

Summary
Rheumatoid arthritis (RA) is a chronic immune inflammatory disease mediated by the influx of immune cells into the synovial joint space. As Tanshinone IIA (TIIA) has potent anti‐oxidant and anti‐inflammatory activities, we used the adjuvant‐induced arthritis (AA) murine model of RA to investigate the impact of TIIA on RA and immune cell activation. The anti‐arthritic activity of TIIA was investigated in an adjuvant‐induced arthritis model of RA in mice. Myeloperoxidase and neutrophil elastase expression levels were assessed in ankle joints by immunohistochemistry analysis. Immune cell infiltration was evaluated in air pouch experiments. Proinflammatory cytokines expression levels were determined by quantitative real‐time polymerase chain reaction (PCR) and enzyme‐linked immunosorbent assays. Neutrophil extracellular traps (NETs) were assessed by immunostaining and confocal microscopy. Treatment with TIIA alleviated cartilage erosion and neutrophil infiltration in the ankle joints of AA mice and reduced proinflammatory cytokine expression levels in sera. TIIA suppressed interleukin‐6 and tumour necrosis factor‐α expression and release in neutrophils and promoted neutrophil apoptosis. TIIA also inhibited the NET formation of neutrophils. Our findings demonstrated that TIIA can ameliorate RA effectively by targeting neutrophils, indicating that TIIA may act as a potential therapeutic for RA.
Keywords: arthritis, inflammation, neutrophil, tanshinone IIA
Introduction
Rheumatoid arthritis (RA) is a chronic immune inflammatory disease characterized by synovial hyperplasia, joint destruction and extra‐articular manifestations, with significant impacts on both morbidity and mortality 1. Although the aetiology and pathogenesis of this disease have not been elucidated fully, it is known that joint inflammation and damage in RA are mediated by the influx of innate and adaptive immune cells into the synovial joint space, such as neutrophils, macrophages, synovial fibroblasts, T cells and B cells 2. Neutrophils are the most abundant immune cells present in synovial fluid from the joints of RA patients and are also abundant at the pannus/cartilage interface, the site of active tissue damage 3.
Neutrophils, terminally differentiated cells with a short lifespan in circulation, are the most abundant leucocytes in the human body, with homeostasis maintained by their continuous release from the bone marrow 4. Complex interactions between receptors on neutrophils and vascular endothelial cells regulate their attachment and subsequent migration from the circulation into tissues during inflammation 5, 6. Various functional abnormalities of neutrophils have been reported in RA 2. Many clinical studies have revealed the pivotal contributions of cytokines in RA pathogenesis 7, 8; the critical roles of tumour necrosis factor (TNF) and interleukin (IL)‐6 therein have been demonstrated unequivocally by successful clinical targeting of these cytokines in RA using biological therapies 9. Neutrophil granule proteins, such as myeloperoxidase (MPO), matrix metalloproteinases (MMPs), neutrophil elastase (NE), cathepsin G and proteinase 3 10, are found in high concentrations in RA synovial fluid and may be responsible for damage to cartilage and tissue, activation of soluble cytokines and receptors, inhibition of chondrocyte proliferation and activation of synoviocyte proliferation and invasion 2. Circulating and synovial fluid neutrophils in RA patients are more prone to form neutrophil extracellular traps (NETs) compared with neutrophils from healthy controls and from patients with osteoarthritis 11. Levels of NETosis correlate with the presence and levels of anti‐citrullinated peptide antibodies (ACPAs) and markers of systemic inflammation 12, 13. Furthermore, in RA, dysregulation of neutrophil apoptosis can lead to their extended survival in inflamed tissues, which prolongs the release of neutrophil‐derived immune regulatory cytokines, chemokines and cytotoxic products and can lead to persistent inflammation 1, 2. Therefore, neutrophils serve as important targets for RA therapy.
