

Abstract
Chordomas are primary malignant bone tumors that arise in the axial skeleton, believed to originate from remnants of embryologic notochordal cell rests. Multicentric origin of chordoma is extremely rare. To our literature search, we found only three cases of multicentric chordoma in adults. We report a first case of multicentric chordoma in pediatric age group. A 14-month-old child presented with torticolis and left upper limb monoparesis, imaging showed expansile bony destructive lesion in clivus and dorsal spine simultaneously. The child underwent laminectomy, decompression of cord, excision of lesion, and histopathology was suggestive of chordoma. Pediatric chordomas are aggressive tumors, require multidisciplinary management with maximal safe resection followed by radiotherapy (conventional and/or proton). Even with multidisciplinary management, pediatric chordomas have high morbidity and mortality.
KEYWORDS: Chordoma, multicentric, pediatric
INTRODUCTION
Chordomas are rare malignant tumors arising from the embryonic remnants of primitive notochord. Chordomas are slow growing, locally invasive tumors with high chances of recurrences and low rate of metastasis. Chordoma at single region with subsequent development at distant site due to metastasis is a well-known entity. Multicentric origin of chordoma is extremely rare, whereas metastatic chordomas are known to occur. We suppose multicentric origin because of different foci of chordomas arising at different sites of the vertebral column simultaneously. Here, we present a pediatric case of multicentric chordoma, which is very rare.
CASE REPORT
A 14-month-old child was brought by parents with complaints of torticollis and decreased movements of left upper limb from 3 months, an inability to sit and stand from 1 week. Birth and developmental history was normal. On examination, the child was conscious, identifying parents, active, playful, restricted neck movements+, power: 5/5 in all limbs except left upper limb monoparesis Grade 3/5, and bilateral plantars were withdrawals. On evaluation, his magnetic resonance imaging (MRI) spine showed an expansile mass lesion involving the clivus with T1/T2 isointense signal and few T2 hyperintense areas. The lesion showed postero-inferior extension to involve the C1, C2 vertebrae with an associated extradural soft-tissue component. Another extradural soft-tissue mass was also noted in the dorsal spine extending from D3 to D5 level causing significant cord compression. Collapse of D4 vertebral body was noted with posterior cortical bulge and diffusely altered marrow signal intensity [Figure 1a–d]. On contrast, lesion showed heterogeneous enhancement of extradural lesions, clivus, C2 and D4 vertebral bodies [Figure 2a and b]. Positron emission tomography (PET) scan showed lytic clivus lesion with diffuse increased tracer uptake in extradural soft-tissue lesion in the upper dorsal spine and D4 vertebral body [Figure 3a and b]. It was reported as multifocal bony expansile lesions with large soft-tissue component features in favor of neoplastic/infiltrative lesions.
Figure 1.
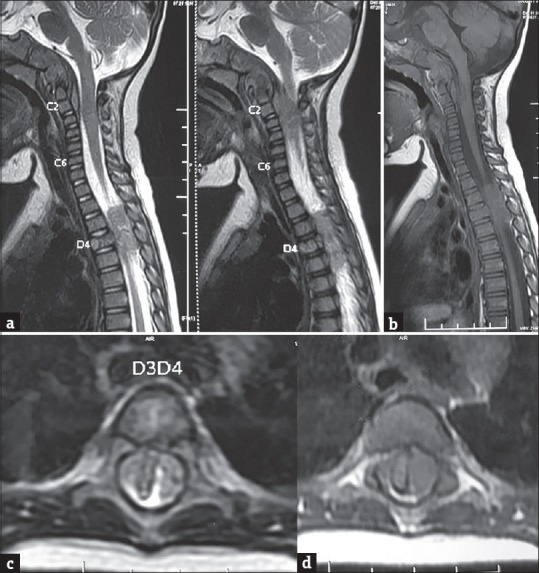
(a) Sagittal T2-weighted magnetic resonance images showing an isointense soft-tissue mass with few hyperintense areas involving the clivus with destruction. Postero-inferior extension into the C1, C2 vertebrae with an associated extradural component. An extradural soft-tissue mass is also noted at D3-D5 level. Collapse of D4 vertebral body is noted with altered signal intensity. (b) Sagittal T1-weighted magnetic resonance image showing the mass to be isointense. Axial T2W (c) and T1W (d) MR image shows an extradural soft tissue mass at D3-D4 level. Cord has been sandwiched between the mass on either side at same level
Figure 2.
