

In the crystal of the title compound, C—H⋯O hydrogen bonds and C—O⋯π interactions form a two-dimensional network lying parallel to the ab plane.
Keywords: crystal structure, theoretical calculations, quinolinic acid imide, hydrogen bonding
Abstract
The title compound, C15H18N2O2S {systematic name: 6-[2-(cyclohexylsulfanyl)ethyl]-5H-pyrrolo[3,4-b]pyridine-5,7(6H)-dione}, was obtained from the reaction of pyridine-2,3-dicarboxylic anhydride (synonym: quinolinic anhydride) with 2-(cyclohexylsulfanyl)ethylamine. The dihedral angle between the mean plane of the cyclohexyl ring and the quinolinic acid imide ring is 25.43 (11)°. In the crystal, each molecule forms two C—H⋯O hydrogen bonds and one weak C—O⋯π [O⋯ring centroid = 3.255 (2) Å] interaction with neighbouring molecules to generate a ladder structure along the b-axis direction. The ladders are linked by weak C—O⋯π [O⋯ring centroid = 3.330 (2) Å] interactions, resulting in sheets extending parallel to the ab plane. The molecular structure is broadly consistent with theoretical calculations performed by density functional theory (DFT).
Chemical context
Quinolinic anhydrides have been used extensively as versatile intermediates in the synthesis of various heterocyclic systems, such as aphthyridines, nicotinamides and isotonic derivatives. Recently, they have been exploited in antiviral, dementia, anti-allergy and antitumor targets (Metobo et al., 2013 ▸). In addition, it is expected that various metal complexes may be formed because they are composed of N/S-donor atoms. In particular, our group reported copper(I) coordination polymers with N/S-donor-atom ligands, which showed their various luminescence and reversible/irreversible structural transformations (Jeon et al., 2014 ▸; Cho et al., 2015 ▸). As part of our ongoing studies in this area, we designed and synthesized a new N/S-donor ligand, namely N-[2-(cyclohexylsulfanyl)ethyl]quinolinic acid imide, which was prepared from the reaction of quinolinic anhydride with 2-(cyclohexylsulfanyl)ethylamine. Herein, we report its crystal structure.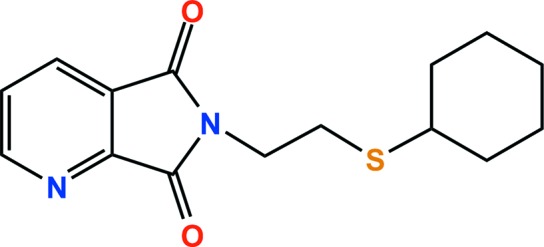
Structural commentary
The crystal structure of the title compound is shown in Fig. 1 ▸. The cyclohexyl ring adopts a chair conformation, with the exocyclic C—S bond in an equatorial orientation; the dihedral angle between the mean plane (r.m.s. deviation = 0.2317 Å) of the cyclohexyl ring and the quinolinic acid imide ring is 25.43 (11)°. All bond lengths and angles are normal and comparable to those observed in similar crystal structures (Garduño-Beltrán et al., 2009 ▸; Inoue et al., 2009 ▸).
Figure 1.

The molecular structure of the title compound, with displacement ellipsoids drawn at the 50% probability level.
Supramolecular features
In the crystal, molecules are linked by C2—H2⋯O1i and C3—H3⋯O1i hydrogen bonds [H⋯O = 2.50 and 2.55 Å, respectively; symmetry code: (i) x, y + 1, z; Table 1 ▸], and weak C6—O1⋯Cg1ii (Cg1 is the centroid of the N1/C1–C5 ring) interactions [O⋯π = 3.255 (2) Å; symmetry code: (ii) 1 − x, − + y, − z], forming a one-dimensional ladder structure along the b axis. The ladders are packed in an ABAB pattern along the c axis (yellow dashed lines in Fig. 2 ▸). In addition, the ladders are linked by C7—O2⋯Cg1iii interactions [O⋯π = 3.330 (2) Å; symmetry code: (iii) −1 + x, y, z], resulting in the formation of a two-dimensional network structure lying parallel to the ab plane (red dashed lines in Fig. 3 ▸).
