinetics in this and six other oocytes. The normalized Itail at 1 second was plotted as a function of time after application of Rg3 and fitted with a single exponential function (Fig. 1E). The calculated τ of 1.9 seconds is only an approximation, given the solution flow rate of 2 ml/min and a chamber volume of 0.2 ml. We previously reported that the onset and recovery from modification of human EAG-like K channel type 1 (hELK1) channel gating by Rg3 (0.3 µM) was also very rapid, with a τ of 6.1 and 19.3 seconds, respectively (Wu et al., 2016). By comparison, hERG1 blockers bind to the central cavity and therefore must cross the cell membrane to gain access to the central pore. The rates of block onset by compounds that must cross the oocyte membrane to reach their putative receptor site (e.g., dofetilide) are much slower and require approximately 10 minutes to reach equilibrium (Ficker et al., 1998). Thus, the effects of Rg3 on hERG1 (and hELK1) channel gating are extremely rapid in onset, indicating that the compound must interact with an extracellular accessible site on these channels.
Fig. 1.

Ginsenoside Rg3 alters activation gating of hERG1 channels. (A) Voltage-clamp pulse protocol (upper left) and whole cell currents (bottom) recorded from a single oocyte expressing WT hERG1 before (control) and after treatment with 3 µM Rg3. Small arrows indicate zero current level. (B) Rg3 shifts the Ipeak–Vt relationship for hERG1 to more negative potentials. (C) Rg3 shifts the voltage dependence of hERG1 activation to more negative potentials. Peak Itail values at −70 mV from currents shown in (A) were plotted as a function of Vt and fitted with a Boltzmann function (smooth curves). For the control, V0.5 = −31.5 mV and k = 8.1; for 3 µM Rg3, V0.5 = −42.6 mV and k = 9.9. (D) Currents recorded in response to repetitive pulses, applied once every 5 seconds to the indicated voltage step shown in the upper image, before (0) and during the first, second, and third steps (1, 2, and 3) after the start of bath perfusion of the oocyte chamber with a solution containing 3 µM Rg3. (E) Onset of Rg3 action is rapid. Itail magnitude (measured at 1 second after return of voltage to −70 mV) was normalized to the peak value for each oocyte, plotted as a function of time after start of Rg3 application, and then fitted with a single exponential function (dashed curve). The time constant for onset of Rg3 action was 1.9 ± 1.1 seconds (n = 7).
Mutation of Ser631 Does Not Prevent Effects of Rg3.
Consistent with the idea that Rg3 binds to an extracellular site of the channel, it was previously reported that a point mutation of a Ser residue located just outside the selectivity filter of the outer pore region abolished the effects of Rg3 on the hERG1 channel. Specifically, the fast deactivation of S631C hERG1 channel currents was not slowed by 3 µM Rg3 and the voltage dependence of activation was unaffected by 30 µM Rg3 (Choi et al., 2011a). This finding is puzzling because binding of Rg3 to Ser631 should result in the block of the outer pore, not activation of the channel. In contrast to this previous report we found that Rg3 affected S631C hERG1 channels in a manner similar to WT hERG1. S631C hERG1 channels were poorly expressed in oocytes and currents were of small magnitude even when oocytes were injected with up to 40 ng cRNA or treated with 5 mM dithiothreitol (in an effort to minimize disulfide formation). In three oocytes that expressed measurable S631C hERG1 currents, the rate of Itail deactivation was dramatically slowed by 3 µM Rg3 (Fig. 2A, left) and the V0.5 for activation was shifted by −14.3 ± 1.9 mV (Fig. 2A, right; n = 3). To further explore the potential importance of Ser631 in the action of Rg3, we also characterized S631A and G628C/S631C hERG1 channels that express well in oocytes. The rate of S631A hERG1 channel deactivation was also greatly slowed (Fig. 2B, left), activation was shifted by −13.0 ± 1.4 mV (Fig. 2B, right; n = 4), and the maximum Itail-peak was increased 1.2-fold (Supplemental Table 2) by 3 µM Rg3. G628C/S631C hERG1 channels also responded to Rg3 (Fig. 2C) with slowed Itail deactivation and a V0.5 for activation shift of −9.1 ± 1.3 mV (n = 5). Note that the tail currents of this mutant channel are inward at a Vret of −70 mV because of its reduced K+ ion selectivity (Smith et al., 1996). Together these findings indicate that Ser631 is unlikely to interact with Rg3.
Fig. 2.

Ser631 is not a structural determinant of the Rg3 binding site. (A-C) Representative current traces recorded during 4 s pulses to 0 mV, followed by 4 s pulses to –70 mV are shown on the left. for oocytes expressing S631C hERG1 (A), S631A hERG1 (B), or G628C/S631C hERG1 (C) mutant channels. Note that 3 µM Rg3 slows the rate of current deactivation at −70 mV for all channels. Plots of g/gmax–Vt relationships for indicated mutant hERG1 channel types are shown on the right. Data were normalized to gmax determined under control conditions and fitted with a Boltzmann function (smooth curves). V0.5 and k values for all fits are presented in Supplemental Table 1. The shift in V0.5 induced by Rg3 was −14.3 ± 1.9 mV for S631C (n = 3), −12.7 ± 1.4 mV for S631A (n = 4), and −9.1 ± 1.3 mV for G628C/S631C (n = 5) hERG1 channels.
Rg3 Does Not Interact With Residues Previously Identified as Important for Other hERG1 Activators.
Site-directed mutagenesis was previously used to identify specific residues that interact with several hERG1 activators. For example, L553A hERG1 channels were shown to be insensitive to RPR (Perry et al., 2007). However, this mutation did not alter the response of hERG1 to Rg3. Itail deactivation was greatly slowed (Fig. 3A) and V0.5 for activation was shifted by – 12.5 ± 1.6 mV (Fig. 3B; n = 3). Two other mutations (F557L, N658A) also prevented the slowing of deactivation of hERG1 channels by RPR (Perry et al., 2007). F557L and L622C abolished the effects of ICA-105574 on inactivation and deactivation (Garg et al., 2011) and F619A prevented the effects of PD-118057 (Perry et al., 2009). None of these five point mutations affected the ability of 3 µM Rg3 to slow channel deactivation or shift V0.5 (Fig. 3C). Thus, it is unlikely that Rg3 binds to the same regions of the hERG1 channel previously characterized for other activators.
