

SUMMARY
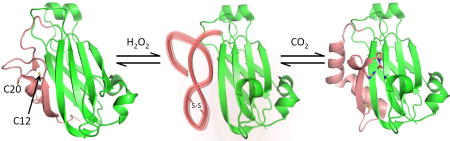
XRCC1, a scaffold protein involved in DNA repair, contains an N-terminal domain (X1NTD) that interacts specifically with DNA polymerase β. It was recently discovered that X1NTD contains a disulfide switch that allows it to adopt either of two metamorphic structures. In the present study we demonstrate that formation of an N-terminal proline carbimate adduct resulting from the non-enzymatic reaction of Pro2 with CO2 is essential for stabilizing the oxidized structure, X1NTDox. The kinetic response of X1NTDred to H2O2, monitored by NMR was determined to be very slow, consistent with involvement of the buried, kinetically trapped Cys12 residue, but was significantly accelerated by addition of protein disulfide isomerase or by Cu2+. NMR analysis of a sample containing the pol β polymerase domain, and both the reduced and oxidized forms of X1NTD indicates that the oxidized form binds to the enzyme 25-fold more tightly than the reduced form.
Keywords: XRCC1, DNA pol beta, oxidative stress, disulfide switch, NMR spectroscopy, carbimate adduct
Graphical abstract
INTRODUCTION
Over the past two decades, multiple examples of metamorphic proteins have been identified for which the relative abundance of the alternately folded states is governed by a redox-sensitive disulfide switch (Fan et al., 2009; Linke and Jakob, 2003; Paget and Buttner, 2003). These metamorphic changes typically involve significant structural remodeling that is much more extensive than the conformational responses characteristic of most proteins. The disulfide switch enhances the ability of the cell to respond to variations in cellular redox conditions that may be associated with cell cycle changes (Sarsour et al., 2009) or result from oxidative stress (Linke and Jakob, 2003; Paulsen and Carroll, 2010). Since DNA damage represents an ongoing process, the maintenance of genomic integrity requires that DNA repair activity be continuous. However, under more extreme conditions of genomic stress, the cell can upregulate some of these repair activities. For example, expression of the DNA repair scaffold X-ray cross complementing group 1 protein (XRCC1), which is involved in the overlapping base excision repair and single strand break repair pathways (Caldecott, 2003; Kubota et al., 1996), is reported to be upregulated during S phase (Jin et al., 2011) and in response to oxidative DNA damage (Quach et al., 2005). Sensitivity to changes in oxidation potential does not necessarily require that XRCC1 itself contain a redox switch and could, for example, result from increased expression levels or as a result of interactions with other redox-sensitive proteins; however, the recent discovery that the XRCC1 N-terminal domain (X1NTD) contains a redox-sensitive disulfide switch (Cuneo and London, 2010) suggests that additional regulation occurs at the structural level. A functional role for the X1NTD disulfide switch was suggested by the higher affinity of the oxidized domain, X1NTDox, for its binding partner, DNA pol β, consistent with the possibility that oxidative stress can result in upregulation of the XRCC1-pol β binding affinity. Further, recent studies of mouse fibroblasts containing the redox-insensitive analog XRCC1(C12A) not capable of forming the disulfide bond exhibited an altered response to the alkylating agent MMS (Horton et al., 2013).
The two structures determined for the XRCC1 N-terminal domain (X1NTD) in complex with the pol β palm/thumb reveal extensive remodeling of the N-terminal 40 residues of the protein, so that the N-terminal β-strand switches from an anti-parallel to a parallel interaction with β-strand 2, and there is a significant increase in helical content (Cuneo and London, 2010). However, these initial studies showed that, in contrast with other model proteins such as OxyR (Lee et al., 2004), addition of hydrogen peroxide is not sufficient to produce a stable X1NTDox structure. As described below, it became clear that an additional, non-enzymatic modification at the N-terminus was also involved. In the present study, we demonstrate that stability of X1NTDox is dependent on the presence of sufficient CO2/bicarbonate to allow formation of an N-terminal carbimate adduct with Pro2. The effects of other factors on the kinetics of the redox transition have also been evaluated, with the acceleration produced by protein disulfide isomerase consistent with the buried location of Cys12 in the reduced X1NTD.
RESULTS
Identification of an N-terminal carbimate adduct
In the crystal structure obtained for the complex formed between the reduced form of the XRCC1 N-terminal domain, X1NTDred and the polymerase domain of DNA pol β (PD), the N-terminal residue is Pro2 (pdb code: 3K75) (Cuneo and London, 2010), consistent with removal of Met1 by methionine aminopeptidase present in the E. coli expression system. However, the electron density in the N-terminus of X1NTD apparent in the complex formed between the oxidized form, X1NTDox and DNA pol β palm/thumb subdomains indicated additional structure preceding Pro2 and this was initially modeled by including an N-terminal methionine residue. Further structural analysis suggested that this might not be correct, since the region near the N-terminal methionine contained three closely positioned electropositive groups arising from Met1, Arg7, and Lys129. This analysis also indicated that electron density initially assigned to the Met1 sidechain was more likely to arise from water molecules. The unexplained additional electron density at the N-terminus was then tentatively assigned to an N-terminal carbimate adduct, formed as a consequence of the non-enzymatic reaction of the N-terminal Pro2 residue with adventitious CO2 (pdb code: 3LQC(Cuneo and London, 2010)).
For X1NTDox, addition of an N-terminal carboxyl group provides a good fit to the electron density, and results in more favorable electrostatic and H-bond interactions with Arg7 and Lys129 (Figure 1) as well as an H-bond interaction with Ser44. In order to verify this possibility, we prepared U-[15N]X1NTD, and converted it to the oxidized form by addition of H2O2 in the presence of 5 mM 13C-bicarbonate. The 1D 13C spectrum shows both a singlet (δ13C = 160.75 ppm) arising from the bicarbonate, and a doublet at δ13C = 162.6 ppm with 1JCN = 19 Hz arising from the adduct with the N-terminal proline (Figure 2A). Since proline is an imino acid, this adduct is more properly referred to as a carbimate, and is to the best of our knowledge, the first example of a carbimate adduct identified in a biological system. The in vitro CO2 adducts of various amino acids including proline have previously been studied by NMR (Dettman et al., 1985; Morrow et al., 1974; Myers and Nelson, 1990; Sherry et al., 1990).
Figure 1.

Identification of an oxidized X1NTD post-translational modification. (A) Expanded view of the N-terminus of the oxidized XRCC1-NTD with methionine modeled as the N-terminal residue. 2FoFc electron density contoured at 1.0 is shown for the modeled Met1 (A), and for the modeled carbimate adduct (B). FoFc electron density contoured at 3 is shown for the CO2 group.
Figure 2.