+ y, − z], forming a one-dimensional ladder structure along the b axis. The ladders are packed in an ABAB pattern along the c axis (yellow dashed lines in Fig. 2 ▸). In addition, the ladders are linked by C7—O2⋯Cg1iii interactions [O⋯π = 3.330 (2) Å; symmetry code: (iii) −1 + x, y, z], resulting in the formation of a two-dimensional network structure lying parallel to the ab plane (red dashed lines in Fig. 3 ▸).
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C2—H2⋯O1i | 0.95 | 2.50 | 3.119 (3) | 123 |
| C3—H3⋯O1i | 0.95 | 2.55 | 3.129 (3) | 119 |
Symmetry code: (i)  .
.
Figure 2.

The crystal packing of the title compound, indicating the C—H⋯O hydrogen bonds and C—O⋯π interactions (yellow dashed lines) [symmetry codes: (i) x, y + 1, z; (ii) 1 − x, − + y, − z], which results in a one-dimensional ladder structure along the b axis.
Figure 3.

The packing diagram, showing the two-dimensional network structure formed by C—O⋯π interactions (red dashed lines) [symmetry code: (iii) −1 + x, y, z]. H atoms and cyclohexanesulfanyl groups not involved in intermolecular interactions have been omitted for clarity.
Theoretical calculations
To support the experimental data based on the diffraction study, computational calculations on the N-[2-(cyclohexylsulfanyl)ethyl]quinolinic acid imide molecule were performed using the GAUSSIAN09 software package (Frisch et al., 2009 ▸). Full geometry optimizations were calculated at the DFT level of theory using a basis set of 6-311++G(d,p). The optimized parameters, such as bond lengths and angles, are in generally good agreement (the largest bond-length deviation is less than 0.03 Å) with the experimental crystallographic data (Table 2 ▸). The calculated and experimental torsion angles for N2—C8—C9—S1 (C8—C9—S1—C10) are 53.64 (65.80) and 64.2 (3)° [97.4 (2)°], respectively. The calculated and experimental dihedral angle between the ring systems were 25.34 and 25.43 (11)°, respectively. However, several relatively large differences between the experimental and theoretical data (see Table 2 ▸) may be due to the packing effects induced by the intermolecular interactions in the crystal.
Table 2. Experimental and calculated bond lengths (Å).
| Bond | X-ray | B3LYP (6–311++G(d,p)) | Difference |
|---|---|---|---|
| S1—C9 | 1.813 (3) | 1.830 | −0.017 |
| S1—C10 | 1.827 (3) | 1.853 | −0.026 |
| O1—C6 | 1.212 (3) | 1.205 | 0.007 |
| O2—C7 | 1.209 (3) | 1.210 | 0.001 |
| N1—C5 | 1.325 (3) | 1.324 | 0.001 |
| N1—C1 | 1.342 (4) | 1.342 | 0.000 |
| N2—C6 | 1.394 (3) | 1.407 | −0.013 |
| N2—C7 | 1.395 (4) | 1.399 | −0.004 |
| N2—C8 | 1.460 (3) | 1.456 | 0.004 |
| C1—C2 | 1.382 (4) | 1.400 | −0.018 |
| C2—C3 | 1.381 (4) | 1.396 | −0.015 |