Fig. 3.

Rg3 does not interact with hERG1 residues important for activity of other activators. (A) Voltage-clamp pulse protocol (upper left) and whole cell currents recorded from an oocyte expressing L553A hERG1 channels before (control) and after treatment with 3 µM Rg3. Small arrows indicate zero current level. (B) g/gmax–Vt relationship for L553A hERG1 channels. Data were normalized to gmax determined under control conditions and fitted with a Boltzmann function (smooth curves). V0.5 and k values for fits are presented in Supplemental Table 1. The shift in V0.5 induced by Rg3 was −12.5 ± 1.6 mV (n = 3). (C) Plot of the shift in V0.5 for activation (black bars) and fold change in τdeact (gray bars) induced by 3 µM Rg3 for WT or mutant hERG1 channels as indicated on the x-axis (n indicated below V0.5 bars). *deactivation in presence of Rg3 too slow to measure.
Ala Scan of the Extracellular Regions of the VSD in hERG1.
Because the onset of Rg3 is extremely fast, we reasoned that it likely binds to a region of the channel that is readily accessible from the extracellular side of the membrane. Moreover, because Rg3 affects voltage dependence of activation, we initially focused our Ala scan on the outer region of the VSD. First, we probed S3 and S4 segments and the interconnecting S3–S4 linker to search for residues that might interact with Rg3. Figure 4A is a plot of the average shift in V0.5 for activation and the fold change in τdeact induced by 3 µM Rg3 for mutant channels containing a single Ala substitution of 21 consecutive residues ranging from Phe508 to Arg528. Two mutations, I521A and K525A, prevented the negative shift in V0.5 and reduced the slowing of channel deactivation compared with WT channels. Moreover, 10 µM Rg3 induced a 12.2 ± 1.1 mV (n = 5) positive shift in V0.5 for activation of I521A channels, an effect that also reduced the magnitude of outward currents during depolarizing test pulses (Fig. 4, B and C). The gating of K525A hERG1 channels was quite abnormal. These channels activated at more negative potentials (V0.5 = −78.3 ± 4.0 mV; n = 8) than WT channels and did not fully close at membrane voltages as negative as −120 mV (Fig. 4D). Similar to I521A channels, the voltage dependence of K525A hERG1 channel activation was shifted in the positive direction (15.2 ± 5.6 mV; n = 7) by 10 µM Rg3 (Fig. 4E). In summary, mutation of two residues (Ile521 and Lys525) located in the outer region of the S4 segment greatly attenuated the response to 3 µM Rg3 and responded in an unusual fashion (positive shift in V0.5 for activation) to 10 µM Rg3.
Fig. 4.

Ala scan of select residues located in the S3–S4 region of hERG1. (A) Plot of the shift in V0.5 for activation (black bars) and fold change in τdeact (gray bars) induced by 3 µM Rg3 for WT or mutant hERG1 channels as indicated on the x-axis (n indicated below V0.5 bars). Point mutations were introduced into the S3 segment, S3–S4 linker (S34L), or S4 segment. (B) Voltage-clamp pulse protocol (upper image) and whole cell currents recorded from an oocyte expressing I521A hERG1 channels before (control) and after treatment with 10 µM Rg3. Small arrows indicate zero current level. (C) Plot of g/gmax–Vt relationships for I521A hERG1 channels. Data were normalized to gmax determined under control conditions and fitted with a Boltzmann function (smooth curves). V0.5 and k values for fits are presented in Supplemental Table 1. Rg3 shifted the V0.5 of I521A hERG1 channels by 2 ± 2.0 mV at 3 µM (n = 8) and by 12.2 ± 1.1 mV at 10 µM (n = 5). (D) Whole cell currents recorded from an oocyte expressing K525A hERG1 channels before (control) and after treatment with 10 µM Rg3. Small arrows indicate zero current level. (E) Plot of g/gmax–Vt relationships for K525A hERG1 channels. Data were normalized to gmax determined under control conditions and fitted with a Boltzmann function (smooth curves). Rg3 shifted V0.5 of K525A hERG1 channels by 1.1 ± 2.7 mV at 3 µM (n = 5) and by −0.2 ± 5.1 mV at 10 µM (n = 8). V0.5 and k values for Boltzmann fits in (C) and (E) are presented in Supplemental Table 1.
We next extended the Ala scan to the S1–S2 extracellular region of the VSD in hERG1. Not all residues in this extensive region were probed by single mutations. Native Ala residues were not mutated to any other residue and Pro and Gly residues were not mutated since these residues are often important for secondary structure. The specific residues that were mutated to Ala are indicated in Fig. 5A. In the outer region of the S1 segment, 11 of 15 residues were mutated to Ala. In the S1–S2 linker, 13 of the 19 residues were mutated, either singly to Ala or to two different combinations of residues as indicated in the legend for Fig. 5A. In the outer region of the S2 segment, five of seven residues were mutated to Ala. In addition, three other key gating residues in S2 were mutated to Ala, including Asp460 and Asp466 (which form charge pairing interactions with basic residues in S4) and Phe463 (which together with Asp466 and Asp501 in S3 form a charge transfer center in the VSD) (Tao et al., 2010; Whicher and MacKinnon, 2016).
Fig. 5.