NMR characterization of the N-terminal carbimate adduct. (A) 1D direct 13C detection of 13C-bicarbonate (5 mM) in the presence of U-[15N]X1NTD after addition of 5 mM H2O2. Inset is a close-up view of the coupling between the 15N of the N-terminal Pro2 and the 13C of the carbimate. (B) Annotated 1H-15N HSQC spectra of U-[15N]X1NTDox (blue spectrum), overlayed with the spectrum of U-[15N]X1NTDred (red spectrum). Assignments of the reduced form were in good agreement with those determined previously by Marintchev et al. (BMRB accession No. 4282) (Marintchev et al., 1999), but are not shown. Spectra were run at 25 °C in 25 mM Tris-d11, pH 7.6 (uncorrected), 140 mM NaCl, 75 mM NaHCO3, 0.25 mM sodium azide in 90%/10% H2O/D2O.
We therefore conclude that the N-terminal residue of the X1NTD in both the reduced and oxidized forms is Pro2, Met1 having been removed by the bacterial methionine aminopeptidase. The oxidized form of X1NTD is further stabilized by formation of an N-terminal carbimate adduct formed from adventitious CO2. Further studies, some of which are described below, have indicated that the oxidized form of X1NTD is considerably less stable in the absence of CO2/dissolved bicarbonate, exhibiting spectral characteristics that are inconsistent with a stable, folded conformation.
Characterization of oxidized X1NTD in solution
The oxidized form of X1NTD (X1NTDox), containing a Cys12-Cys20 disulfide bond, was previously characterized in complex with the palm/thumb subdomains of DNA pol β (Cuneo and London, 2010). NMR studies were designed to characterize X1NTDox in solution without its pol β binding partner. Based on the importance of N-terminal carbimate adduct formation for stabilization of the X1NTDox, we prepared U-[13C,15N]X1NTD, oxidized it using H2O2 in the presence of bicarbonate, and then assigned the backbone resonances. An overlay of the 1H-15N HSQC spectra of the oxidized and reduced forms is shown in Figure 2B. Consistent with expectations based on the two crystal structures (pdb codes: 3K75, 3LQC), the spectra differ very significantly, particularly for resonances corresponding to the N-terminal 40 or so residues of the protein. Extreme amide 1H shifts are observed for Ser17 and Ser75, which form double H-bond interactions with the carboxylate groups of Glu53 and Asp126, respectively. The amide shifts for Ser17 in the X1NTDox (δ1H,15N = 10.24, 117.9), relative to the shifts in X1NTDred, (8.04,115.9) (BMRB code: 4282 (Marintchev et al., 1999)), provide confirmation that a large structural change has occurred which is at least similar to that observed in the crystal structure of the complex.
The metamorphic transition of X1NTD involves substantial variation of the secondary structure of the N-terminal 40 residues of the protein (Cuneo and London, 2010), and it was expected that if the isolated X1NTDox undergoes a similar structural transformation, these changes should be apparent from analysis of the chemical shift changes. First, the chemical shifts of the Cys12 and Cys20 Cβ carbons change from 28.7 to 37.3 and from 29.7 to 39.5 ppm, respectively, which are characteristic of disulfide formation in the oxidized state (Sharma and Rajarathnam, 2000). Second, in the reduced form, the only helical segment in the N-terminal 40 residues is a single turn helix involving residues 22–24. An analysis of the backbone shift data for X1NTDox with Talos+ (Shen et al., 2009) predicts that residues 17–24 and 30–36 exhibit backbone chemical shifts characteristic of alpha helices. This result agrees well with the positions of the two α-helical segments that are observed in the N-terminal 40 residues of X1NTDox in complex with the pol β palm/thumb. Assignments for residues 28 and 29, which are also helical in the crystal structure, are missing, most probably as a consequence of conformational exchange. Third, the phi and psi angles derived from the X1NTDox NMR data for residues 7–37 agrees much more closely with the values obtained from the crystal structure of the X1NTDox-pol β complex than with the values obtained from the X1NTDred•pol β complex (Supplemental Figure S1). In summary, the chemical shift analysis is consistent with disulfide formation and with secondary structure that is essentially identical with that observed in the crystal structure of the X1NTDox:pol β complex. This structure can be characterized by NMR if sufficient bicarbonate is present to support formation of the N-terminal carbimate adduct.
Stabilization of X1NTDox by carbimate adduct formation
The role of the carbimate adduct in stabilizing the structure of X1NTDox can be elucidated by removing the bicarbonate buffer from a sample of U-[15N]X1NTDox. The resulting 1H-15N HSQC spectrum shows significantly fewer resolved resonances, and these exhibit a more uneven intensity distribution (Figure 3A). Although there is some increase in the intensity of random coil resonances particularly near δ(1H,15N)=8.3,120 ppm, the resonance characteristics are consistent with the conclusion that portions of the protein remain well folded while other regions are experiencing motion on a micro-millisecond time scale, resulting in resonance broadening. In sharp contrast with this result, the addition of bicarbonate to a sample of U-[15N]X1NTDred resulted in no significant changes in the 1H-15N HSQC spectrum (Supplemental Figure S2).
Figure 3.

Effects of bicarbonate on the 1H-15N HSQC spectrum of X1NTD. A) 1H-15N HSQC spectrum obtained for U-[15N]X1NTDox after removal of bicarbonate from the buffer. B) Overlay of the spectrum in A (black) with the spectrum of U-[15N]X1NTDox obtained in the presence of bicarbonate (blue) and with the spectrum of X1NTDred (red). Well-resolved resonances in the blue spectrum that correspond to residues in the N-terminal segment of the protein are annotated, and it is seen that nearly all of these are no longer observed after removal of bicarbonate. The buffer also contained 50 mM Tris-d11, pH 7.0, 140 mM NaCl. Sample temperature was 25 °C.
An overlay of the spectrum shown in Figure 3A with the spectra corresponding to both X1NTDred and X1NTDox-CO2, the latter obtained in the presence of bicarbonate (Figure 3B), indicates limited agreement with either spectrum. Well-resolved resonances that correspond to residues in the N-terminal segment of X1NTDox in the bicarbonate buffer are annotated, and it is clear that these are not present in the spectrum of X1NTDox after removal of the bicarbonate. In general, the strongest resonances correspond to residues that have similar shifts in both the reduced and oxidized species. Overall, the spectrum shown in Figure 3A appears somewhat more similar to that of X1NTDred, as seen most directly from the region at low δ15N shifts that corresponds primarily to glycine residues. This result indicates that the perturbations experienced by residues 41–155 due to the metamorphic changes in the N-terminal 40 residues are largely, although incompletely, removed when these residues adopt a less defined, more dynamic structure.