| C3—C4 | 1.380 (4) | 1.385 | −0.005 |
| C4—C5 | 1.376 (4) | 1.392 | −0.016 |
| C4—C7 | 1.490 (4) | 1.492 | −0.002 |
| C5—C6 | 1.497 (4) | 1.508 | −0.011 |
| C8—C9 | 1.522 (4) | 1.536 | −0.014 |
| C10—C11 | 1.516 (4) | 1.534 | −0.018 |
| C10—C15 | 1.530 (4) | 1.536 | −0.006 |
| C11—C12 | 1.523 (4) | 1.539 | −0.016 |
| C12—C13 | 1.523 (4) | 1.534 | −0.011 |
| C13—C14 | 1.514 (4) | 1.535 | −0.021 |
| C14—C15 | 1.524 (5) | 1.537 | −0.013 |
Synthesis and crystallization
A mixture of quinolinic anhydride (0.67 g, 5.0 mmol) and 2-(cyclohexylsulfanyl)ethylamine (0.83 g, 5.3 mmol) in toluene (15 ml) was heated at 433 K with stirring for 8 h. The crude product was extracted with dichloromethane. The dichloromethane layer was dried with anhydrous Na2SO4 and evaporated to give a crude solid. The reaction mixture was then concentrated and purified by chromatography on silica gel (MeCOOEt/n-C6H14 = 30/70 v/v, R F = 0.28) (Kang et al., 2015 ▸). Colourless plates were obtained by slow evaporation of a hexane solution of the title compound. 1H NMR (300 MHz, CDCl3): δ 7.40 (dd, H, Py), 8.02 (t, H, Py), 7.52 (dd, H, Py), 3.74 (t, 2H, NCH2), 2.64 (t, 2H, CH2S), 2.56 (d, H, SCH), 1.82–1.04 [m, 10H, (CH2)5]; 13C NMR (75.4 MHz, CDCl3): δ 166.84, 166.47, 155.60, 144.65, 139.31, 125.76, 116.76, 42.95, 37.89, 33.36, 27.71, 25.91, 25.68
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. All H atoms were positioned geometrically and refined using a riding model, with C—H = 0.95 Å and U iso(H) = 1.2U eq(C) for aromatic C—H groups, C—H = 0.99 Å and U iso(H) = 1.2U eq(C) for CH2 groups, and C—H = 1.00 Å and U iso(H) = 1.2U eq(C) for Csp 3—H groups.
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | C15H18N2O2S |
| M r | 290.37 |
| Crystal system, space group | Orthorhombic, P212121 |
| Temperature (K) | 173 |
| a, b, c (Å) | 5.5322 (2), 7.8707 (3), 32.9092 (14) |
| V (Å3) | 1432.94 (10) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.23 |
| Crystal size (mm) | 0.28 × 0.10 × 0.09 |
| Data collection | |
| Diffractometer | Bruker APEXII CCD |
| Absorption correction | Multi-scan (SADABS; Bruker, 2014 ▸) |
| T min, T max | 0.690, 0.746 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 11035, 2536, 2302 |
| R int | 0.046 |
| (sin θ/λ)max (Å−1) | 0.595 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.034, 0.072, 1.04 |
| No. of reflections | 2536 |
| No. of parameters | 181 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.20, −0.19 |
| Absolute structure | Flack x determined using 839 quotients [(I +) − (I −)]/[(I +) + (I −)] (Parsons et al., 2013 ▸) |
| Absolute structure parameter | 0.05 (5) |
Supplementary Material