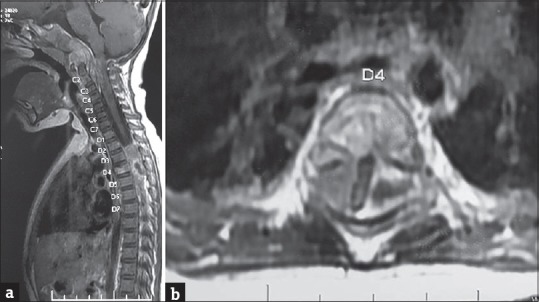
(a) Sagittal T1-weighted magnetic resonance image with gadolinium contrast showing heterogeneous enhancement of the clival and extradural lesions. Also note the heterogeneous enhancement of C2, D4 vertebral bodies. (b) Axial T1-weighted magnetic resonance image with gadolinium contrast showing heterogeneous enhancement of the D4 vertebral body, extradural soft-tissue component. Note the compressed cord
Figure 3.
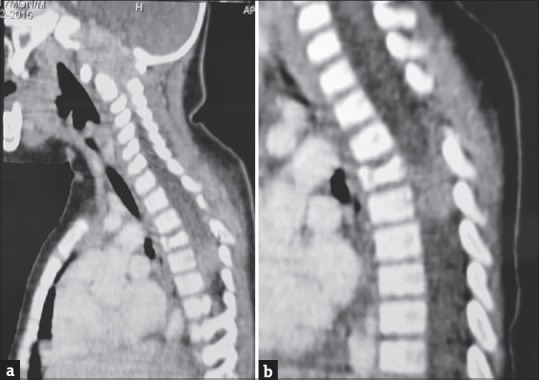
Postlaminectomy status sagittal contrast computed tomography images of cervical (a) and dorsal (b) spine showing a heterogeneously enhancing soft-tissue mass causing expansion and destruction of the clivus, a lobulated enhancing extradural mass at D4–D6 level. Collapse of D4 vertebral body is noted
The child underwent D4 spine-sparing hemilaminectomy, excision of tumor, and decompression of cord under general anesthesia. Bone marrow aspiration was done at the same setting. Intraoperatively, the lesion was soft, vascular, and suckable. Dura was free from the lesion. Postoperative period was uneventful and neurologically same. Histopathological examination [Figure 4a] showed polygonal cells with vacuolated cytoplasm (physalipherous cells) over chondroid background suggestive of chondroid chordoma. On immunohistochemistry, tumor was strongly positive for cytokeratin, vimentin, epithelial membrane antigen [Figure 4b–d], and S100. Tumor tissue had high proliferative index (50%–60%). Bone marrow cytology showed normal myeloid and erythroid differentiation. Radiotherapy was deferred in view of very young age and potential complications. He was started on chemotherapy with vincristine, doxorubicin, and cyclophosphamide. His interval MRI done [Figure 5] after 1 month showed collapse of D4 vertebral body, altered marrow signal in the D5 vertebral body, new epidural lesion at D5, D6 levels compressing the cord, and no significant increase in clival lesion.
Figure 4.
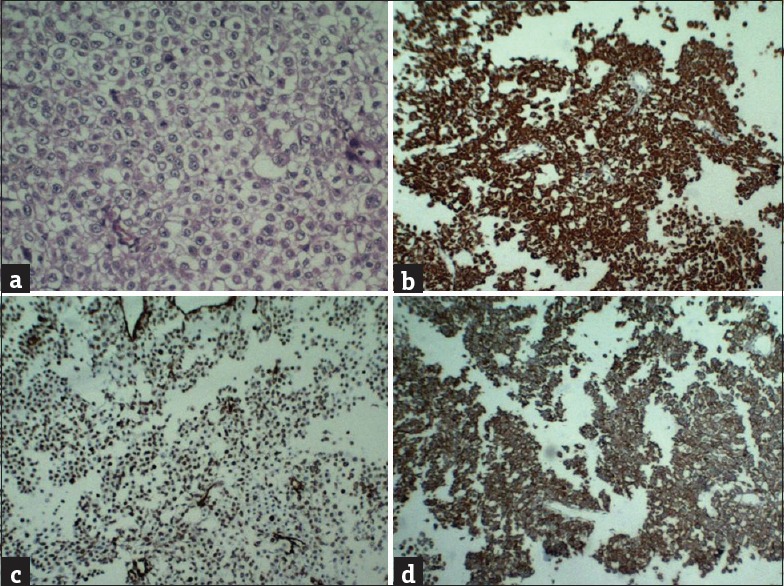
(a) Histopathological examination showing polygonal cells with vacuolated cytoplasm (physalipherous cells) over chondroid background, suggestive of chondroid chordoma. On immuno histochemistry, tumor was strongly positive for cytokeratin, vimentin and epithelial membrane antigen (b-d)
Figure 5.