Monitoring the transition to the oxidized state
As is apparent from Figure 2B, there are multiple amide resonances that could serve as monitors of the fraction of reduced vs. oxidized X1NTD. However, the upfield shifted methyl resonance of Val72 provides a relatively convenient probe that combines the high methyl group sensitivity with the ease of a 1D assay. The upfield 1H region X1NTD spectrum corresponding to three different degrees of oxidation is shown in Figure 4. This spectral region includes the Ile43 Hδ resonance of the reduced X1NTD (−0.58 ppm), the reduced Val72 methyl resonance (−0.66 ppm), and the oxidized Val72 γ2 methyl resonance (−0.74 ppm). In X1NTDred, the Ile43 sidechain is sandwiched between residues Trp33 and Phe142. As the sample becomes oxidized, these residues move away from Ile43, which becomes positioned near Arg34 resulting in a large, downfield shift. Alternatively, the Val72, which is positioned near Trp125, undergoes only a subtle change of environment upon oxidation, resulting in a small upfield shift of −0.08 ppm. The two Val72 resonances thus provide a direct readout of the oxidized fraction of the X1NTD. Further, since Val72 is not positioned in the region of major structural remodeling, the relaxation properties are expected to be very similar, facilitating a quantitative comparison of resonance intensities. Time constants were determined from the resonance intensities as described in Experimental Procedures.
Figure 4.

Sensitivity of upfield 1H resonances to redox state. The region of the 1D 1H NMR spectrum of X1NTD containing the Ile43 Cδ and Val72 Cγ2 methyl resonances: a) reduced X1NTD; b) partially oxidized X1NTD; c) primarily oxidized X1NTD. D) Typical series of spectra used for the analysis of time-dependent oxidation or reduction. Upon formation of X1NTDox, the Ile43 resonance is shifted out of the spectral region of interest, while the upfield-shifted Val72 methyl resonance undergoes a small additional upfield shift of ~ 0.08 ppm. The time under each spectrum corresponds to the completion of the accumulation period after the initial addition of 500 µM H2O2. The sample contained 25 µM X1NTD in 25 mM Tris-d11, pH 7.6, 140 mM NaCl, 75 mM NaHCO3, 0.5 mM EDTA, 0.25 mM sodium azide in D2O. Each spectrum corresponds to a 30 m accumulation; T = 30 °C.
In order to study how various parameters affected the oxidation rate, experimental conditions were selected to allow a slow, time-dependent oxidation after addition of 500 µM H2O2 to 25 µM X1NTD. The studies were performed at 30 °C in 50 mM Tris-d11, pH*=7.6 (uncorrected pH meter reading in D2O), 140 mM NaCl, 0.5 mM EDTA, 0.25 mM NaN3, 75 mM bicarbonate. D2O was used for optimal sensitivity of the 1H assay. In the presence of bicarbonate there was always evidence of 5 – 10 % of X1NTDox prior to the addition of H2O2, Thus, dissolved oxygen is sufficient to produce low levels of X1NTDox if bicarbonate is present to stabilize this form of the protein. Representative studies are shown in Figure 5, and the results summarized in Table 1. In general, the accuracy of the rate determinations was limited by two factors: 1) incomplete resolution of the three Ile43red, Val72ox and Val72red resonances (Figure 4), and 2) strong sensitivity of the rate constant to pH variations and transition metal ion contaminants. The oxidation rate increases rapidly with pH as a function of the greater reactivity of ionized cysteine. Several transition metal ions were found to increase the oxidation rate, with Cu2+ having the greatest effect (Table 1 and Figure 5D), with measurable effects on the oxidation rate at 10 nM, and the sample fully oxidized prior to the first accumulation period in the presence of 10 µM Cu2+.
Figure 5.

Time-dependent oxidation of X1NTD by H2O2. The fraction of oxidized X1NTD, fox, determined from the Val72 methyl resonance using Equation 1, is plotted as a function of time after the addition of H2O2. The traces correspond to: A) wt X1NTD (TC = 7.6 h); B) X1NTD(H8Y) (TC = 5.9 h); C) X1NTD plus 100 nM CuCl2 (TC = 3.0 h); D) X1NTD plus 5.8 µM PDI (TC = 1.9 h). For the sample shown in panel C, the initial fiox = 0.36, for the remaining samples, fiox < 0.1. The mean time constants are reported in Table 1. All samples contained 25 µM X1NTD in 50 mM Tris-d11, pH 7.6, 140 mM NaCl, 0.5 mM EDTA, 0.25 mM NaN3, and 75 mM bicarbonate. Experiments were run at 30 °C on a Varian INOVA 600.
Table 1.
NMR-derived kinetic parameters
| Protein | Fitted parametersa | Mean TCb | Other conditions | |
|---|---|---|---|---|
| fiOx | TC h | |||
| X1NTD | .08 | 6.33 | Standard bufferc | |
| .08 | 7.09 | |||
| .08 | 9.8 | |||
| .08 | 6.72 | 7.49±0.79 | ||
| H8Y | 0.8 | 4.31 | ||
| .08 | 6.90 | |||
| .08 | 6.85 | |||
| .08 | 5.81 | |||
| .08 | 5.46 | 5.87±0.48 | ||
| S17T | .16 | 9.92 | ||
| .12 | 10.33 | |||
| .12 | 11.32 | |||
| .15 | 11.68 | 10.81±0.41 | ||
| Y30W | .20 | 8.43 | ||
| .16 | 9.84 | |||
| .25 | 10.12 | 9.46±0.91 | ||
| X1NTD | .08 | 7.80 | 25 mM HCO3− | |
| .08 | 9.20 | |||
| wt | .08 | 5.92 | 20% D2O | |
| wt | 0.17 | 4.55 | 10 nM CuCl2 | |
| 0.36 | 3.03 | 100 nM CuCl2 | ||
| 1.0 | - | 10 µM CuCl2 | ||
| wt | 0.18 | 2.99 | 2.9 µM PDI | |
| 0.15 | 1.88 | 5.8 µM PDI | ||
| 0.06 | 0.70 | 29 µM PDI | ||
Data for the wt human X1NTD and for each of the four mutants were fit setting fiOx = 0.08, as discussed in methods. Time constants are given in hours. PDI concentration calculated using MW = 57 kD.
Includes standard error calculation.
Unless otherwise indicated studies were done in 25 mM Tris-d11, pH 7.6 (uncorrected), 140mM NaCl, 75mM NaHCO3, 0.5 mM EDTA, 0.25 mM sodium azide in D2O.