Ala scan of select residues located in the S1–S2 region of hERG1. (A) Amino acid sequence (single letter code) of extracellular regions of S1 and S2 segments and S1–S2 extracellular linker (S12L). Single residues that were mutated to Ala are shown in bold. Underlined residues designate the multiple residues that were mutated to construct 4E2C/6A (E435A/E437A/E438A/E444A/C445A/C449A) or 3E1P/4R (E435R/E437R/E438R/P440R) multimutation channels. (B) Plot of the shift in V0.5 for activation (black bars) and fold change in τdeact (gray bars) induced by 3 µM Rg3 for WT or mutant hERG1 channels as indicated on the x-axis (n indicated below V0.5 bars). Point mutations were introduced into the S1 segment, S1–S2 linker (S12L), or S2 segment. 4E2C/6A, E435A/E437A/E438/E444A/C445A/C449A (Fernandez et al., 2005); 3E1P/4R, E435R/E437R/E438R/P440R. (C–E) Voltage-clamp pulse protocol (upper left) and whole cell currents recorded from an oocyte expressing Y420A (C), L452A (D), and F463A (E) hERG1 channels before (control) and after treatment with 3 and 30 µM Rg3. Small arrows indicate zero current level. (F–H) Plot of g/gmax–Vt relationships for Y420A (F), L452A (G), and F463A (H) hERG1 channels. Data were normalized to gmax determined under control conditions and fitted with a Boltzmann function (smooth curves). V0.5 and k values for fits are presented in Supplemental Table 1. Rg3 shifted the V0.5 of Y420A hERG1 channels by −2 ± 2.4 mV at 3 µM (n = 9) and by −9.6 ± 1.9 mV at 30 µM (n = 7). Rg3 shifted the V0.5 of L452A hERG1 channels by −6.1 ± 1.2 mV at 3 µM (n = 4) and by −7.3 ± 0.9 mV at 30 µM (n = 3). Rg3 shifted the V0.5 of F463A hERG1 channels by −3.5 ± 2.4 mV at 3 µM (n = 7) and by 4.7 ± 2.6 mV at 30 µM (n = 4).
A plot of the average shift in V0.5 for activation and the fold change in τdeact induced by 3 µM Rg3 for S1–S2 mutant channels are summarized in Fig. 5B. Notably, combined neutralization of four Glu residues plus two Cys to Ala mutations (E435A/E437A/E438/E444A/C445A/C449A) did not appreciably change gating properties of the channel (Fernandez et al., 2005) or its response to Rg3. To further probe the potential importance of the acidic charges in the S1–S2 linker, three of the Glu residues and one Pro residue were all mutated to Arg (E435R/E437R/E438R/P440R). This mutant channel also responded normally to Rg3. These findings strongly suggest that Rg3 does not interact with residues in the S1–S2 linker of hERG1. However, four mutations (Y420A and V423A in the S1 segment and L452A and F463A in the S2 segment) attenuated both the negative shift in V0.5 and reduced the slowing of channel deactivation compared with WT channels. Representative currents conducted by Y420A, L452A, and F463A hERG1 channels under control conditions and after treatment of oocytes with 3 and 30 µM Rg3 are illustrated in Fig. 5, C–E, and the corresponding g/gmax–Vt relationships are plotted in Fig. 5, F–H. At 3 and 30 µM, Rg3 caused a −2.0- and −9.6-mV shift in the V0.5 for activation but did not alter current magnitude of Y420A hERG1 channels (Fig. 5F). Rg3 enhanced Itail-peak by 25%–30% and shifted the V0.5 for activation of L452A hERG1 channels by −6.1 and −7.3 mV at 3 and 30 µM, respectively (Fig. 5G). F463A hERG1 channels deactivated much faster than normal and Rg3 increased Itail-peak by 84% and shifted the V0.5 for activation by −3.5 mV at 3 µM. At 30 µM, Rg3 increased Itail-peak by 41%, decreased peak outward current during test pulses by approximately 50% and shifted V0.5 in a positive direction by 4.7 mV (Fig. 5H).
Mutation of Leu417 within the S1 segment resulted in a particularly enhanced response to Rg3 (Fig. 6A). Although L417A channels deactivated very rapidly under control conditions, Rg3 still induced a dramatic slowing of this gating process (Fig. 6B). At a Vret of −70 mV, τdeact was increased by a factor of 16.1 ± 3.8 (n = 5), more than three times that observed for WT channels. Rg3 also shifted the V0.5 for activation by −37.4 ± 9.2 mV (n = 5; Fig. 6C), four times that observed for WT channels.
Fig. 6.

Rg3 dramatically slows deactivation and causes a large negative shift in the voltage dependence of activation of L417A hERG1 channels. (A) Voltage-clamp pulse protocol (upper left) and whole cell currents recorded from an oocyte expressing WT hERG1 before (control) and after treatment with 3 µM Rg3. Small arrows indicate zero current level. (B) Rg3 slows rate of Itail deactivation measured at a Vret of −70 mV. In seven oocytes, τdeact before and after treatment with 3 µM Rg3 was 43 ± 13 milliseconds and 696 ± 336 milliseconds, respectively. (C) g/gmax–Vt relationship for L417A hERG1 channels. Data were normalized to gmax determined under control conditions and fitted with a Boltzmann function (smooth curves). For control conditions: V0.5 = −5.7 ± 6.9 mV, k = 12.2 ± 1.1; after 3 µM Rg3: V0.5 = −43.1 ± 7.8 mV, k = 5.03 ± 0.42 (n = 5). The shift in V0.5 induced by Rg3 was −37.4 ± 9.2 mV (n = 5).
Ala Scan of the Extracellular Region of the Pore Domain.
We next probed the outer portion of the S5 segment and the adjoining, extracellular turret to search for residues that might interact with Rg3. Supplemental Figure 1 is a plot of the average shift in V0.5 for activation and the fold change in τdeact induced by 3 µM Rg3 for mutant channels containing a single substitution in 25 of the 45 residues between Ile567 and Tyr611. We did not mutate the single Pro or seven Gly residues in this region and several mutant channels (K595C, Y597C, S599C) did not express. Although not a complete scan, none of the mutant channels examined had a reduced response to Rg3 compared with WT channels.
Effect of Mutations on Rate of Current Deactivation.
It is important to note that there was no correlation between the deactivation rate of multiple mutant channels under control conditions and the fold change in τdeact caused by 3 µM Rg3 (Supplemental Fig. 2). Mutations that induced fast deactivation had a variable response to Rg3. For example, D509A and L510A channels deactivated very rapidly (τdeact = 37 and 31 milliseconds at −70 mV, respectively) and the Rg3 induced slowing of these channels was about half that observed for WT channels. However, other mutations that induced fast deactivation did not alter (e.g., D460A, τdeact = 67 milliseconds; D466A, τdeact = 59 milliseconds), or even increased (e.g., L417A, τdeact = 43 milliseconds; Y569A, τdeact = 104 milliseconds; N658A, τdeact = 128 milliseconds), the fold change in τdeact induced by Rg3.
Double Mutations Nearly Eliminate the Action of Rg3.