Tanshinone IIA (TIIA), a phenanthrenequinone derivative extracted from Salvia miltiorrhiza Bunge, has been used clinically to manage many diseases, such as angina pectoris, myocardial infarction and stroke 14. Previous studies have demonstrated that TIIA may exert anti‐inflammatory effects 15, 16, 17. TIIA inhibits the production of proinflammatory mediators, such as nitric oxide (NO), TNF‐α, IL‐1β and IL‐6 via the inhibition of nuclear factor kappa B (NF‐κB) activation in RAW 264.7 cells stimulated with lipopolysaccharide (LPS) 15. In brain microvascular endothelial cells, TIIA inhibits vascular cell adhesion protein 1 (VCAM‐1) and intercellular adhesion molecule 1 (ICAM‐1) expression concentration‐dependently through the inhibition of NF‐κB activation and reactive oxygen species (ROS) generation induced by TNF‐α 16. TIIA also induces inflammation resolution in vivo by both the induction of neutrophil apoptosis and the promotion of neutrophil reverse migration 17. However, whether or not TIIA can ameliorate chronic inflammation in RA remains largely unknown.
We hypothesized that TIIA reduced neutrophil infiltration and activation, which may eventually attenuate the inflammatory response in RA. We investigated whether treatment with TIIA could ameliorate adjuvant‐induced arthritis in a murine model of RA.
Materials and methods
Animals
C57BL/6 female mice, 8 weeks of age, were purchased from Academy of Military Medical Sciences (Beijing, China). The mice were maintained in standard housing cages under specific pathogen‐free conditions. All experimental procedures were reviewed and approved by the Beijing University of Chinese Medicine Animal Care and Use Committee and were in accordance with the institutional guidelines for the Care and Use of Laboratory Animals.
Reagents
LPS, phorbol 12‐myristate 13‐acetate (PMA), Freund's complete adjuvant (FCA) and TIIA were purchased from Sigma (St Louis, MO, USA). Anti‐myeloperoxidase, anti‐neutrophil elastase, anti‐B cell lymphoma 2 (Bcl‐2), anti‐Bax and anti‐rabbit‐horseradish peroxidase (HRP) and immunoglobulin (Ig)G were purchased from Abcam (Cambridge, MA, USA) and anti‐cleaved caspase‐3 was purchased from Cell Signaling Technology (Danvers, MA, USA). Goat anti‐rabbit IgG (H + L) secondary antibody, Alexa Fluor® 488 conjugate, goat anti‐rabbit IgG (H + L) secondary antibody, Alexa Fluor® 555 conjugate, TNF‐α and the IL‐6 enzyme‐linked immunosorbent assay (ELISA) kit were purchased from Invitrogen (Carlsbad, CA, USA).
Murine model of adjuvant‐induced arthritis
Chronic arthritis was induced by injection of FCA, as described previously 18. Briefly, 20 and 80 μl of FCA (10 mg/ml heat‐killed Mycobacterium tuberculosis, Sigma) was injected into the joint space and periarticular in mice, respectively. Joint swelling was assessed by measuring ankle joint diameter using a pocket thickness gauge (Mitutoyo, Kawasaki, Japan). Arthritis severity was graded on a scale of 0–4, as described previously 19.
Histological analysis
For histological analysis, the ankle joints from mice were fixed in 4% paraformaldehyde, decalcified in 10% ethylenediamine tetraacetic acid (EDTA) solution, processed and embedded in paraffin. Four‐μm‐thick tissue sections were stained with haematoxylin and eosin (H&E) to characterize ankle joint injury under a microscope.
Immunohistochemistry
For the immunostaining of paraffin sections, 4‐μm‐thick ankle joint sections were deparaffinized with xylene and rehydrated with ethanol and water. Endogenous peroxidase activity was blocked with 3% H2O2 for 15 min at room temperature. Antigen retrieval was accomplished in a microwave oven at 600 W for 15 min in 0·01 M citrate buffer at pH 6·0, which was rinsed later with phosphate‐buffered saline (PBS). Sections were incubated with primary anti‐MPO antibody and anti‐NE antibody at 4°C overnight. The sections were incubated with biotinylated HRP‐conjugated goat anti‐rabbit antibodies. The coloured product was developed by incubation with 3,3′‐diaminobenzidine tetrahydrochloride dihydrate (DAB). Immunohistochemical staining was photographed with a light microscope. The intensity of the staining signal was measured and documented using the Image‐Pro Plus version 6.0 image analysis software.