We evaluated the effects of several mutations, including C20R, H8Y, S17T and Y30W. The first substitution replaces Cys20 with an arginine residue, as is found in rat XRCC1 (Supplemental Table S1). The NMR spectra confirm that this form of the protein does not adopt the oxidized structure. The H8Y substitution was based on the XRCC1 sequence in O. Orca (Supplemental Table S1), and was predicted to favor the oxidized form due to the greater solvent exposure of residue 8 in the reduced form. The H8Y substitution does appear to result in a small enhancement of the oxidation rate. The S17T and Y30W mutations were selected based on molecular modeling. Unfortunately, the resolution of the upfield-shifted Val72 methyl resonances were degraded in the spectra of both of these mutants. This resulted in apparently greater values for fiOx, but lower rate constants. The accuracy of both of these values is significantly compromised by the poorer resolution characteristics of the mutants, so we conclude only that the mutations produce no major change.
Based on the above studies, the rate of the redox transition would likely be too slow to provide a response to conditions of oxidative stress. However, as also shown in Table 1, the oxidation rate was significantly enhanced in the presence of either Cu2+ ions or protein disulfide isomerase (PDI). In both cases, the rate enhancement was dependent on the concentration of these agents, consistent with a catalytic effect. For Cu2+ ions, there was a significant increase in the fraction of oxidized X1NTD, fiox, prior to the addition of H2O2, indicating that Cu2+ is able to catalyze formation of the disulfide species using dissolved oxygen (Figure 5C). In the presence of 10 µM Cu2+, the sample was found to be fully oxidized prior to the addition of H2O2, while addition of 10 nM Cu2+ decreased the time constant by almost a factor of 2, compared with the rate measured in the presence of EDTA. The strong catalytic effect observed for PDI is consistent with its role in catalyzing disulfide bond formation from solvent-inaccessible cysteine residues, such as the Cys12 residue of XRCC1 (pdb: 3K75, 3K77).
Effect of X1NTD oxidation/reduction on intrinsic fluorescence
The fluorescence emission spectrum of X1NTD is strongly dependent on the redox state of the protein (Figure 6). Transformation from the reduced to the oxidized state results in quenching of 60% of the fluorescence signal, and in a shift of the fluorescence emission maximum from 344 to 354 nm (Figure 6A). This effect was largely, although incompletely reversed by addition of 0.5 mM DTT (Figure 6B). This effect is consistent with the significant change in environment of Trp33, which in the oxidized state is positioned next to His8. Fluorescence quenching can result from electron transfer to the protonated imidazole sidechain.(McMillan et al., 2013; Shinitzky and Goldman, 1967)
Figure 6.

Dependence of intrinsice fluorescence of X1NTD on redox state. The fluorescence emission spectrum of 2 µM X1NTD is shown as a function of time: A) Oxidation of X1NTDred. 0.5 mM H2O2 was added at t = 0, and spectra were recorded at 30 m intervals. A fully oxidized spectrum was obtained by addition of excess H2O2 after 4 h. or B) reduction of X1NTDox after addition of 0.5 mM DTT. Spectra were recorded at 15 m intervals, as indicated. The buffer for both the oxidation and reduction studies was 50 mM Tris-HCl, pH 7.65, 140 mM NaCl, 75 mM sodium bicarbonate, 0.1 mM EDTA. Spectra were run at 25 °C.
Ratio of pol β binding affinity for X1NTDred/X1NTDox
As shown previously, pol β has a higher affinity for X1NTDox than for X1NTDred, consistent with expectations based on structural data for the reduced and oxidized complexes (Cuneo and London, 2010). Since the previous determination utilized several potentially perturbing modifications of both pol β and X1NTD, we utilized NMR to obtain a more accurate value for the dissociation constant ratio. Although the sensitivity of the NMR method limits the range of KD values that can be directly determined, accurate ratios, such as that for KDβ•X1red/KDβ•X1ox, can be determined by comparison of four resonance intensities that correspond to the uncomplexed and pol β-complexed X1NTDox and X1NTDred species (Levy et al., 1996). As outlined in Methods, the desired ratio can be expressed as:
The measurements shown in Figure 7 utilized a sample containing a mixture of the pol β polymerase as well as both reduced and oxidized X1NTD: PD(V303M):X1NTDred:X1NTDox. The need for utilizing the PD(V303M) mutation arose because we were unable to identify a single set of well resolved resonances in the 1H-15N HSQC spectra of X1NTD that showed sufficient sensitivity to both the redox status of X1NTD and to PD binding. The positions of the PD M303 resonances were determined directly from studies of apo pol β, pol β•X1NTDred, and pol β•X1NTDox (Supplemental Figure S3). In the spectrum of the polymerase domain, an additional intense methionine resonance arising from the N-terminal tag overlaps the Met303 resonance of uncomplexed pol β, however resonances from the uncomplexed PD do not appear in Eq. 2. The proportions of the three components were initially set to 1:1:1. However, the resonances for uncomplexed X1NTDox and complexed X1NTDred were very weak and difficult to measure accurately, so that the final mixture utilized a ratio of 1:1.44:0.8. The ratio KDβ •X1red/KDβ •X1ox Based on the observations of the M303 methyl resonances in the PD, and of Ser57, Gly64, Gly73, Ser75, and Gly143 amide resonances in X1NTD (Supplemental Table S2), we obtained a mean ratio KDβ•X1red:KDβ•X1ox = 25.2.
Figure 7.

Relative affinities of reduced and oxidized X1NTD for pol β. NMR spectra were obtained on a sample containing 250 µM [13CH3-Met]PD(V303M), 360 µM U[15N]X1NTDred, and 200 µM U-[15N]X1NTDox, corresponding to a ratio of 1.0:1.44:0.8. A) 1H-13C HSQC of spectral region showing the methionine methyl resonances of [13CH3-Met]PD(V303M); B) 1H-15N HSQC spectrum of the region containing the Ser75 resonances of X1NTD. The spectrum shown in panel A does not include the M282 resonance, Met18 is not present in the PD construct, but an additional, intense methionine resonance arising from the His-tag is observed near the random coil position and overlaps the resonance of uncomplexed [13CH3-Met]PD(V303M). The sample was in 20 mM phosphate, pH 7.5, 75 mM NaHCO3, 70 mM NaCl, 80 µM DSS and the studies performed at T=25°C.
