

Abstract
Dopa-responsive dystonia also known as “Segawa's syndrome” was first described in 1976. The dystonia typically shows diurnal variations and is more marked toward the end of the day and improves in sleep. This entity is often misdiagnosed in the clinical setting, mostly due to the lack of awareness, and these patients are exposed to various treatment regimens and nonpharmacological measures. We present a boy being treated as dystonic cerebral palsy who showed significant improvement in dystonic symptoms with L-dopa therapy.
KEYWORDS: Dopa-responsive dystonia, GTP cyclohydrolase 1, L-dopa therapy
INTRODUCTION
Segawa et al. first reported the entity of dopa-responsive dystonia (DRD) in 1976 when they described two cousins with gait disturbance and dystonia with the onset in the first decade. Their symptoms showed diurnal variations with worsening in the evening and significant improvement with L-dopa treatment.[1] Since then, there have been many reports describing the typical and atypical features of DRD. We report a 14-year-old boy with DRD misdiagnosed as dystonic cerebral palsy and emphasize the importance of recognition of this entity in young onset dystonias.
CASE REPORT
A 14-year-old boy, third born of a nonconsanguineous parentage at term with normal development, had walking difficulty from 5 years of age. It started with an abnormal lower limb posture and limping followed by neck and limb posturing with truncal deviation to one side while walking. With time, the abnormality persisted at rest and worsened on activity. There was mild diurnal variation and sleep improved his symptoms considerably. He was evaluated for the complaints elsewhere and was being treated as dystonic cerebral palsy with trihexyphenidyl and physiotherapy.
Examination revealed a mentally normal boy with no Kayser–Fleischer rings or hepatosplenomegaly. He had generalized dystonia involving the neck, trunk, and upper limbs at rest [Figure 1a] with worsening on gait examination and additional lower limb dystonia on walking. His routine blood investigations were normal. In addition, peripheral smear examination for acanthocytes, serum ceruloplasmin levels, and thyroid tests were within normal limits. Magnetic resonance imaging brain did not show any abnormality. Generalized dystonia at this young age with diurnal variations and no other abnormality led us to suspect DRD, and a trial of L-dopa was given; after 3 days of therapy, his dystonia completely improved [Figure 1b]. He is on regular follow-up and maintaining improvement while on low-dose L-dopa therapy.
Figure 1.
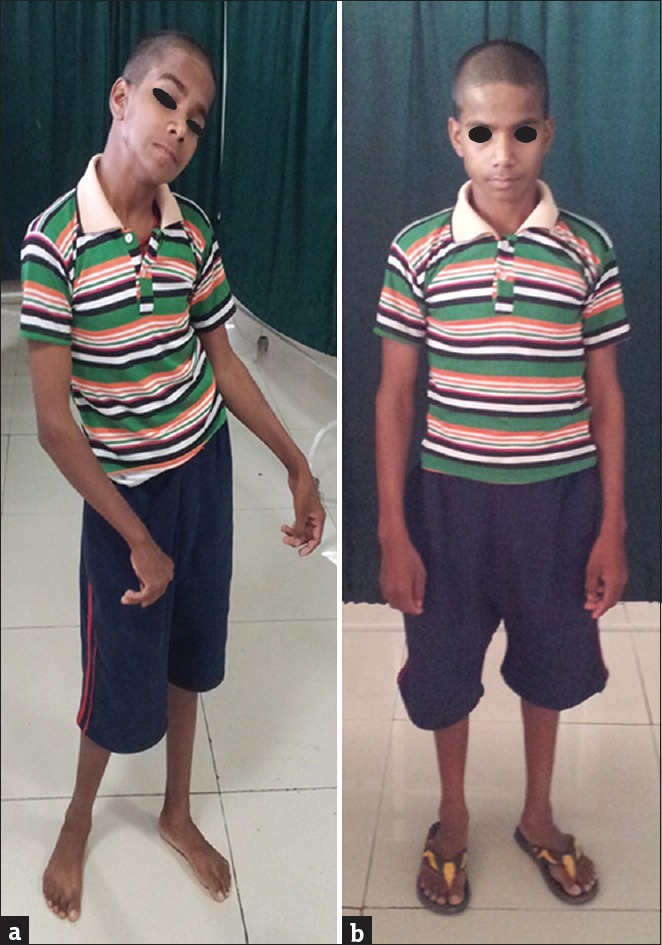
(a) Generalized dystonia involving the neck, trunk, upper, and lower limbs at rest. (b) Complete disappearance of dystonic posturing after L-dopa trial
DISCUSSION
DRD typically presents around 4–6 years of age with a slight female preponderance. The dystonia commonly starts in lower extremities and gradually becomes generalized. The hallmark of the disease is marked diurnal variations, improvement with sleep, and a dramatic response to low-dose L-dopa without motor fluctuations or dyskinesias.[2] With the increasing recognition of this not so uncommon entity, the age at presentation may vary, and in fact, Furuya et al. have described the onset of DRD in a patient at 46 years age.[3] Atypical presentations include early onset in infancy, psychomotor retardation, convulsions, systemic symptoms, and cerebellar dysfunction.[2] DRD may be misdiagnosed as cerebral palsy, as was the case in our scenario, particularly with no other symptomatology except dystonia. Important differentials that need to be considered in these individuals are young onset Parkinsonism, Wilson's disease, idiopathic torsion dystonia, and other genetic/hereditary dystonias of childhood.[4] DRD is inherited in an autosomal dominant fashion though recessive inheritance is also seen in some cases. The enzyme responsible for DRD is GTP cyclohydrolase 1 (GCH), which is a rate-limiting enzyme in the synthesis of dopamine. Tyrosine hydroxylase makes dopamine from tyrosine using tetrahydrobiopterin (BH4) as a cofactor. GCH takes the initial step in the synthesis of BH4. The defect in GCH activity causes decreased dopamine synthesis and is responsible for the symptoms in DRD.[3,4] Our case highlights the often seen delay in diagnosing DRD, and these patients are usually subjected to unnecessary interventions and vague therapies. Tadic et al. have reported an average delay of 13–15 years in the diagnosis of DRD, thus emphasizing the need to have a high index of suspicion in detecting DRD.[5]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Segawa M, Hosaka A, Miyagawa F, Nomura Y, Imai H. Hereditary progressive dystonia with diurnal fluctuations. In: Eldrige R, Fahn S, editors. Advances in Neurology. New York: Raven Press; 1976. pp. 215–53. [PubMed] [Google Scholar]
- 2.Lee WW, Jeon BS. Clinical spectrum of dopa-responsive dystonia and related disorders. Curr Neurol Neurosci Rep. 2014;14:461. doi: 10.1007/s11910-014-0461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Furuya H, Murai H, Takasugi K, Ohyagi Y, Urano F, Kishi T, et al. Acase of late-onset Segawa syndrome (autosomal dominant dopa-responsive dystonia) with a novel mutation of the GTP-cyclohydrase I (GCH1) gene. Clin Neurol Neurosurg. 2006;108:784–6. doi: 10.1016/j.clineuro.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Mittal R, Goraya JS, Basu S. Dopa-responsive dystonia. Indian Pediatr. 2001;38:1056–8. [PubMed] [Google Scholar]
- 5.Tadic V, Kasten M, Brüggemann N, Stiller S, Hagenah J, Klein C. Dopa-responsive dystonia revisited: Diagnostic delay, residual signs, and nonmotor signs. Arch Neurol. 2012;69:1558–62. doi: 10.1001/archneurol.2012.574. [DOI] [PubMed] [Google Scholar]