Air pouch experiments
Air pouch experiments were performed as described previously 20. In brief, C57BL/6 mice (five mice per group) were anaesthetized with chloral hydrate and sterilized air (3 ml filtered through a 0·22 μm Millipore filter; Millipore, Billerica, MA, USA) was given via subcutaneous injection in the back using a 26‐gauge needle to make an air pouch on days 0 and 3. On day 6, 1 ml of buffer (Ctrl) and 1 μg/ml of LPS (positive control) with or without 25 μM TIIA was injected into the air pouches of mice 6 h before the mice were killed by CO2 asphyxiation. The air pouches were washed once with 1 ml and then twice with 2 ml of Hanks's balanced salt solution (HBSS) containing 10 mM EDTA, and the exudates were centrifuged at 100 g for 10 min at room temperature. The cells were resuspended in 1 ml of HBSS containing 10 mM EDTA and were stained with Wright's stain to quantify the cell populations.
Neutrophil preparation and culture
Neutrophils were prepared as described previously 21. In brief, peritoneal exudate cells were obtained from mice inoculated intraperitoneally (i.p.) 10–12 h previously with 1 ml of 10% protease peptone. The cells were resuspended in 1 ml of RPMI‐1640 with 10% fetal bovine serum (FBS), layered onto a two‐step (54·8%/70·2%) discontinuous Percoll gradient, and centrifuged at 1500 g for 30 min at 22°C. The cells (> 95% neutrophils, 1 × 107 neutrophils/mouse) were recovered from the lower interface. Neutrophils were cultured in RPMI‐1640 with 10% FBS at 37°C with 5% CO2 with or without LPS (10 ng/ml) and TIIA. After treatment, the cells were collected for quantitative real‐time PCR (qRT–PCR) and Western blot analysis and the medium was collected for ELISA analysis.
RNA extraction and qRT–PCR analysis
Neutrophils were plated at the density of 5 × 106 cells in a six‐well plate with 2 ml of culture medium per well. Cells were treated with TIIA at different concentrations and stimulated with LPS (10 ng/ml) at 37°C for 2 h. Total RNA was extracted from neutrophils with Trizol (Invitrogen) and 1 μg total RNA was reverse‐transcribed to cDNA with ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan), according to the manufacturer's instructions. The qRT–PCR was performed with SYBR Green real‐time PCR Master Mix (Toyobo) and pairs of oligonucleotide primers: IL‐6 (sense: CTGCAAGAGACTTCCATCCAG, anti‐sense: AGTGGTATAGACAGGTCTGTTGG); TNF‐α (sense: ACAGAAAGCATGATCCGCG, anti‐sense: GCCCCCCATCTTTTGGG); β‐actin (sense: AGAGGGAAATCGTGCGTGAC, anti‐sense: CAATAGTGATGACCTGGCCGT). The specificity of the amplified PCR products was assessed by a melting curve analysis. The 2−ΔΔCT method was used for the calculation of relative expression and the fold induction of gene expression was normalized to beta actin levels.
ELISA analysis
The concentrations of IL‐6 and TNF‐α were measured using an ELISA kit (Invitrogen), according to the manufacturer's instructions.
Immunofluorescence assay
Neutrophils were seeded at a density of 105 cells/well in a 24‐well tissue culture plate with flame‐sterilized coverslips. The cells were incubated with or without TIIA and phorbol 12‐myristate 13‐acetate (PMA) (25 nM) for 4 h. The cells were fixed for 15 min in 4% (w/v) paraformaldehyde/(PBS and made permeable by the addition of 0·25% Triton X‐100/PBS for 15 min. Cells were incubated with 1% bovine serum albumin (BSA) in PBS with Tween‐20 (PBST) for 30 min to block no specific binding of the antibodies and were then incubated with anti‐NE and anti‐MPO (diluted in 1% BSA in PBST) in a humidified chamber overnight at 4°C. After washing three times, secondary antibodies conjugated with fluorescein were added and incubated for 1·5 h. The cells were then incubated with 4',6‐diamidino‐2‐phenylindole (DAPI) for 5 min to stain the nuclei. The cells were imaged using a confocal microscope (BX60, Olympus, Tokyo, Japan) and charge‐coupled device system.