DISCUSSION
The N-terminal domain of XRCC1 contains a disulfide switch that can allow it to respond to changes in cellular oxidation potential (Cuneo and London, 2010). In the present study, we determined that: 1) a stable, folded X1NTDox can exist in solution in the absence of its pol β binding partner, but requires sufficient CO2/bicarbonate to support formation of an N-terminal carbimate adduct with Pro2; 2) in the absence of catalytic factors such as Cu2+ or PDI, the transition from the reduced to the oxidzed state is an extremely slow process, occurring on a time scale of hours; 3) the transition rate is subject to significant catalytic acceleration by protein disulfide isomerase, and can be directly monitored by NMR or fluorescence measurements. The transition rate is significantly increased in the presence of Cu2+ ions, which also lead to formation of the X1NTDox even in the absence of added hydrogen peroxide, consistent with the reported ability of Cu2+ to catalyze oxygen-dependent oxidation of cysteine to cysteine(Cavallini et al., 1969). The effect of PDI is consistent with formation of the Cys12-Cys20 dislufide bond from an initially buried Cys12 residue. Although disulfide isomerase is generally considered to function primarily in facilitating disulfide isomerization reactions, the enzyme has been shown to play a critical role in facilitating disulfide bond formation in proteins containing inaccessible, kinetically trapped cysteine residues (Walker and Gilbert, 1995; Weissman and Kim, 1993). The disulfide isomerase ERp57 has been reported to exist in the nucleus (Coppari et al., 2002), and Grillo et al. (Grillo et al., 2006) have reported an association of this enzyme with AP-endonuclease. In view of the important role of APE1 in BER, the redox function may also be important as a regulator of the XRCC1 disulfide switch. APE1 appears to act as a mediator transmitting the redox status of the nucleus to transcription factors and other nuclear proteins (Bhakat et al., 2009; Luo et al., 2008).
The previous structural comparison of the pol β interface with X1NTDred and X1NTDox suggested a somewhat higher binding affinity of the oxidized form and this prediction was verified using a fluorescent assay (Cuneo and London, 2010). This initial study utilized an X1NTD(I4D) mutant, a pol β(P300C) analog containing a fluorophore located at the binding interface, as well as an undetermined concentration of CO2/bicarbonate. In the present study, we have utilized NMR to determine the relative binding affinities on a mixture containing approximately equal concentrations of pol β polymerase domain (PD), X1NTDox, and X1NTDred. This study confirms the earlier conclusion and provides a more accurate value for the KdβX1red/KDβX1dox = 25. Previous determinations of the KdβX1red gave values of 0.3 µM (Marintchev et al., 2003) and 0.1 µM (Cuneo and London, 2010). These values indicate binding sufficient to allow pol β-dependent affinity capture of XRCC1 and XRCC1-complexed BER proteins (Prasad et al., 2012), but probably insufficient to result in constitutive complex formation. Thus, the redox-dependent gain in affinity is probably sufficient to result in an increased level of binding that is physiologically significant. As a result of the greater affinity of pol β for X1NTDox, the ratio of the oxidized/reduced forms will probably be enhanced in the presence of nuclear pol β. Importantly, we note that the initial crystal structure of the pol β:X1NTDox complex was obtained from a solution containing no added bicarbonate, indicating that low levels of bicarbonate are sufficient to support the formation of X1NTDox in the presence of pol β. Alternatively, the Cys12 and Cys20 residues are a commonly but not universally present in the reported XRCC1 sequences (Supplemental Table S1). This could reflect a variety of factors, e.g. variations in X1NTD binding affinity resulting from other residue differences or related to differences in the nuclear concentrations of pol β or XRCC1. Interestingly, one of the species that lacks Cys20, D. rerio, also is reported to lack a redox-active APE1 (Georgiadis et al., 2008).
The reaction of biological amines with carbon dioxide is probably the most important non-enzymatic reaction in cells, and plays a central role in plant CO2 fixation by RUBISCO (Hartman and Harpel, 1994; O'Leary et al., 1979), CO2 transport by hemoglobin (Morrow et al., 1976), in formation of metal ion binding sites in enzymes such as urease (Jabri et al., 1995), in the mechanisms of some enzymes (Leonard et al., 2013), and in the formation of neutrotoxins (Myers and Nelson, 1990) among other functions. Although protein N-terminal amino groups represent relatively favorable cases for formation of carbamate adducts due to the depressed pK value of ~ 7.5, relatively few examples of protein N-terminal carbamate adducts have been studied (Fauman et al., 1994; Griffey et al., 1988; Morrow et al., 1974; Morrow et al., 1976; Roberts et al., 2006; Rothgeb et al., 1978; Wittebort et al., 1978). Presumably, some degree of N-terminal carbamate adduct formation is present in all proteins studied under physiological conditions for which the N-terminal amino group is not structurally inaccessible. Bacterial thymidylate synthase (Fauman et al., 1994; Roberts et al., 2006) and X1NTDox appear to be the only proteins in this group characterized by a sufficient favorable set of interactions to select the N-terminal carba(i)mate species as the predominant form of the protein. Although it is conceivable that the sensitivity to dissolved CO2 or pH has regulatory significance, in our view it is more probable that the adduct just makes use of available CO2 to achieve a desired structure, much as a Zn2+ finger incorporates available Zn2+ ions as a useful structural element.
The response of X1NTD to H2O2 differs dramatically from that of the bacterial transcription factor OxyR, which requires only seconds to become oxidized (Lee et al., 2004). Based on the slower kinetics and dependence on PDI catalysis, the X1NTD redox transition likely represents a slower adaptation to sustained changes in the nuclear redox environment. Furthermore, reduction of X1NTDox was also found to be a sluggish process. The DTT reduction shown in Figure 6 transpired over a few hours, and other reducing agents produced even slower results (data not shown). In combination with the significantly greater affinity of DNA pol β for X1NTDox, these results suggest that once formed, the pol β•X1NTDox complex may persist for significant periods of time. This conclusion is supported by recently reported mass spectrometric studies, which evaluated the extent of intracellular XRCC1 oxidation based on the ability of the disulfide bond to protect the cysteine residues from reaction with iodoacetamide (Horton et al., 2013). This analysis indicated that approximately half of the intracellular X1NTD contains the Cys12-Cys20 disulfide bond. Although this result is somewhat surprising given the reducing environment of the cell, we believe that greater stability of X1NTDox in complex with pol β, combined with the stabilizing effect of the carbimate adduct, provide a basis for the significant intracellular accumulation of X1NTDox. The greater tendency to form X1NTDox in the presence of pol β is also indicated by the initial crystallization of this species as a pol β complex utilizing only the available dissolved CO2, as no bicarbonate was added to the buffer (Cuneo and London, 2010).
Horton et al. recently evaluated XRCC1−/− mouse fibroblast cells expressing an oxidation-blocked form of XRCC1, the C12A mutant (Horton et al., 2013). Among other deficiencies, over-accumulation of cellular poly(ADP-ribose) after DNA base damage was consistent with a disruption of the interaction between pol β, XRCC1, and PARP-1. These results are consistent with functionally significant oxidation of intra-nuclear XRCC1. Blocking formation of X1NTDox with the C12A mutation would restrict the N-terminal domain to the lower affinity X1NTDred structure, which may be insufficient to fully support the function of the XRCC1 repair complex. Regardless of the basis for these effects, these results are consistent with a significant role for XRCC1 oxidation in the cellular response to DNA damage. The present study reveals the conditions under which the oxidized form of the X1NTD can be prepared and studied, and so should provide a basis for clarification of the role of the XRCC1 disulfide switch.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise indicated, chemicals were obtained from Sigma-Aldrich, Inc. [methyl-13C]-L-methionine was obtained from CIL, Cambridge, MA. Bovine hepatic protein disulfide isomerase (PDI), 100–400 units/mg, was obtained as a lyophilized powder, and used without further purification. The concentration of H2O2 was determined spectrophotometrically (Tietjen and Mancott, 1971).