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S2056989017012142/hb7685sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989017012142/hb7685Isup2.hkl
CCDC reference: 1570205
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The main calculations were carried out by the Supercomputing Center/Korea Institute of Science and Technology Information (KISTI) (KSC-2017-C1-0002).
supplementary crystallographic information
Crystal data
| C15H18N2O2S | Dx = 1.346 Mg m−3 |
| Mr = 290.37 | Mo Kα radiation, λ = 0.71073 Å |
| Orthorhombic, P212121 | Cell parameters from 2024 reflections |
| a = 5.5322 (2) Å | θ = 2.5–27.2° |
| b = 7.8707 (3) Å | µ = 0.23 mm−1 |
| c = 32.9092 (14) Å | T = 173 K |
| V = 1432.94 (10) Å3 | Plate, colourless |
| Z = 4 | 0.28 × 0.10 × 0.09 mm |
| F(000) = 616 |
Data collection
| Bruker APEXII CCD diffractometer | 2302 reflections with I > 2σ(I) |
| φ and ω scans | Rint = 0.046 |
| Absorption correction: multi-scan (SADABS; Bruker, 2014) | θmax = 25.0°, θmin = 1.2° |
| Tmin = 0.690, Tmax = 0.746 | h = −6→6 |
| 11035 measured reflections | k = −8→9 |
| 2536 independent reflections | l = −39→39 |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.034 | w = 1/[σ2(Fo2) + (0.0215P)2 + 0.3497P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.072 | (Δ/σ)max < 0.001 |
| S = 1.04 | Δρmax = 0.20 e Å−3 |
| 2536 reflections | Δρmin = −0.19 e Å−3 |
| 181 parameters | Absolute structure: Flack x determined using 839 quotients [(I+)-(I-)]/[(I+)+(I-)] (Parsons et al., 2013) |
| 0 restraints | Absolute structure parameter: 0.05 (5) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.53243 (13) | 0.93377 (11) | 0.09181 (3) | 0.0414 (2) | |
| O1 | 0.5825 (3) | 0.9636 (2) | 0.20137 (6) | 0.0362 (5) | |
| O2 | 0.1331 (3) | 1.3885 (3) | 0.14389 (6) | 0.0360 (5) | |
| N1 | 0.8186 (4) | 1.2919 (3) | 0.22791 (7) | 0.0292 (6) | |
| N2 | 0.3211 (4) | 1.1461 (3) | 0.16766 (7) | 0.0268 (5) | |
| C1 | 0.8794 (5) | 1.4552 (4) | 0.23364 (8) | 0.0306 (7) | |
| H1 | 1.0179 | 1.4779 | 0.2498 | 0.037* | |
| C2 | 0.7561 (5) | 1.5929 (4) | 0.21783 (8) | 0.0320 (7) | |
| H2 | 0.8111 | 1.7050 | 0.2233 | 0.038* | |
| C3 | 0.5532 (5) | 1.5678 (3) | 0.19413 (8) | 0.0301 (6) | |
| H3 | 0.4639 | 1.6597 | 0.1829 | 0.036* | |
| C4 | 0.4884 (5) | 1.4006 (3) | 0.18778 (7) | 0.0235 (6) | |
| C5 | 0.6241 (5) | 1.2727 (3) | 0.20487 (8) | 0.0234 (6) | |
| C6 | 0.5174 (5) | 1.1058 (3) | 0.19241 (8) | 0.0262 (6) | |
| C7 | 0.2900 (5) | 1.3212 (4) | 0.16376 (8) | 0.0281 (7) | |
| C8 | 0.1681 (5) | 1.0227 (4) | 0.14678 (9) | 0.0335 (7) | |
| H8A | −0.0035 | 1.0559 | 0.1499 | 0.040* | |
| H8B | 0.1895 | 0.9095 | 0.1594 | 0.040* | |
| C9 | 0.2300 (5) | 1.0119 (4) | 0.10179 (9) | 0.0369 (8) | |
| H9A | 0.2130 | 1.1263 | 0.0896 | 0.044* | |
| H9B | 0.1120 | 0.9361 | 0.0883 | 0.044* | |