We next paired Y420A with either L452A or I521A into a single construct. Each of these single mutations reduced the response to Rg3 (Figs. 4 and 5). Y420A/L452A hERG1 channels activated at more positive potentials (V0.5 = 5.2 ± 6.7 mV, n = 14) and deactivated more rapidly than WT channels (Fig. 7A). The double mutant channel current also displayed an unusual tendency to decrease in magnitude (“run-down”) during repetitive pulsing soon after impaling an oocyte with recording microelectrodes. Run-down typically reduced currents to about half their initial amplitude before reaching a steady-state level. Currents for this mutant channel were not recorded until after run-down was complete. Rg3 at 3 and 30 µM increased Ipeak and Itail-peak for Vt >0 mV but did not significantly alter the voltage dependence of Y420A/L452A hERG1 channel activation (Fig. 7, A–C). V0.5 for activation was shifted by 2.4 ± 4.9 mV (n = 14) at 3 µM Rg3 and −1.3 ± 3.6 mV (n = 9) at 30 µM Rg3 (Fig. 7B).
Fig. 7.

Effect of Rg3 on Y420A/L452A hERG1 channel currents. (A) Voltage-clamp pulse protocol (upper left) and whole cell currents recorded from an oocyte expressing Y420A/L452A hERG1 channels before (control) and after treatment with 3 and 30 µM Rg3. Small arrows indicate zero current level. (B) Plot of g/gmax–Vt relationship for Y420A/L452A hERG1 channels. Data were normalized to gmax determined under control conditions and fitted with a Boltzmann function (smooth curves). V0.5 and k values are presented in Supplemental Table 1. Rg3 shifted V0.5 by 2.4 ± 4.9 mV at 3 µM (n = 14) and −1.3 ± 3.6 mV at 30 µM (n = 9). (C) Ipeak–Vt relationship for Y420A/L452A hERG1 channels. Data were normalized to the maximum Ipeak determined under control conditions. (D–G) Rg3 has only minor effects on voltage-dependent gating of Y420A/L452A hERG1 channels. (D) Voltage-clamp pulse protocol (upper left) and tail currents recorded from an oocyte expressing Y420A/L452A hERG1 channels before (control) and after treatment with 3 and 30 µM Rg3. For these experiments, the holding potential was –70 mV and tail currents were measured at the indicated Vret after a 1-second prepulse to 40 mV. Pulses were applied once every 12 seconds. Currents recorded at a Vret ranging from 20 to –30 mV are indicated by dashed curves. Currents recorded at a Vret ranging from –40 to –130 mV are indicated by solid curves. (E) Plot of the average normalized Itail-peak versus Vret before (control) and after treatment of oocytes with 3 and 30 µM Rg3. (F) Effect of Rg3 on the two time constants of deactivation of hERG1 current deactivation as a function of Vret. (G) Effect of Rg3 on the relative magnitude of the slow component of current deactivation as a function of Vret. For (E) to (G), n = 8.
Y420A/L452A hERG1 channels were fully activated by applying a prepulse to 40 mV for 1 second before repolarization to a Vret that was varied from 20 to −130 mV in 10-mV increments (Fig. 7D). For a Vret of –40 to –130 mV, deactivation of current was sufficient for accurate kinetic measurements. Rg3 had only minimal effects on the fully activated Itail-peak–Vt relationship (Fig. 7E). Rg3 at 3 µM slightly slowed (by 1.14-fold at −70 mV) the rate of Itail deactivation, whereas 30 µM Rg3 had no effect on τdeact (Fig. 7F). Rg3 had little or no effect on the relative amplitude of the slowly deactivating component of Itail, including at return potentials (i.e., −40 mV) where τdeact was slowest (Fig. 7G). By contrast, 10 µM Rg3 increased τf and τs for WT hERG1 channel deactivation at a Vret of −70 mV by 6-fold and 11-fold, respectively, and increased the relative amplitude of the slowly deactivating component of Itail (Wu et al., 2016). The negligible effects of Rg3 on τdeact of Y420A/L452A hERG1 channels were not simply due to the fact that these mutant channels deactivate rapidly in the absence of Rg3. As shown in Supplemental Fig. 2, there is no correlation between the control τdeact and the fold change in this parameter induced by Rg3.
Y420A/I521A hERG1 channel currents (Fig. 8A) activated at more negative potentials (V0.5 = −38.2 ± 4.2, n = 6) than WT channels. In response to 3 and 30 µM Rg3, V0.5 was shifted by −1.8 ± 1.5 mV (n = 6) and 8.2 ± 0.9 mV (n = 3), respectively (Fig. 8B). The maximum Itail-peak for Y420A/I521A hERG1 channels was increased by 3 µM Rg3 (Fig. 8B), whereas 30 µM Rg3 caused a reduction in Ipeak (Fig. 8C). Although Rg3 had almost no effect on V0.5 or τdeact of Y420A/L452A channels, it increased the maximum value of Itail-peak by a factor of approximately 1.3 (Supplemental Table 2). For comparison, the fold change in Itail-peak was 0.9–1.2 for the majority of other mutant channels and 1.1 for WT channels (Supplemental Fig. 3). The relatively large increase in Itail-peak observed for Y420A/L452A channels may be related to the fast rate of deactivation for these channels under control conditions, especially considering that the fold increase in Itail-peak was 1.02 and 1.23 for the single mutant (Y420A or L452A) hERG1 channels, respectively. The increase in Itail-peak was >1.6-fold in most other fast deactivating mutant channels, including D456A, F463A, D509A, L510A, and F619A hERG1 (Supplemental Fig. 3). We suspect that Itail-peak for these channels may have been underestimated under control conditions because deactivation was too fast to permit adequate recovery of channels from an inactivated state. Another possibility is that Rg3 activates channels by multiple mechanisms, possibly by binding to multiple sites including one that is not altered by the double mutation Y420A/L452A. Finally, the reduced efficacy of Rg3 on the gating of the double mutant channels is not simply the sum of the effects of single mutations (Fig. 8, D and E). The combination of Y420A and L452A mutations was most effective in suppressing the action of Rg3.
Fig. 8.