Cloning, Expression, Purification
The N-terminal domain of human XRCC1 (X1NTD) consisted of amino acids 1–155 with an added hexa-histidine C-terminal tag. This construct was cloned into a pET-21a plasmid. Three additional mutated constructs (H8Y, S17T and Y30W) were produced using QuickChange II XL site directed mutagenesis kit (Agilent).
Plasmids were transformed into BL21-RIL(DE3) cells. Cells were grown in 2×YT media to an OD~0.6, induced with 1mM IPTG, then expressed overnight at 20°C. Cells were then pelleted, re-suspended in buffer (150 mM NaCl, 20mM imidazole, 20 mM Tris pH 7.5, protease inhibitor cocktail tablet (Roche)) and lysed by sonication. Samples were centrifuged, the lysate was loaded onto a Ni-NTA resin column, then eluted using a stepped imidazole gradient (20, 75, 400 mM). The protein was further purified using a Superdex 26/60 S75 preparative grade gel filtration column (GE Amersham). For 15N-labeled NTD, the protein was expressed using M9 minimal media containing 15N-labeled NH4Cl and supplemented with 15N Bioexpress cell growth media (Cambridge Isotope Laboratories).
The pol β polymerase domain (PD) used for the Kd determinations consisted of amino acids 88–335 with an N-terminal His-tag of sequence: MGSSHHHHHHSSGLVPRGSHM preceding residue 88, and a V303M mutation produced using Quikchange II XL site directed mutagenesis kit (Agilent). The pET28-aderived expression vector generates a polypeptide with two more methionine residues than the native PD, one of which is not processed during expression and contributes to the observed 1H-13C HMQC NMR spectrum of the 13C methionine-labeled domain. The plasmid was transformed into BL21-RIL(DE3) and [methyl-13C]methionine-labeled PD was expressed by growth of the cells on PAG medium. containing [methyl-13C]methionine to an OD~1.0, followed by addition of IPTG to 1 mM and shaking overnight at 25°C (Studier, 2005). The purification protocol was identical to that used for X1NTD.
Assignment of X1NTDox
Standard NMR assignment methods were used to assign the backbone resonances of the XRCC1NTDox. The backbone chemical shifts were assigned from a combined analysis of HNCA (Ikura et al., 1990), HNCACB (Wittekind and Mueller, 1993), and CBCA(CO)NH spectra (Grzesiek and Bax, 1992), C(CO)NH(Grzesiek et al., 1993) spectra. The 3D spectra were acquired at 25 °C on a Varian Inova 600, equipped with a 1H/13C/15N Varian Coldprobe, using Agilent’s BioPack versions of the triple resonance experiments. At 25°C and high concentrations, the oxidized samples precipitate slowly. In order to deal with this behavior, each triple resonance experiment, requiring 20–40 hour acquisitions was collected on successive, freshly prepared samples. The 2D 15N-1H HSQC spectra were compared prior to data acquisition to insure consistency from sample to sample. All spectra were processed using NMRPipe (Delaglio et al., 1995), and the chemical shift assignments were made using the RunAbout module within NMRView (One Moon Scientific, Westfield NJ) (Johnson and Blevins, 1994).
Fluorescence Measurements
Fluorescence emission spectra were obtained on a Fluorolog-3 fluorometer (Horiba). The excitation wavelength was set at 280 nm, while emission wavelength was scanned from 290–400 nm. The X1NTD concentration was 2 µM in a buffer containing 50 mM Tris-HCl, pH 7.6, 140 mM NaCl, 75 mM sodium bicarbonate, 0.1 mM EDTA. Studies were run at 25°C.
1H NMR kinetic studies
Kinetic data were determined from observation of the upfield shifted 1H resonances of Val72 Cγ2 corresponding to the reduced (δ1H = −0.66 ppm) and oxidized (δ1H = −0.74 ppm) species. Time-dependent spectra, typically in 30 m increments, were run on a Varian Unity Inova 600 MHz spectrometer with a 5 mm triple resonance probe. Temperature was set to 30°C. Unless otherwise indicated, NMR studies were performed on 25 µM X1NTD in the NMR buffer: 25 mM Tris-d11, pH 7.6, 140 mM NaCl, 75 mM NaHCO3, 0.5 mM EDTA, 0.25 mM sodium azide in D2O. Oxidation was initiated by addition of 0.5 mM H2O2.
Kinetic data were fit to an exponential function using either two parameters, corresponding to the time constant and initial fraction of oxidized species, or using a fixed value for :
| [1] |
The resonances of the reduced and oxidized species were not baseline resolved at 14.1 T (600 MHz 1H frequency), and a careful examination of the spectra revealed that most of the variation in the initial fraction of oxidized X1NTD resulted from incomplete resolution rather than from differences in the fractions of X1NTDox present prior to the addition of H2O2. Furthermore, a further loss of resolution was found to characterize two of the mutations studied: S17T and Y30W, making these data even more subject to error. A slight improvement in the consistency of the results was obtained unless the data clearly indicated a significant deviation from this value, e.g. the studies of Cu2+ oxidation.
KD ratio determination
NMR provides an extremely useful basis for the determination of KD ratios when the four resonances corresponding to the free and bound forms of both ligands can be observed simultaneously (Levy et al., 1996). Dissociation constants of pol β with the reduced and oxidized forms of X1NTD were defined by:
Elimination of [pol β] from the two equations and rearranging terms gives:
| [2] |
An important feature of this approach is that nearly all of the different relaxation characteristics of the smaller uncomplexed X1NTD and the larger pol β•X1NTD complexes will cancel out when the ratios used in the above equation are determined. The results also are not dependent on the exact ratios of the three species: pol β, X1NTDred and X1NTDox in the mixture, although it is necessary to select ratios that produce reasonable signal/noise ratios for the four resonances that are measured. Typically, ratios near 1:1:1 are optimal. We were unable to identify isolated resonances in X1NTD sensitive to both pol β binding and redox status, so that we used sequential measurements on a sample containing a methionine-labeled polymerase domain, [13CH3-Met]PD(V303M), and U-[15N]-labeled X1NTD. The V303M mutation was introduced based on the location of the methionine ε-methyl group at the pol β-X1NTD binding interface, and proximity to Pro2 in X1NTDox, so that shift sensitivity to both binding and X1NTD redox status could be achieved. The perturbation resulting from this conservative substitution is expected to be minimal. Several amide resonances of the U-[15N]X1NTD were used to evaluate the ratio of uncomplexed reduced and oxidized X1NTD, since resonances arising from the complexed species were severely broadened.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Jason Williams for input related to the mass spectrometric characterization of XRCC1, and to Dr. Sam Wilson and Dr. Natalie Gassman for helpful comments on this manuscript.