| C10 | 0.4745 (5) | 0.7095 (3) | 0.08142 (8) | 0.0278 (6) | |
| H10 | 0.3597 | 0.6645 | 0.1023 | 0.033* | |
| C11 | 0.3645 (5) | 0.6857 (4) | 0.03962 (8) | 0.0318 (7) | |
| H11A | 0.4706 | 0.7395 | 0.0191 | 0.038* | |
| H11B | 0.2057 | 0.7435 | 0.0387 | 0.038* | |
| C12 | 0.3313 (6) | 0.4988 (4) | 0.02908 (10) | 0.0448 (9) | |
| H12A | 0.2117 | 0.4476 | 0.0478 | 0.054* | |
| H12B | 0.2676 | 0.4887 | 0.0011 | 0.054* | |
| C13 | 0.5693 (6) | 0.4026 (4) | 0.03229 (10) | 0.0463 (8) | |
| H13A | 0.5408 | 0.2803 | 0.0271 | 0.056* | |
| H13B | 0.6831 | 0.4450 | 0.0114 | 0.056* | |
| C14 | 0.6795 (6) | 0.4253 (4) | 0.07406 (10) | 0.0438 (8) | |
| H14A | 0.8382 | 0.3672 | 0.0750 | 0.053* | |
| H14B | 0.5734 | 0.3716 | 0.0946 | 0.053* | |
| C15 | 0.7133 (5) | 0.6123 (4) | 0.08458 (9) | 0.0388 (8) | |
| H15A | 0.8329 | 0.6633 | 0.0658 | 0.047* | |
| H15B | 0.7773 | 0.6223 | 0.1126 | 0.047* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0268 (4) | 0.0445 (5) | 0.0529 (5) | −0.0066 (4) | 0.0022 (3) | −0.0203 (4) |
| O1 | 0.0412 (12) | 0.0199 (11) | 0.0474 (12) | 0.0036 (10) | −0.0046 (10) | 0.0019 (10) |
| O2 | 0.0354 (11) | 0.0345 (12) | 0.0380 (11) | 0.0093 (10) | −0.0109 (9) | −0.0026 (10) |
| N1 | 0.0318 (13) | 0.0227 (14) | 0.0332 (13) | 0.0016 (11) | −0.0039 (11) | −0.0014 (11) |
| N2 | 0.0258 (12) | 0.0212 (13) | 0.0333 (13) | −0.0014 (11) | −0.0006 (11) | −0.0047 (10) |
| C1 | 0.0334 (15) | 0.0274 (17) | 0.0310 (16) | 0.0000 (14) | −0.0036 (12) | −0.0047 (13) |
| C2 | 0.0432 (17) | 0.0200 (17) | 0.0329 (16) | −0.0009 (14) | −0.0028 (13) | −0.0022 (13) |
| C3 | 0.0408 (15) | 0.0212 (15) | 0.0282 (14) | 0.0072 (14) | −0.0074 (13) | −0.0001 (12) |
| C4 | 0.0302 (14) | 0.0186 (14) | 0.0217 (13) | 0.0009 (13) | −0.0013 (11) | −0.0016 (11) |
| C5 | 0.0263 (13) | 0.0186 (15) | 0.0252 (14) | 0.0002 (12) | 0.0010 (12) | −0.0010 (12) |
| C6 | 0.0275 (14) | 0.0215 (15) | 0.0297 (14) | 0.0013 (13) | 0.0031 (12) | −0.0012 (12) |
| C7 | 0.0304 (15) | 0.0289 (17) | 0.0251 (14) | 0.0027 (14) | 0.0033 (12) | −0.0035 (13) |
| C8 | 0.0253 (14) | 0.0272 (17) | 0.0478 (18) | −0.0047 (13) | 0.0013 (14) | −0.0099 (14) |
| C9 | 0.0290 (14) | 0.0363 (18) | 0.0455 (18) | 0.0024 (13) | −0.0079 (13) | −0.0159 (14) |
| C10 | 0.0245 (14) | 0.0323 (16) | 0.0265 (14) | −0.0028 (13) | 0.0030 (12) | −0.0022 (12) |
| C11 | 0.0361 (16) | 0.0329 (18) | 0.0265 (15) | 0.0029 (14) | −0.0032 (13) | −0.0027 (13) |
| C12 | 0.0435 (19) | 0.039 (2) | 0.052 (2) | 0.0007 (16) | −0.0084 (16) | −0.0145 (16) |
| C13 | 0.0491 (19) | 0.0342 (19) | 0.056 (2) | 0.0052 (17) | 0.0069 (16) | −0.0118 (16) |
| C14 | 0.0379 (17) | 0.042 (2) | 0.051 (2) | 0.0103 (18) | 0.0047 (15) | 0.0076 (17) |