Effect of Rg3 on Y420A/I521A hERG1 channel currents. (A) Voltage-clamp pulse protocol (upper left) and whole cell currents recorded from an oocyte expressing Y420A/I521A hERG1 channels before (control) and after treatment with 3 and 30 µM Rg3. (B) g/gmax–Vt relationships for Y420A/I521A hERG1 channels. V0.5 and k values are presented in Supplemental Table 1. Rg3 shifted V0.5 by −1.8 ± 1.5 mV at 3 µM (n = 6) and by 8.2 ± 0.9 mV at 30 µM (n = 3). (C) Ipeak–Vt relationship for Y420A/I521A hERG1 channels. Data were normalized to the maximum Ipeak determined under control conditions [n is the same as indicated in (B)]. (D) Plot of the shift in V0.5 for activation induced by 3 µM Rg3 (black bars), 10 µM Rg3 (control, gray bars), or 30 µM Rg3 (mutants, gray bars) for indicated WT or mutant channels (n indicated for each bar). (E) Plot of the fold change in τdeact induced by 3 µM Rg3 (black bars), 10 µM Rg3 (control, gray bars), or 30 µM Rg3 (gray bars) for indicated WT or mutant hERG1 channels [n is the same as indicated in (D)]. Asterisks indicate controls.
In summary, mutation to Ala of Leu417 and Tyr420 in the S1 segment, Leu452 and Phe463 in the S2 segment, and Ile521 and Lys525 in the S4 segment produced the most dramatic changes in the effects of Rg3 on hERG1 channel gating. These findings suggest that these residues are either structural determinants of the Rg3 binding site or that mutation of these particular residues alters the response to Rg3 by an indirect, allosteric mechanism. We employed molecular modeling to help distinguish between these possibilities.
Docking and MD Simulations Suggest That the VSD of hERG1 Can Accommodate Rg3.
Simulated docking of Rg3 to molecular models of the hERG1 voltage sensor module in two different activated states was performed. It is not clear whether the VSD in the rEAG1 structure template represents a fully activated state (Whicher and MacKinnon, 2016), since only the first three positive charges are above the charge transfer center. Thus, we additionally built a model, where the alignment of helix S4 was shifted upward by three residues (denoted activated1), to obtain a model with four positive charges above the charge transfer center. This activated1 model is consistent with experimental work from Zhang et al. (2005), suggesting a direct interaction between residues D456 from helix S2 and Arg528 from the S4 segment. In both models, docking predicts that Rg3 can protrude deeply into the VSD modules (Fig. 9; Supplemental Fig. 4), where they remain stably bound during 50 nanoseconds of unbiased MD simulations (Supplemental Fig. 5, A and B). Detailed views of the putative drug-binding regions in the activated and activated1 states are depicted in Fig. 9, in which residues identified by scanning mutagenesis and a few other residues in close contact (within 4 Å) with the drug molecule are shown in stick representation. In both activated states, residues Tyr420, Thr421, Leu452, Asp460, Asp509, Ile512, Ile521, Gly522, Lys525, and Arg528 are within 4 Å of the Rg3 molecule. A second class of high-impact residues (as determined by mutagenesis) is not in direct contact with Rg3 in either model; however, a downward movement of the drug is already apparent in the activated model, where the distance between Rg3 and Phe463 is approaching 3.5 Å, compared with >5 Å in the activated1 model (measured at the end of the 50-nanosecond runs). Thus, it is plausible that residues Phe463, Val418, and Leu417 might come in close contact with the drug upon voltage sensor downward movement. The third group of residues is unlikely to form direct interactions according to our homology models, even when taking into consideration conformational rearrangements upon gating. Most notable are residues Ile419 (S1) and Leu510 (S3) that are lipid interacting, facing the opposite site of the predicted binding site. Ser428 is predicted to form a stable hydrogen bond to Rg3; however S428A hERG1 channels responded normally to Rg3. Furthermore, loop residues including Glu435 can be within 4 Å of the drug, but due to the high flexibility of these regions (see Supplemental Fig. 6) might not contribute to binding. Hydrogen bond contacts between Rg3 and hERG1 in the activated and activated1 model are summarized in Supplemental Table 3. Finally, during the revision of this manuscript, the Wang and MacKinnon (2017) published the first cryo-EM structure of the hERG1 channel in the open state. A structural alignment of the VSDs of hERG1 with rEAG1 reveals similar conformations in this domain with a root-mean-square deviation of 1.25 Å (see Supplemental Fig. 7). However, the hERG1 VSD has a lower resolution (approximately 4–5 Å) than the EAG1 structure and is also missing the S1–S2 and S3–S4 extracellular loops. For these reasons the new structure provided no advantage for our simulations.
Fig. 9.

Docking interactions of Rg3 with the voltage sensor models in two activated state models. (A and B) The side view of the rEAG1-based activated VSD module of the hERG1 channel is shown. Rg3 is shown in green stick representation. Key residues identified in scanning mutagenesis are labeled in bold and shown in stick representation. Additional residues shown in plain text (e.g., S428, F424, E435, D456) are also predicted to be in close contact (within 4 Å) with Rg3, but exhibited normal drug response when mutated to Ala. (C and D) Activated1 VSD module with residue labeling the same as in (A) and (B).
Discussion
Rg3 Interacts With the VSD of hERG1.