Funding: This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences under Research Project Number Z01-ES050111 to R.E.L. E.F.D. is supported by the National Institutes of Health, NIEHS, under Delivery Order HHSN273200700046U.
Abbreviations
- pol β
DNA polymerase β
- PD
DNA polymerase β polymerase domain
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- XRCC1
X-ray cross complementing group 1 protein
- X1NTD
XRCC1 N—terminal domain = XRCC1(1–155)
- X1NTDox
oxidized form of X1NTD containing the C12–C20 disulfide bond
- X1NTDred
reduced form of X1NTD with reduced cysteine residues
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Information. Supplemental Material includes Tables showing sequence comparisons of the N-terminal XRCC1 cysteine-containing sequence (Table S1), resonance intensities determined in the study of the pol β PD, X1NTDred, and X1NTDox mixture (Table S2), a Figure demonstrating the better consistency of the NMR-derived ϕ values with the oxidized crystal structure (Figure S1), a Figure demonstrating the lack of an effect of bicarbonate on the 1H-15N HSQC spectrum of X1NTDred (Figure S2), and figures supporting the assignments of the M303 resonances in X1NTDred- and X1NTDox-complexes of pol β(V303M) (Figure S3).
References
- Bhakat KK, Mantha AK, Mitra S. Transcriptional Regulatory Functions of Mammalian AP-Endonuclease (APE1/Ref-1), an Essential Multifunctional Protein. Antioxid Redox Sign. 2009;11:621–637. doi: 10.1089/ars.2008.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair. 2003;2:955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- Cavallini D, Demarco C, Dupre S, Rotilio G. Copper Catalyzed Oxidation of Cysteine to Cystine. Archives of Biochemistry and Biophysics. 1969;130:354–361. doi: 10.1016/0003-9861(69)90044-7. [DOI] [PubMed] [Google Scholar]
- Coppari S, Altieri F, Ferraro A, Chichiarelli S, Eufemi M, Turano C. Nuclear localization and DNA interaction of protein disulfide isomerase ERp57 in mammalian cells. Journal of Cellular Biochemistry. 2002;85:325–333. doi: 10.1002/jcb.10137. [DOI] [PubMed] [Google Scholar]
- Cuneo MJ, London RE. Oxidation state of the XRCC1 N-terminal domain regulates DNA polymerase beta binding affinity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6805–6810. doi: 10.1073/pnas.0914077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. Journal of Biomolecular Nmr. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dettman HD, Weiner JH, Sykes BD. A F-19 Nuclear Magnetic-Resonance Study of the Interaction of Carbon-Dioxide with Fluoro-Amino Acids. Can J Biochem Cell B. 1985;63:1120–1126. [Google Scholar]
- Fan SW, George RA, Haworth NL, Feng LL, Liu JY, Wouters MA. Conformational changes in redox pairs of protein structures. Protein Science. 2009;18:1745–1765. doi: 10.1002/pro.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauman EB, Rutenber EE, Maley GF, Maley F, Stroud RM. Water-Mediated Substrate/Product Discrimination - the Product Complex of Thymidylate Synthase at 1.83-Angstrom. Biochemistry. 1994;33:1502–1511. doi: 10.1021/bi00172a029. [DOI] [PubMed] [Google Scholar]
- Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffey RH, Scavini M, Eaton RP. Characterization of the Carbamino Adducts of Insulin. Biophys J. 1988;54:295–300. doi: 10.1016/S0006-3495(88)82959-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo C, D'Ambrosio C, Scaloni A, Maceroni M, Merluzzi S, Turano C, Altieri F. Cooperative activity of Ref-1/APE and Erp57 in reductive activation of transcription factors. Free Radical Bio Med. 2006;41:1113–1123. doi: 10.1016/j.freeradbiomed.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Anglister J, Bax A. Correlation of Backbone Amide and Aliphatic Side-Chain Resonances in C-13/N-15-Enriched Proteins by Isotropic Mixing of C-13 Magnetization. J Magn Reson Ser B. 1993;101:114–119. [Google Scholar]
- Grzesiek S, Bax A. Correlating Backbone Amide and Side-Chain Resonances in Larger Proteins by Multiple Relayed Triple Resonance Nmr. Journal of the American Chemical Society. 1992;114:6291–6293. [Google Scholar]
- Hartman FC, Harpel MR. Structure, Function, Regulation, and Assembly of D-Ribulose-1,5-Bisphosphatecarboxylase Oxygenase. Annual Review of Biochemistry. 1994;63:197–234. doi: 10.1146/annurev.bi.63.070194.001213. [DOI] [PubMed] [Google Scholar]
- Horton JK, Stefanick DF, Gassman NR, Williams JG, Gabel SA, Cuneo MJ, Prasad R, Kedar PS, Derose EF, Hou EW, et al. Preventing oxidation of cellular XRCC1 affects PARP-mediated DNA damage responses. DNA Repair (Amst) 2013;12:774–785. doi: 10.1016/j.dnarep.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M, Kay LE, Bax A. A Novel-Approach for Sequential Assignment of H-1, C-13, and N-15 Spectra of Larger Proteins - Heteronuclear Triple-Resonance 3-Dimensional Nmr-Spectroscopy - Application to Calmodulin. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- Jabri E, Carr MB, Hausinger RP, Karplus PA. The Crystal-Structure of Urease from Klebsiella-Aerogenes. Science. 1995;268:998–1004. [PubMed] [Google Scholar]
- Jin RH, Sun Y, Qi XD, Zhang HH, Zhang YL, Li N, Ding W, Chen DX. E2F1 is involved in DNA single-strand break repair through cell-cycle-dependent upregulation of XRCC1 expression. DNA Repair. 2011;10:926–933. doi: 10.1016/j.dnarep.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. Nmr View - a Computer-Program for the Visualization and Analysis of Nmr Data. Journal of Biomolecular Nmr. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase beta and the XRCC1 protein. Embo Journal. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- Lee CJ, Lee SM, Mukhopadhyay P, Kim SJ, Lee SC, Ahn WS, Yu MH, Storz G, Ryu SE. Redox regulation of OxyR requires specific disulfide bond formation involving a rapid kinetic reaction path. Nature Structural & Molecular Biology. 2004;11:1179–1185. doi: 10.1038/nsmb856. [DOI] [PubMed] [Google Scholar]
- Leonard DA, Bonomo RA, Powers RA. Class D beta-Lactamases: A Reappraisal after Five Decades. Accounts Chem Res. 2013;46:2407–2415. doi: 10.1021/ar300327a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LA, Gabel SA, London RE. Synthesis and characterization of two improved NMR indicators for cytosolic Ca2+:3FBAPTA and 35FBAPTA. Magn Reson Chem. 1996;34:440–446. [Google Scholar]