| C15 | 0.0307 (15) | 0.049 (2) | 0.0372 (17) | 0.0036 (15) | −0.0018 (13) | −0.0023 (16) |
Geometric parameters (Å, º)
| S1—C9 | 1.813 (3) | C8—H8B | 0.9900 |
| S1—C10 | 1.827 (3) | C9—H9A | 0.9900 |
| O1—C6 | 1.212 (3) | C9—H9B | 0.9900 |
| O2—C7 | 1.209 (3) | C10—C11 | 1.516 (4) |
| N1—C5 | 1.325 (3) | C10—C15 | 1.530 (4) |
| N1—C1 | 1.342 (4) | C10—H10 | 1.0000 |
| N2—C6 | 1.394 (3) | C11—C12 | 1.523 (4) |
| N2—C7 | 1.395 (4) | C11—H11A | 0.9900 |
| N2—C8 | 1.460 (3) | C11—H11B | 0.9900 |
| C1—C2 | 1.382 (4) | C12—C13 | 1.523 (4) |
| C1—H1 | 0.9500 | C12—H12A | 0.9900 |
| C2—C3 | 1.381 (4) | C12—H12B | 0.9900 |
| C2—H2 | 0.9500 | C13—C14 | 1.514 (4) |
| C3—C4 | 1.380 (4) | C13—H13A | 0.9900 |
| C3—H3 | 0.9500 | C13—H13B | 0.9900 |
| C4—C5 | 1.376 (4) | C14—C15 | 1.524 (5) |
| C4—C7 | 1.490 (4) | C14—H14A | 0.9900 |
| C5—C6 | 1.497 (4) | C14—H14B | 0.9900 |
| C8—C9 | 1.522 (4) | C15—H15A | 0.9900 |
| C8—H8A | 0.9900 | C15—H15B | 0.9900 |
| C9—S1—C10 | 101.54 (14) | H9A—C9—H9B | 107.7 |
| C5—N1—C1 | 113.2 (2) | C11—C10—C15 | 110.3 (2) |
| C6—N2—C7 | 112.0 (2) | C11—C10—S1 | 111.1 (2) |
| C6—N2—C8 | 125.1 (2) | C15—C10—S1 | 108.6 (2) |
| C7—N2—C8 | 122.8 (2) | C11—C10—H10 | 109.0 |
| N1—C1—C2 | 125.0 (3) | C15—C10—H10 | 109.0 |
| N1—C1—H1 | 117.5 | S1—C10—H10 | 109.0 |
| C2—C1—H1 | 117.5 | C10—C11—C12 | 112.0 (2) |
| C3—C2—C1 | 120.1 (3) | C10—C11—H11A | 109.2 |
| C3—C2—H2 | 120.0 | C12—C11—H11A | 109.2 |
| C1—C2—H2 | 120.0 | C10—C11—H11B | 109.2 |
| C4—C3—C2 | 115.7 (3) | C12—C11—H11B | 109.2 |
| C4—C3—H3 | 122.2 | H11A—C11—H11B | 107.9 |
| C2—C3—H3 | 122.2 | C13—C12—C11 | 111.1 (3) |
| C5—C4—C3 | 119.6 (2) | C13—C12—H12A | 109.4 |
| C5—C4—C7 | 108.2 (2) | C11—C12—H12A | 109.4 |
| C3—C4—C7 | 132.2 (2) | C13—C12—H12B | 109.4 |
| N1—C5—C4 | 126.4 (2) | C11—C12—H12B | 109.4 |
| N1—C5—C6 | 125.3 (2) | H12A—C12—H12B | 108.0 |
| C4—C5—C6 | 108.3 (2) | C14—C13—C12 | 110.6 (3) |
| O1—C6—N2 | 125.7 (2) | C14—C13—H13A | 109.5 |
| O1—C6—C5 | 128.7 (2) | C12—C13—H13A | 109.5 |
| N2—C6—C5 | 105.5 (2) | C14—C13—H13B | 109.5 |
| O2—C7—N2 | 124.8 (3) | C12—C13—H13B | 109.5 |
| O2—C7—C4 | 129.2 (3) | H13A—C13—H13B | 108.1 |
| N2—C7—C4 | 106.0 (2) | C13—C14—C15 | 111.7 (3) |
| N2—C8—C9 | 111.4 (2) | C13—C14—H14A | 109.3 |
| N2—C8—H8A | 109.4 | C15—C14—H14A | 109.3 |
| C9—C8—H8A | 109.4 | C13—C14—H14B | 109.3 |
| N2—C8—H8B | 109.4 | C15—C14—H14B | 109.3 |
| C9—C8—H8B | 109.4 | H14A—C14—H14B | 107.9 |
| H8A—C8—H8B | 108.0 | C14—C15—C10 | 111.2 (3) |
| C8—C9—S1 | 113.7 (2) | C14—C15—H15A | 109.4 |
| C8—C9—H9A | 108.8 | C10—C15—H15A | 109.4 |
| S1—C9—H9A | 108.8 | C14—C15—H15B | 109.4 |
| C8—C9—H9B | 108.8 | C10—C15—H15B | 109.4 |
| S1—C9—H9B | 108.8 | H15A—C15—H15B | 108.0 |