Ginsenoside Rg3 is a large steroid glycoside that slows the rate of deactivation and shifts the voltage dependence of hERG1 channel activation to more negative potentials (Choi et al., 2011a; Wu et al., 2016). These effects are similar to a few other hERG1 activators, including mallotoxin (Zeng et al., 2006) and NS1643 [1,3-bis(2-hydroxy-5-trifluoromethylphenyl)urea] (Xu et al., 2008) and related analogs (Guo et al., 2014). However, unlike these other compounds the onset of Rg3 activity upon extracellular application to cells is extremely rapid, suggesting that it likely binds to an extracellular accessible site on the hERG1 channel. S631C hERG1 channels were reported to be insensitive to Rg3 (Choi et al., 2011a), suggesting that the alkaloid might interact directly or indirectly with Ser631, a residue located within the outer vestibule of the channel in close proximity to its selectivity filter. However, our finding that Rg3 altered the gating of channels harboring Ser631 mutations (i.e., S631C, S631A, or G628C/S631C) in a manner similar to WT channels indicated to us that this residue does not interact with Rg3. Therefore, we mutated 84 other residues located in the extracellular facing region of the VSD to search for regions of the hERG1 subunit that may interact directly or indirectly (allosterically) with Rg3. We focused our attention on mutations that reduced both of the primary effects of Rg3. Our scanning mutagenesis identified five mutations (Y420A, L452A, F463A, I521A, K525A) that highly diminished the slowing of deactivation and the negative shift in V0.5 for activation normally induced by Rg3 and one mutation (L417A) that greatly enhanced these effects of Rg3. In addition, mutation of 11 additional residues enhanced the ability of Rg3 to either slow deactivation or shift V0.5 for activation and four other mutations caused a reduced deactivation response. The combined point mutations of Y420A and L452A abolished the effects of Rg3 on V0.5 or τdeact at the high concentration of 30 µM. The double mutation also induced fast channel deactivation (τdeact = 73 milliseconds at −70 mV), but this does not explain why Rg3 did not slow deactivation as illustrated in Supplemental Fig. 2, which shows no correlation between deactivation rate of multiple mutant channels under control conditions and the fold change in τdeact caused by 3 µM Rg3. The side chains of most, but not all, of these “high-impact” residues face toward a cleft located between the transmembrane segments of the VSD. Simulated molecular docking of Rg3 to a homology model of the hERG1 VSD based on the cryo-EM structure of the rat EAG1 channel (Whicher and MacKinnon, 2016) corroborated some, but not all, of the scanning mutagenesis findings. For example, a stable H-bond was predicted between Ser428 and Rg3, but the S428A mutant channel responded normally to Rg3. The residue interactions predicted by the docking model were primarily hydrophobic in nature and the binding mode for Rg3 was very stable, with a root-mean-square deviation of approximately 0.15 nm during repeated MD simulations of 50 nanoseconds. Together these findings suggest that Rg3 modifies the gating of hERG1 channels by binding to the outer region of the VSD. Presumably this binding mode favors the activated state of the VSD and shifts the V0.5 for activation to more negative potentials. Further studies are required to determine whether slow deactivation results from a slower rate of repolarization-induced S4 charge displacement or from a functional uncoupling between S4 movement and closure of the activation gate formed by the S6 bundle crossing. We previously found that the profound slowing of hERG1 deactivation caused by RPR was not associated with a change in the kinetics of gating current. The putative binding site for RPR is located on the pore domain, not in the VSD as proposed here for Rg3. Further evidence that RPR and Rg3 do not interact at a common site on hERG1 was our finding that mutations that drastically alter the response to RPR (i.e., L553A, F557L, N658A) did not alter the response to Rg3. Thus, it is uncertain whether the molecular mechanism of slowed channel deactivation caused by RPR and Rg3 is similar or entirely different.
Study Limitations.
We used a site-directed mutagenesis approach to identify the molecular determinants of Rg3-induced changes in hERG1 channel gating. This indirect approach of identifying the putative binding site for a ligand has several important limitations. Perhaps the most significant is the underlying assumption that an altered response to Rg3 induced by a single or multiple point mutations can be used to identify specific residues that interact with Rg3. It is not possible to determine whether an altered response to Rg3 induced by a point mutation results directly from a decreased binding affinity or from an indirect, allosteric mechanism unrelated to a change in binding affinity. For example, although mutation to Ala of Ile419 in the S1 segment and Leu510/Leu511 in the S3 segment altered the response of hERG1 to Rg3, these residues are not predicted to make contact with the alkaloid in the dockings shown in Fig. 9. A second limitation is that many mutations significantly alter the properties of hERG1 channel gating (Supplemental Tables 1 and 2), perhaps in a manner that would limit or accentuate the effects of Rg3. However, of the 18 mutations that shifted the V0.5 for activation by >+10 mV, only three mutations (Y420A, L452A, F463A) showed a reduced V0.5 shift in response to Rg3. Of the eight mutations that shifted V0.5 to a more negative potential by >−10 mV compared with WT channels, only three mutations (I521A, G522A, K525A) exhibited a reduced V0.5 shift in response to Rg3. Many mutations also altered the rate of channel deactivation. However, there was no correlation between τdeact measured under control conditions and the fold change in this parameter induced by Rg3 (Supplemental Fig. 2). Thus, although many mutations significantly altered channel gating properties, these changes cannot account for the finding that specific mutations altered the response to Rg3. These findings argue against the proposal that activators must “work harder to overcome mutation induced loss of baseline channel function” (Pearlstein et al., 2017). Third, for some mutant channels, fitting Itail deactivation with a single exponential function led to an underestimation of the fold change in τdeact. This is the case for WT channels in which Rg3 increases the τ for the fast and slow components of deactivation by 5-fold and 10-fold, respectively (Wu et al., 2016). Finally, most residues were only mutated to Ala and not all residues were mutated. Mutation to a residue other than Ala may have identified additional native residues as structural determinants of Rg3 activity. We also chose not to mutate most native Ala, Pro, or Gly residues (e.g., in the S1–S2 region) and therefore would have missed if any of these residues interact with Rg3.
Therapeutic Implications.
Activators of hERG1 channels have been proposed as a pharmacological approach for prevention of arrhythmia associated with congenital or acquired LQTS (Kang et al., 2005; Sanguinetti, 2014; Wu and Sanguinetti, 2016). However, excessive activation of hERG1 would cause early repolarization of action potentials, essentially mimicking congenital short QT syndrome, a disorder of ventricular repolarization that also increases the risk of life-threatening arrhythmia (Gaita et al., 2003; Shah, 2010). Rg3 and related ginsenosides have been extensively studied and are claimed to exert a multitude of therapeutic effects (Weber et al., 2012; Nah, 2014; Ahn et al., 2016). Given the plethora of activities associated with Rg3, it is not surprising that this ginsenoside is not a hERG1-specific compound. Rg3 also inhibits Na+, 5-hydroxytryptamine type 3A, and nicotinic acetylcholine receptor channels (Jeong et al., 2004), L-type Ca2+ (Choi et al., 2009), and KCNQ1 channels (Choi et al., 2010). Rg3 modifies gating to activate KCNQ1/KCNE1 (Choi et al., 2010), KCa1.1 (Choi et al., 2011b) as well as EAG1, ERG3, and ELK1 channels (Wu et al., 2016). Some of these channel activities occur over a range of concentrations similar to those that affect hERG1 channels. Thus, although Rg3 provides a structural starting point for design of novel compounds that can activate hERG1, its nonspecific activities make it an inappropriate agent for evaluating the efficacy or safety of hERG1 activators in LQTS.