- Linke K, Jakob U. Not every disulfide lasts forever: Disulfide bond formation as a redox switch. Antioxid Redox Sign. 2003;5:425–434. doi: 10.1089/152308603768295168. [DOI] [PubMed] [Google Scholar]
- Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland RL, Borch RF, Qiao X, Georgiadis MM, et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: Small-molecule inhibition of the redox function of Ape1. Antioxid Redox Sign. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marintchev A, Gryk MR, Mullen GP. Site-directed mutagenesis analysis of the structural interaction of the single-strand-break repair protein, X-ray cross-complementing group 1, with DNA polymerase beta. Nucleic Acids Research. 2003;31:580–588. doi: 10.1093/nar/gkg159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marintchev A, Maciejewski MW, Mullen GP. Letter to the Editor: H-1, N-15, and C-13 resonance assignments for the N-terminal 20 kDa domain of the DNA single-strand break repair protein XRCC1. Journal of Biomolecular Nmr. 1999;13:393–394. doi: 10.1023/a:1008381624318. [DOI] [PubMed] [Google Scholar]
- McMillan AW, Kier BL, Shu I, Byrne A, Andersen NH, Parson WW. Fluorescence of Tryptophan in Designed Hairpin and Trp-Cage Miniproteins: Measurements of Fluorescence Yields and Calculations by Quantum Mechanical Molecular Dynamics Simulations. J Phys Chem B. 2013;117:1790–1809. doi: 10.1021/jp3097378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JS, Keim P, Gurd FRN. Co2 Adducts of Certain Amino-Acids, Peptides, and Sperm Whale Myoglobin Studied by Carbon 13 and Proton Nuclear Magnetic-Resonance. Journal of Biological Chemistry. 1974;249:7484–7494. [PubMed] [Google Scholar]
- Morrow JS, Matthew JB, Wittebort RJ, Gurd FRN. C-13 Resonances of Co-13(2) Carbamino Adducts of Alpha and Beta Chains in Human Adult Hemoglobin. Journal of Biological Chemistry. 1976;251:477–484. [PubMed] [Google Scholar]
- Myers TG, Nelson SD. Neuroactive Carbamate Adducts of Beta-N-Methylamino-L-Alanine and Ethylenediamine - Detection and Quantitation under Physiological Conditions by C-13 Nmr. Journal of Biological Chemistry. 1990;265:10193–10195. [PubMed] [Google Scholar]
- O'Leary MH, Jaworski RJ, Hartman FC. C-13 Nuclear Magnetic-Resonance Study of the CO2 Activation of Ribulosebisphosphate Carboxylase from Rhodospirillum-Rubrum. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:673–675. doi: 10.1073/pnas.76.2.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget MSB, Buttner MJ. Thiol-basedregulatory switches. Annual Review of Genetics. 2003;37:91–121. doi: 10.1146/annurev.genet.37.110801.142538. [DOI] [PubMed] [Google Scholar]
- Paulsen CE, Carroll KS. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. Acs Chem Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Williams JG, Hou EW, Wilson SH. Pol beta associated complex and base excision repair factors in mouse fibroblasts. Nucleic Acids Research. 2012;40:11571–11582. doi: 10.1093/nar/gks898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach N, Chan T, Lu TA, Schreiber SS, Tan ZQ. Induction of DNA repair proteins, Ref-1 and XRCC1, in adult rat brain following kainic acid-induced seizures. Brain Research. 2005;1042:236–240. doi: 10.1016/j.brainres.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Roberts SA, Hyatt DC, Honts JE, Changchien LM, Maley GF, Maley F, Montfort WR. Structure of the Y94F mutant of Escherichia coli thymidylate synthase. Acta Crystallogr F. 2006;62:840–843. doi: 10.1107/S1744309106029691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothgeb TM, England RD, Jones BN, Gurd RS. Physical Characterization of S-Methylglucagon and Quantitation of Carbamino Adduct Formation. Biochemistry. 1978;17:4564–4571. doi: 10.1021/bi00614a031. [DOI] [PubMed] [Google Scholar]
- Sarsour EH, Kumar MG, Chaudhuri L, Kalen AL, Goswami PC. Redox Control of the Cell Cycle in Health and Disease. Antioxid Redox Sign. 2009;11:2985–3011. doi: 10.1089/ars.2009.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Rajarathnam K. C-13 NMR chemical shifts can predict disulfide bond formation. Journal of Biomolecular Nmr. 2000;18:165–171. doi: 10.1023/a:1008398416292. [DOI] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS plus : a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. Journal of Biomolecular Nmr. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry AD, Malloy CR, Jeffrey FMH, Chavez F, Srere HK. Formation of Carbamates of Taurine and Other Amino-Acids during Neutralization of Tissue-Extracts with Potassium Carbonate Bicarbonate. Journal of Magnetic Resonance. 1990;89:391–398. [Google Scholar]
- Shinitzky M, Goldman R. Fluorometric Detection of Histidine-Tryptophan Complexes in Peptides and Proteins. Eur J Biochem. 1967;3:139–144. doi: 10.1111/j.1432-1033.1967.tb19508.x. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expression and Purification. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Tietjen J, Mancott A. Rapid Assay of Hydrogen Peroxide Solution (Usp Xviii) Via Uv Spectrophotometry. J Pharm Sci. 1971;60:460–461. doi: 10.1002/jps.2600600326. [DOI] [PubMed] [Google Scholar]
- Walker KW, Gilbert HF. Oxidation of Kinetically Trapped Thiols by Protein Disulfide-Isomerase. Biochemistry. 1995;34:13642–13650. doi: 10.1021/bi00041a045. [DOI] [PubMed] [Google Scholar]
- Weissman JS, Kim PS. Efficient Catalysis of Disulfide Bond Rearrangements by Protein Disulfide-Isomerase. Nature. 1993;365:185–188. doi: 10.1038/365185a0. [DOI] [PubMed] [Google Scholar]
- Wittebort RJ, Hayes DF, Rothgeb TM. Quantitation of Carbamino Adduct Formation of Angiotensin-Ii and Bradykinin. Biophys J. 1978;24:765–778. doi: 10.1016/S0006-3495(78)85419-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittekind M, Mueller L. Hncacb, a High-Sensitivity 3d Nmr Experiment to Correlate Amide-Proton and Nitrogen Resonances with the Alpha-Carbon and Beta-Carbon Resonances in Proteins. J Magn Reson Ser B. 1993;101:201–205. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.