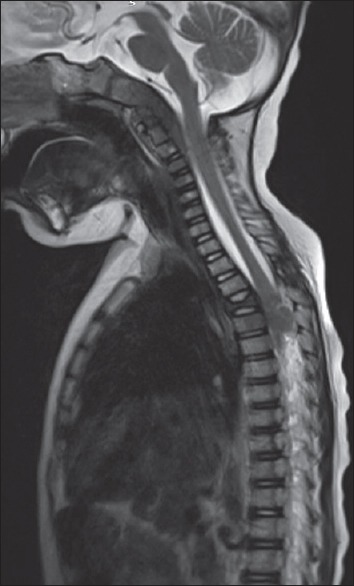
Sagittal T2-weighted magnetic resonance image showing mild decrease in bulk of the clival lesion with increase in the extent of the extradural component extending up to C4 level. A lobulated extradural soft-tissue mass is noted at D4–D7 level. Collapse of D4 vertebral body is noted with altered marrow signal intensity and bulging of the posterior cortex of the D5 vertebral body
The child gradually developed quadriparesis and abnormal breathing pattern. His repeat imaging showed [Figure 6a–d] significant increase in lesion with involvement of multiple vertebral bodies in cervical and dorsal region. The patient was initially managed with oxygen mask ventilation later requiring intubation and ventilation. The patient had an episode of bradycardia and cardiorespiratory arrest for which cardiopulmonary resuscitation was done and revived. At present, the child is on ventilator, opening eye spontaneously, with paucity of movement in limbs.
Figure 6.
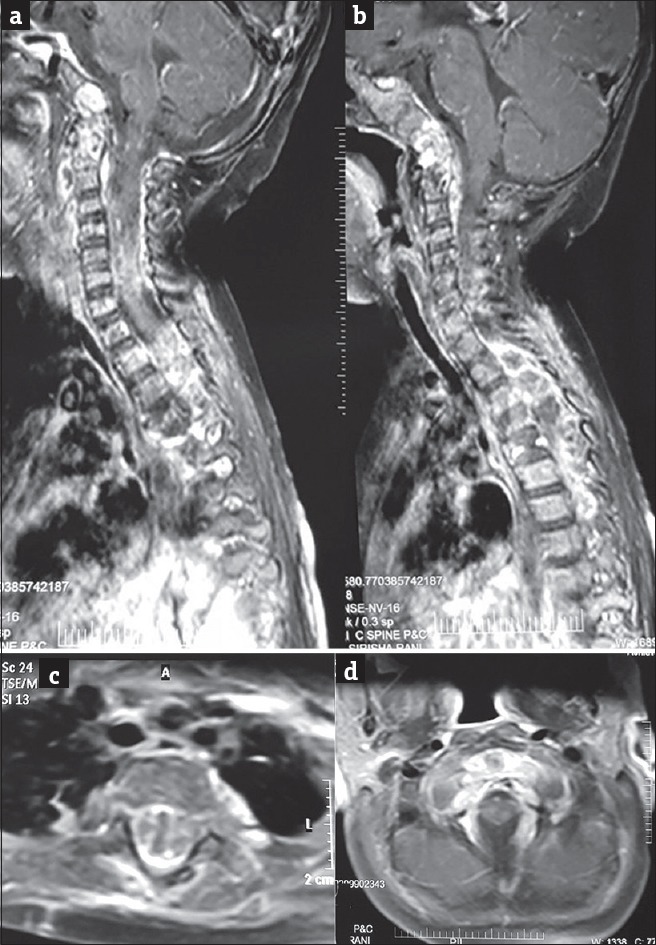
(a and b) Sagittal T1W MR with gadolinium contrast shows heterogeneous multilevel, extensive vertebral, dural enhancement. Enhancement of the clival lesion, extradural lesions in the cervical and dorsal spine seen. (c and d) Axial T1W MR with gadolinium images at dorsal (c) and cervical (d) levels show heterogeneous enhancement of the D4 vertebral body and extradural soft tissue component which is significantly compressing cord. No cord compression is seen at the cervical level
DISCUSSION
Pediatric chordomas are rare; <5% of chordomas present in the first two decades.[1] The youngest case described was in a neonate with a tumor of the clivus.[2] Around 54% of pediatric chordomas are intracranial and characteristically centered on the spheno-occipital synchondrosis. Characteristic feature of pediatric chordoma is localized destruction of clivus with extradural compression of neuroaxis.[3]
Children <5 years usually present with features of raised intracranial pressure, long tract signs, lower cranial nerve palsy, and torticolis whereas older children usually present with diplopia or isolated headache. Sacrococcygeal lesion usually presents with perineal pain, radiculopathy, or weakness of limbs, and cauda equine symptoms such as bowel and bladder disturbances. Vertebral and paravertebral lesions usually present with compression of cord.[4]
Chordomas are malignant entities due to their local invasiveness, high recurrence rate, and potential for metastasis. Most common sites of metastasis include the lymph nodes, lung, liver, brain, and bone. Metastasis along the neuroaxis is extremely rare. The incidence of metastasis in chordoma ranges from 5% to 43%.[5] PET scan should be done to rule out metastasis at distant site. In our case, PET scan at initial presentation ruled out metastasis and active lesions at distant site other than clivus and dorsal spine.
Chordomas usually show epidural space and pre- and para-vertebral regions. Chordomas can cause dural compression, meningeal sheath invasion, and cerebrospinal fluid spreading. Multicentric origin of chordoma is extremely rare. There are no specific criteria for differentiating multicentric from metastatic tumors.[6] Chordoma usually involves vertebral body sparing intervertebral disc. Posterior elements are rarely involved.