Other compounds that interact with the VSD of voltage-gated K+ or Na+ channels have been reported to activate or inhibit channels in a specific manner. For example, ICA-27243 [N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide] selectively binds to the VSD and shifts the voltage dependence of KCNQ2/3 (Kv7.2/.3) channels to more negative potentials (Padilla et al., 2009). GX-936 (3-cyano-4-[2-[1-(1-ethyl-3-azetidinyl)-1H-pyrazol-5-yl]-4-(trifluoromethyl)phenoxy]-N-1,2,4-thiadiazol-5-yl-benzenesulfonamide) and related aryl sulfonamides selectively inhibit Nav1.7 channels by binding to and trapping the fourth VSD in its activated state. This action prevents deactivation of the VSD and thereby stabilizes the inactivated state of the Nav1.7 channel (Ahuja et al., 2015). Future studies are required to determine whether structural modifications of Rg3 might result in a compound that specifically interacts with the VSD of hERG1 channels.
Acknowledgments
The authors thank Gea-Ny Tseng for providing several constructs used for mutagenesis scanning of the turret region of hERG1.
Abbreviations
- EM
electron microscopy
- g/gmax
relative conductance
- GX-936
3-cyano-4-[2-[1-(1-ethyl-3-azetidinyl)-1H-pyrazol-5-yl]-4-(trifluoromethyl)phenoxy]-N-1,2,4-thiadiazol-5-yl-benzenesulfonamide
- hERG1
human ether-à-go-go–related gene type 1
- ICA-27243 [N-(6-chloro-pyridin-3-yl)-3
4-difluoro-benzamide
- ICA-105574
3-nitro-N-(4-phenoxyphenyl)benzamide
- Ipeak
peak outward current during test pulse
- Itail
tail current
- Itail-peak
peak tail current
- LQTS
long QT syndrome
- MD
molecular dynamics
- NS1643
1,3-bis(2-hydroxy-5-trifluoromethylphenyl)urea
- PD-118057
2-({4-[2-(3,4-dichlorophenyl)ethyl]phenyl}amino)benzoic acid
- Rg3
20(S)-ginsenoside Rg3 [(2S,3R,4S,5S,6R)-2-[(2R,3R,4S,5S,6R)-4,5-dihydroxy-2-[[(3S,5R,8R,9R,10R,12R,13R,14R,17S)-12-hydroxy-17-[(2S)-2-hydroxy-6-methylhept-5-en-2-yl]-4,4,8,10,14-pentamethyl-2,3,5,6,7,9,11,12,13,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-yl]oxy]-6-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol]
- RPR
RPR-260243 [(3R,4R)-4-[3-(6-methoxy-quinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5 trifluorophenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid]
- τdeact
time constant of current deactivation
- V0.5
half-point of Boltzmann function
- Vret
return potential
- VSD
voltage-sensing domain
- Vt
test potential
- WT
wild type
Authorship Contributions
Participated in research design: Gardner, Sanguinetti.
Conducted experiments: Gardner, Wu, Thomson, Sanguinetti.
Performed data analysis: Gardner, Zangerl-Plessl, Stary-Weinzinger, Sanguinetti.
Wrote or contributed to the writing of the manuscript: Stary-Weinzinger, Sanguinetti.
Footnotes
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01-HL055236 (to M.C.S.)], the Austrian Science Fund [Grant W1232 (to E.-M.Z.-P. and A.S.-W.)], and Wiener Hochschuljubiläumsstiftung [Grant H-304013/2014 (to A.S.-W.)].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Ahn EJ, Choi GJ, Kang H, Baek CW, Jung YH, Woo YC, Bang SR. (2016) Antinociceptive effects of ginsenoside Rg3 in a rat model of incisional pain. Eur Surg Res 57:211–223. [DOI] [PubMed] [Google Scholar]
- Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H, Shriver S, Young C, Lin S, Johnson JP, Jr, et al. (2015) Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 350:aac5464. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Grigera JR, Straatsma TP. (1987) The missing term in effective pair potentials. J Phys Chem 91:6269–6271. [Google Scholar]
- Choi SH, Lee JH, Pyo MK, Lee BH, Shin TJ, Hwang SH, Kim BR, Lee SM, Oh JW, Kim HC, et al. (2009) Mutations Leu427, Asn428, and Leu431 residues within transmembrane domain-I-segment 6 attenuate ginsenoside-mediated L-type Ca(2+) channel current inhibitions. Biol Pharm Bull 32:1224–1230. [DOI] [PubMed] [Google Scholar]
- Choi SH, Shin TJ, Hwang SH, Lee BH, Kang J, Kim HJ, Jo SH, Choe H, Nah SY. (2011a) Ginsenoside Rg(3) decelerates hERG K(+) channel deactivation through Ser631 residue interaction. Eur J Pharmacol 663:59–67. [DOI] [PubMed] [Google Scholar]
- Choi SH, Shin TJ, Lee BH, Chu DH, Choe H, Pyo MK, Hwang SH, Kim BR, Lee SM, Lee JH, et al. (2010) Ginsenoside Rg3 activates human KCNQ1 K+ channel currents through interacting with the K318 and V319 residues: a role of KCNE1 subunit. Eur J Pharmacol 637:138–147. [DOI] [PubMed] [Google Scholar]
- Choi SH, Shin TJ, Lee BH, Hwang SH, Lee SM, Lee BC, Park CS, Ha TS, Nah SY. (2011b) Ginsenoside Rg3 enhances large conductance Ca2+-activated potassium channel currents: a role of Tyr360 residue. Mol Cells 31:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. (1993) Particle mesh Ewald: an N-log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. [Google Scholar]
- Fernandez D, Ghanta A, Kinard KI, Sanguinetti MC. (2005) Molecular mapping of a site for Cd2+-induced modification of human ether-à-go-go-related gene (hERG) channel activation. J Physiol 567:737–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM. (1998) Molecular determinants of dofetilide block of HERG K+ channels. Circ Res 82:386–395. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, and Scalmani G (2009) Gaussian 09, Revision A.1. Gaussian, Inc., Wallingford, CT.
- Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M. (2003) Short QT syndrome: a familial cause of sudden death. Circulation 108:965–970. [DOI] [PubMed] [Google Scholar]
- Garg V, Sachse FB, Sanguinetti MC. (2012) Tuning of EAG K(+) channel inactivation: molecular determinants of amplification by mutations and a small molecule. J Gen Physiol 140:307–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Stary-Weinzinger A, Sachse F, Sanguinetti MC. (2011) Molecular determinants for activation of human ether-à-go-go-related gene 1 potassium channels by 3-nitro-N-(4-phenoxyphenyl) benzamide. Mol Pharmacol 80:630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach AC, Stoehr SJ, Castle NA. (2010) Pharmacological removal of human ether-à-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574). Mol Pharmacol 77:58–68. [DOI] [PubMed] [Google Scholar]
- Guo J, Durdagi S, Changalov M, Perissinotti LL, Hargreaves JM, Back TG, Noskov SY, Duff HJ. (2014) Structure driven design of novel human ether-a-go-go-related-gene channel (hERG1) activators. PLoS One 9:e105553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472. [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SM, Lee JH, Kim JH, Lee BH, Yoon IS, Lee JH, Kim DH, Rhim H, Kim Y, Nah SY. (2004) Stereospecificity of ginsenoside Rg3 action on ion channels. Mol Cells 18:383–389. [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC. (1995) Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol 245:43–53. [DOI] [PubMed] [Google Scholar]
- Kang J, Chen XL, Wang H, Ji J, Cheng H, Incardona J, Reynolds W, Viviani F, Tabart M, Rampe D. (2005) Discovery of a small molecule activator of the human ether-a-go-go-related gene (HERG) cardiac K+ channel. Mol Pharmacol 67:827–836. [DOI] [PubMed] [Google Scholar]
- Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291–325. [DOI] [PubMed] [Google Scholar]
- Nah SY. (2014). Ginseng ginsenoside pharmacology in the nervous system: involvement in the regulation of ion channels and receptors. Frontiers Physiol 5:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys 81:511–519. [Google Scholar]
- Padilla K, Wickenden AD, Gerlach AC, McCormack K. (2009) The KCNQ2/3 selective channel opener ICA-27243 binds to a novel voltage-sensor domain site. Neurosci Lett 465:138–142. [DOI] [PubMed] [Google Scholar]
- Parrinello M, Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. [Google Scholar]
- Pearlstein RA, Dickson CJ, Hornak V. (2017) Contributions of the membrane dipole potential to the function of voltage-gated cation channels and modulation by small molecule potentiators. Biochim Biophys Acta 1859:177–194. [DOI] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Abbruzzese J, Sanguinetti MC. (2009) PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc Natl Acad Sci USA 106:20075–20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Sanguinetti MC. (2007) Structural basis of action for a human ether-a-go-go-related gene 1 potassium channel activator. Proc Natl Acad Sci USA 104:13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC. (2014) HERG1 channel agonists and cardiac arrhythmia. Curr Opin Pharmacol 15:22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreibmayer W, Lester HA, Dascal N. (1994) Voltage clamping of Xenopus laevis oocytes utilizing agarose-cushion electrodes. Pflugers Arch 426:453–458. [DOI] [PubMed] [Google Scholar]
- Shah RR. (2010) Drug-induced QT interval shortening: potential harbinger of proarrhythmia and regulatory perspectives. Br J Pharmacol 159:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. (1996) The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379:833–836. [DOI] [PubMed] [Google Scholar]
- Stühmer W. (1992) Electrophysiological recording from Xenopus oocytes. Methods Enzymol 207:319–339. [DOI] [PubMed] [Google Scholar]
- Tao X, Lee A, Limapichat W, Dougherty DA, MacKinnon R. (2010) A gating charge transfer center in voltage sensors. Science 328:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, MacKinnon R. (2017) Cryo-EM structure of the open human ether-à-go-go-related K+ channel hERG. Cell 169:422–430 e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber DA, Wheat JM, Currie GM. (2012). Cancer stem cells and the impact of Chinese herbs, isolates and other complementary medical botanicals: a review. Zhong Xi Yi Jie He Xue Bao 10:493–503. [DOI] [PubMed] [Google Scholar]
- Whicher JR, MacKinnon R. (2016) Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 353:664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Gardner A, Sachse FB, Sanguinetti MC. (2016) Ginsenoside Rg3, a gating modifier of EAG family K+ channels. Mol Pharmacol 90:469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Gardner A, Sanguinetti MC. (2015) Concatenated hERG1 tetramers reveal stoichiometry of altered channel gating by RPR-260243. Mol Pharmacol 87:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Sachse FB, Gardner A, Sanguinetti MC. (2014) Stoichiometry of altered hERG1 channel gating by small molecule activators. J Gen Physiol 143:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Sanguinetti MC. (2016) Molecular basis of cardiac delayed rectifier potassium channel function and pharmacology. Card Electrophysiol Clin 8:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Recanatini M, Roberti M, Tseng GN. (2008) Probing the binding sites and mechanisms of action of two human ether-a-go-go-related gene channel activators, 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643) and 2-[2-(3,4-dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid (PD307243). Mol Pharmacol 73:1709–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Lozinskaya IM, Lin Z, Willette RN, Brooks DP, Xu X. (2006) Mallotoxin is a novel human ether-a-go-go-related gene (hERG) potassium channel activator. J Pharmacol Exp Ther 319:957–962. [DOI] [PubMed] [Google Scholar]
- Zhang M, Liu J, Jiang M, Wu DM, Sonawane K, Guy HR, Tseng GN. (2005) Interactions between charged residues in the transmembrane segments of the voltage-sensing domain in the hERG channel. J Membr Biol 207:169–181. [DOI] [PubMed] [Google Scholar]
- Zhou J, Augelli-Szafran CE, Bradley JA, Chen X, Koci BJ, Volberg WA, Sun Z, Cordes JS. (2005) Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol 68:876–884. [DOI] [PubMed] [Google Scholar]