| C5—N1—C1—C2 | 0.3 (4) | C6—N2—C7—C4 | 1.1 (3) |
| N1—C1—C2—C3 | 0.1 (4) | C8—N2—C7—C4 | −177.0 (2) |
| C1—C2—C3—C4 | −0.3 (4) | C5—C4—C7—O2 | −179.3 (3) |
| C2—C3—C4—C5 | 0.2 (4) | C3—C4—C7—O2 | −0.8 (5) |
| C2—C3—C4—C7 | −178.2 (3) | C5—C4—C7—N2 | −0.6 (3) |
| C1—N1—C5—C4 | −0.4 (4) | C3—C4—C7—N2 | 177.9 (3) |
| C1—N1—C5—C6 | 178.4 (3) | C6—N2—C8—C9 | −103.0 (3) |
| C3—C4—C5—N1 | 0.2 (4) | C7—N2—C8—C9 | 74.9 (3) |
| C7—C4—C5—N1 | 179.0 (2) | N2—C8—C9—S1 | 64.2 (3) |
| C3—C4—C5—C6 | −178.8 (2) | C10—S1—C9—C8 | 97.4 (2) |
| C7—C4—C5—C6 | 0.0 (3) | C9—S1—C10—C11 | 75.0 (2) |
| C7—N2—C6—O1 | 178.8 (3) | C9—S1—C10—C15 | −163.5 (2) |
| C8—N2—C6—O1 | −3.1 (4) | C15—C10—C11—C12 | 55.5 (3) |
| C7—N2—C6—C5 | −1.1 (3) | S1—C10—C11—C12 | 175.9 (2) |
| C8—N2—C6—C5 | 176.9 (2) | C10—C11—C12—C13 | −56.0 (4) |
| N1—C5—C6—O1 | 1.7 (4) | C11—C12—C13—C14 | 55.3 (4) |
| C4—C5—C6—O1 | −179.3 (3) | C12—C13—C14—C15 | −55.8 (4) |
| N1—C5—C6—N2 | −178.4 (2) | C13—C14—C15—C10 | 56.0 (3) |
| C4—C5—C6—N2 | 0.6 (3) | C11—C10—C15—C14 | −55.2 (3) |
| C6—N2—C7—O2 | 179.9 (3) | S1—C10—C15—C14 | −177.1 (2) |
| C8—N2—C7—O2 | 1.8 (4) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C2—H2···O1i | 0.95 | 2.50 | 3.119 (3) | 123 |
| C3—H3···O1i | 0.95 | 2.55 | 3.129 (3) | 119 |
Symmetry code: (i) x, y+1, z.
Funding Statement
This work was funded by National Research Foundation of Korea grants 2015R1D1A3A01020410 and 2016R1D1A1B03934376.
References
- Brandenburg, K. (2010). DIAMOND. Crystal Impact GbR, Bonn, Germany.
- Bruker (2014). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Cho, S., Jeon, Y., Lee, S., Kim, J. & Kim, T. H. (2015). Chem. Eur. J. 21, 1439–1443. [DOI] [PubMed]
- Frisch, M. J., et al. (2009). GAUSSIAN09. Gaussian Inc., Wallingford, CT, USA. http://www.gaussian.com.
- Garduño-Beltrán, O., Román-Bravo, P., Medrano, F. & Tlahuext, H. (2009). Acta Cryst. E65, o2581. [DOI] [PMC free article] [PubMed]
- Inoue, S., Shiota, H., Fukumoto, Y. & Chatani, N. (2009). J. Am. Chem. Soc. 131, 6898–6899. [DOI] [PubMed]
- Jeon, Y., Cheon, S., Cho, S., Lee, K. Y., Kim, T. H. & Kim, J. (2014). Cryst. Growth Des. 14, 2105–2109.
- Kang, G., Jeon, Y., Lee, K. Y., Kim, J. & Kim, T. H. (2015). Cryst. Growth Des. 15, 5183–5187.
- Metobo, S. E., Jabri, S. Y., Aktoudianakis, E., Evans, J., Jin, H. & Kim, C. U. (2013). Tetrahedron Lett. 54, 6782–6784.
- Parsons, S., Flack, H. D. & Wagner, T. (2013). Acta Cryst. B69, 249–259. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, New_Global_Publ_Block. DOI: 10.1107/S2056989017012142/hb7685sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989017012142/hb7685Isup2.hkl
CCDC reference: 1570205
Additional supporting information: crystallographic information; 3D view; checkCIF report