In our case, the patient had clival and dorsal lesions at presentation. The child underwent spine-sparing laminectomy and decompression at D4 level. Chordoma had been pathologically confirmed at dorsal spine. Metastatic spread of chordomas along the neuroaxis is rare; if it occurs, there should be widely disseminated disease. In our case, at presentation, each lesion was independent of neoplasms without extensive spread as confirmed by PET scan. Hence, multicentric origin of chordoma has been proposed. Follow-up MRI showed extensive involvement of cervical spine. To our literature search, we found only three cases of multicentric chordoma in adults.[6,7,8] We report the first case of multicentric chordoma in pediatric age group.
Management of chordoma is multidisciplinary. Given high risk of recurrence, it should be aggressively managed.[9] Chordomas are usually treated with maximal safe reduction followed by radiotherapy either conventional or proton radiotherapy. Chemotherapy has been tried by some authors but with disappointing results.[10]
Ridenour showed better survival in children with complete excision versus incomplete excision without statistical significance.[11] Complete excision is possible only in minority of cases due to location of lesion, complexity of their extension, and proximity to neural structures (brain stem, cranial nerves, and sacral nerves) or vascular structures. Even though radiotherapy is currently an integral part of adult chordomas management, issues regarding both the timing and optimal type of radiotherapy in pediatric chordoma are unresolved.[12] Given the significant complications of radiotherapy in growing children, proton therapy is considered a modality of choice for pediatric chordomas as dose requirement decreases by 2–3 times.[13]
The overall survival rate in the major pediatric series in literature ranges from 56.8% to 81%.[11,14,15] Major prognostic factors are age of the child, location of the tumor, and histological type of the tumor. Children <5 years of age have a poor prognosis. Intracranial location of tumor has a good prognosis when compared to sacral and spinal lesions. Atypical and undifferentiated forms have a worst outcome when compared to classic and chondroid chordomas. The majority of children <5 years of age reported expired within 18 months of diagnosis in spite of surgery, radiotherapy, and/or chemotherapy.[16]
CONCLUSION
Chordomas in pediatric age group are rare and aggressive. Pediatric chordomas have more local invasiveness, high recurrence rate, and potential for metastasis. Even though rare, chordomas should also be considered in the basket of differential diagnosis when multiple neuroaxis is involved in pediatric age group. Multidisciplinary management is the key as they have high morbidity and mortality.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Borba LA, Al-Mefty O, Mrak RE, Suen J. Cranial chordomas in children and adolescents. J Neurosurg. 1996;84:584–91. doi: 10.3171/jns.1996.84.4.0584. [DOI] [PubMed] [Google Scholar]
- 2.Probst EN, Zanella FE, Vortmeyer AO. Congenital clivus chordoma. AJNR Am J Neuroradiol. 1993;14:537–9. [PMC free article] [PubMed] [Google Scholar]