Western blot analysis
Samples were subjected to electrophoresis on 10% sodium dodecyl sulphate–polyacrylamide gel (SDS‐PAGE) and were transferred to nitrocellulose membranes. The membranes were blocked with 5% non‐fat dried milk in Tris‐buffered saline, 0·1% Tween 20 (TBST) buffer (137 mM NaCl, 20 mM Tris‐HCl, pH 7·6 and 0·1% Tween 20) for 2 h and then incubated with primary antibodies at 4°C overnight. After washing with TBST three times, the membranes were incubated with HRP‐conjugated secondary antibodies (1 : 7500 dilutions) for 1 h at room temperature. Enhanced chemiluminescence (ECL) reagent (GE Healthcare, Chicago, IL, USA) was used for protein detection. Following another triple wash, the proteins were visualized using a chemiluminescence system. The intensities of protein bands in the Western blot were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Flow cytometry analysis
Neutrophils were plated at a density of 2 × 106 cells in a six‐well plate with 2 ml of culture medium per well. Cells were washed twice with cold PBS and then resuspended in 1× binding buffer at a concentration of 1 × 106 cells/ml. One hundred microlitres of the solution (1 × 105 cells) was then transferred to a 5‐ml culture tube. Fluorescein isothiocyanate (FITC)‐annexin V and propidium iodide (PI) (5 µl each) were then added. The cells were then gently vortexed and incubated for 15 min at room temperature (RT) (25°C) in the dark, after which 400 μl of 1× binding buffer was added to each tube. The cells were analysed by flow cytometry within 1 h.
Statistical analysis
Data are depicted as the means ± standard error of the mean (s.e.m.). Two‐way analyses of variance (anova) were used to analyse the joint diameters and arthritis scores of the two groups. For the air pouch experiments, qPCR and ELISA experiments in vitro, the statistical significance of differences was determined by one‐way anova. One‐way anova [post hoc, least significant difference (LSD)] was then applied to compare the statistical significance levels of differences in quantifications of immunohistochemistry, ELISA analyses of mouse plasma and assessments of apoptosis. P‐values less than 0·05 were considered significant.
Results
TIIA alleviated RA in mice
In this study, we used a murine model challenged with FCA to examine whether TIIA could prevent joint inflammation. The adjuvant‐induced paw oedema was compared with the control mice; it was noted that i.p. injection of TIIA (30 mg/kg/d) reduced paw oedema (Fig. 1a) (n = 10), ankle joint diameter (Fig. 1b) and arthritis scores significantly (Fig. 1c). Histological analysis of ankle joints showed that FCA challenge resulted in typical inflammation, characterized by cartilage erosion, synovial hyperplasia and infiltration of multiple types of inflammatory cells compared with normal mice. Treatment with TIIA alleviated the arthritis in mice significantly (Fig. 1d).
Figure 1.

Alleviation of adjuvant‐induced arthritis following the treatment of Tanshinone IIA (TIIA) in mice. (a) Macroscopic observation of severity of inflammation of paw joints from each group on day 30 after Freund's complete adjuvant (FCA) immunization. (b) Joint swelling was assessed by measuring ankle joint diameter. Values are the mean ± standard error of the mean (s.e.m.). **P < 0·01 when comparing antigen‐induced arthritis treated with the TIIA (RA + TIIA) group with antigen‐induced arthritis (RA) group (n = 10). (c) The severity of arthritis was graded on a scale of 0–4. Values are the mean ± s.e.m. **P < 0·01 when comparing the RA + TIIA group with the RA group (n = 10). (d) Histological assessment was performed using haematoxylin and eosin‐stained ankle joint sections from each treatment groups. Representative sections are shown. [Colour figure can be viewed at wileyonlinelibrary.com].