- 3.Coffin CM, Swanson PE, Wick MR, Dehner LP. Chordoma in childhood and adolescence. A clinicopathologic analysis of 12 cases. Arch Pathol Lab Med. 1993;117:927–33. [PubMed] [Google Scholar]
- 4.Matsumoto J, Towbin RB, Ball WS., Jr Cranial chordomas in infancy and childhood. A report of two cases and review of the literature. Pediatr Radiol. 1989;20:28–32. doi: 10.1007/BF02010629. [DOI] [PubMed] [Google Scholar]
- 5.Lountzis NI, Hogarty MD, Kim HJ, Junkins-Hopkins JM. Cutaneous metastatic chordoma with concomitant tuberous sclerosis. J Am Acad Dermatol. 2006;55(2 Suppl):S6–10. doi: 10.1016/j.jaad.2005.08.061. [DOI] [PubMed] [Google Scholar]
- 6.Lim JJ, Kim SH, Cho KH, Yoon DH, Kim SH. Chordomas involving multiple neuraxial bones. J Korean Neurosurg Soc. 2009;45:35–8. doi: 10.3340/jkns.2009.45.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson WB, Meyers HI. Multicentric chordoma. Report of a case. Cancer. 1968;21:126–8. doi: 10.1002/1097-0142(196801)21:1<126::aid-cncr2820210120>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 8.Aydin AL, Sasani M, Oktenoglu T, Solaroglu I, Ozer AF. A case of chordoma invading multiple neuraxial bones: Report of ten years follow up. Turk Neurosurg. 2013;23:551–6. doi: 10.5137/1019-5149.JTN.5666-11.2. [DOI] [PubMed] [Google Scholar]
- 9.Carpentier A, Polivka M, Blanquet A, Lot G, George B. Suboccipital and cervical chordomas: The value of aggressive treatment at first presentation of the disease. J Neurosurg. 2002;97:1070–7. doi: 10.3171/jns.2002.97.5.1070. [DOI] [PubMed] [Google Scholar]
- 10.Dhall G, Traverso M, Finlay JL, Shane L, Gonzalez-Gomez I, Jubran R. The role of chemotherapy in pediatric clival chordomas. J Neurooncol. 2011;103:657–62. doi: 10.1007/s11060-010-0441-0. [DOI] [PubMed] [Google Scholar]
- 11.Ridenour RV, 3rd, Ahrens WA, Folpe AL, Miller DV. Clinical and histopathologic features of chordomas in children and young adults. Pediatr Dev Pathol. 2010;13:9–17. doi: 10.2350/09-01-0584.1. [DOI] [PubMed] [Google Scholar]
- 12.Di Maio S, Yip S, Al Zhrani GA, Alotaibi FE, Al Turki A, Kong E, et al. Novel targeted therapies in chordoma: An update. Ther Clin Risk Manag. 2015;11:873–83. doi: 10.2147/TCRM.S50526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rombi B, Ares C, Hug EB, Schneider R, Goitein G, Staab A, et al. Spot-scanning proton radiation therapy for pediatric chordoma and chondrosarcoma: Clinical outcome of 26 patients treated at Paul Scherrer Institute. Int J Radiat Oncol Biol Phys. 2013;86:578–84. doi: 10.1016/j.ijrobp.2013.02.026. [DOI] [PubMed] [Google Scholar]
- 14.Benk V, Liebsch NJ, Munzenrider JE, Efird J, McManus P, Suit H. Base of skull and cervical spine chordomas in children treated by high-dose irradiation. Int J Radiat Oncol Biol Phys. 1995;31:577–81. doi: 10.1016/0360-3016(94)00395-2. [DOI] [PubMed] [Google Scholar]
- 15.Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE. Base of skull chordomas in children and adolescents: A clinicopathologic study of 73 cases. Am J Surg Pathol. 2006;30:811–8. doi: 10.1097/01.pas.0000209828.39477.ab. [DOI] [PubMed] [Google Scholar]
- 16.Beccaria K, Sainte-Rose C, Zerah M, Puget S. Paediatric chordomas. Orphanet J Rare Dis. 2015;10:116. doi: 10.1186/s13023-015-0340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]