TIIA inhibits MPO and NE expression in the ankle joints of RA mice
The measurement of MPO and NE enzymatic activity is a well‐established and defined method for the determination of neutrophil inflammatory function and recruitment to tissue. MPO is a key component of oxidant production in neutrophils, and is also prominent in NETs. Plasma and synovial MPO and its products have been associated strongly with RA severity 22. Aberrant MPO expression from activated neutrophils amplifies inflammation and causes tissue damage through the formation of reactive intermediates. NE, a type of serine protease, also localizes in NETs via its high affinity for DNA. NE promotes cartilage degradation and plays a role in the proteolytic activation of cytokines and chemokines 23. We performed immunohistochemical analyses to investigate whether TIIA can reduce MPO and NE expression in RA mice. The MPO and NE expression levels were significantly higher in RA mice compared with the control group. However, TIIA treatment suppressed significantly the MPO and NE up‐regulation induced by FCA (Fig. 2).
Figure 2.

Immunohistochemistry analyses of myeloperoxidase (MPO) and neutrophil elastase (NE) in ankle joints of antigen‐induced arthritis mice. Immunohistochemistry analysis was used to assess the expression of MPO and NE in the ankle joints of mice treated with phosphate‐buffered saline (PBS) (control, a,d), antigen‐induced arthritis group (RA, b,e) and antigen‐induced arthritis treated with Tanshinone IIA (TIIA) group (RA + TIIA, c,f). The arrow indicated positive signal. The intensity of the staining signal was measured (g,f). [Colour figure can be viewed at wileyonlinelibrary.com].
TIIA inhibited LPS‐induced neutrophil influx in vivo
Infiltration of neutrophils into the joints plays important roles in bone erosion and articular destruction in RA 2. Air pouch experiments were used to investigate the impact of TIIA on LPS‐induced neutrophil infiltration. As shown in Fig. 3, TIIA treatment reduced total leucocyte and neutrophil numbers significantly in the air pouch.
Figure 3.

Tanshinone IIA (TIIA) inhibited lipopolysaccharide (LPS)‐induced neutrophil influx in vivo. Air pouches were created and TIIA or phosphate‐buffered saline (PBS) was administered intraperitoneally 30 min prior to the injection of LPS into pouches, exudates were harvested 6 h later and the number and identification of leucocytes were determined by cytology as described in Materials and methods. (a) The number of total leucocytes in the air pouch are expressed as means ± standard error of the mean (s.e.m.) (n = 5). **P < 0·01 versus corresponding group. (b) The number of neutrophil in the air pouch are expressed as means ± s.e.m. (n = 5). **P < 0·01 versus corresponding group.
TIIA induced the apoptosis of LPS‐treated neutrophils
Apoptosis is essential for the removal of neutrophils from inflamed tissues and the timely resolution of inflammation 24. However, neutrophils that have migrated into RA joints display a delay in apoptosis and have an enhanced potential to cause host tissue damage due to their extended lifespan 2. The Bcl‐2 protein family, which is divided into pro‐apoptotic proteins, such as Bax, and anti‐apoptotic proteins, such as Bcl‐2, is a central mediator of the intrinsic pathway 25. Caspase 3 is a critical executor of apoptosis, which is important for both the extrinsic and intrinsic apoptosis pathways, as caspase 3 cleavage is a key step in the activation of apoptosis 26. To investigate whether the anti‐inflammatory effect of TIIA results from promoting neutrophil apoptosis, we detected the expression levels of Bcl‐2, Bax and cleaved caspase 3 by Western blot. TIIA increased Bax and cleaved caspase 3 expression significantly, while TIIA treatment reduced Bcl‐2 expression (Fig. 4a,b). Thus, TIIA treatment promoted neutrophil apoptosis. The neutrophil apoptosis was also examined in an annexin V/PI staining assay and via flow cytometry. As shown in Fig. 4c,d, TIIA treatment increased the number of late apoptotic cells significantly.
Figure 4.
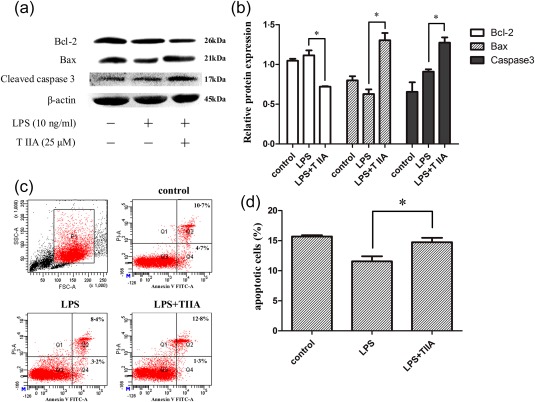
Tanshinone IIA (TIIA) induces the apoptosis of lipopolysaccharide (LPS)‐treated neutrophils. (a) Western blot analyses of B‐cell lymphoma 2 (Bcl‐2), Bax and cleaved caspase 3. (b) Density analyses of Bcl‐2, Bax and cleaved caspase 3 expressions in (a). (c) The effects of TIIA on neutrophils apoptosis by flow cytometry. (d) The percentage of cells labelled as annexin V (+), propidium iodide (PI) (−) and annexin V (+) PI (+) was investigated. The data were obtained from three independent experiments. Data are expressed as means ± standard error of the mean (s.e.m.). *P < 0·05, LPS‐treated cells versus TIIA‐treated cells. [Colour figure can be viewed at wileyonlinelibrary.com].
Effect of TIIA on cytokine production in LPS‐activated neutrophils
Neutrophils can express and secrete large varieties of cytokines after activation, such as TNF‐α, IL‐1β and IL‐6. TNF‐α has been detected in high concentrations in the serum and synovial fluid of RA patients and plays a pivotal role in RA pathogenesis 8. IL‐6 is a multi‐functional cytokine with key roles in inflammation and bone metabolism through the regulation of osteoclast differentiation. Elevated levels of IL‐6 have been reported in the serum and synovial fluid of patients with RA and correlate with inflammation and disease severity 8. We next studied whether TIIA could regulate the cytokine expression profile in neutrophils in vitro. Mouse peritoneal neutrophils were purified and treated with LPS or LPS/TIIA for 2 h. The qRT–PCR analysis showed that TIIA down‐regulated TNF‐α and IL‐6 mRNA expression levels significantly in LPS‐activated neutrophils (Fig. 5a,b). Consistently, ELISA analysis showed that TIIA treatment also reduced TNF‐α and IL‐6 protein secretion significantly in a dose‐dependent manner (Fig. 5c,d). In addition, peritoneal injection of TIIA reversed the elevation of TNF‐α and IL‐6 in the plasma of RA mice (Fig. 5e,f).
Figure 5.

Tanshinone IIA (TIIA) decreases cytokine productions in lipopolysaccharide (LPS)‐treated neutrophils in vitro. (a,b) Quantitative real time–polymerase chain reaction (qRT–PCR) analyses of tumour necrosis factor (TNF)‐α (a) and interleukin (IL)‐6 (b) mRNA level in neutrophils. (c,d) Enzyme‐linked immunosorbent assay (ELISA) analyses of TNF‐α (c) and IL‐6 (d) proteins in the culture media of neutrophil. (e,f) ELISA analyses of TNF‐α (e) and IL‐6 (f) proteins in the plasma of antigen‐induced arthritis (RA) mice and adjuvant‐induced arthritis treated with TIIA (RA + TIIA) mice (n = 10). Values are the means ± standard error of the mean (s.e.m.).*P < 0·05; **P < 0·001.
TIIA inhibited NETs formation and MPO and NE release by neutrophils in vitro
A recently revealed novel feature of neutrophils is their capacity to generate NETs via a distinct process of cell death, termed NETosis. The molecular pathways leading to NETosis include calcium mobilization, generation of reactive oxygen species (ROS) and nuclear delobulation involving the enzymatic activities of MPO and NE. A number of studies have implicated NETs as a source of tissue damage and autoantibody production in RA 13. NETs externalize various autoantigens and granule enzymes, which may perpetuate a vicious cycle leading to the generation of specific autoantibodies and inflammatory responses. In our study, PMA‐induced NETs formation was visualized by staining with DAPI (blue) and anti‐MPO (red) or anti‐NE (green) and was observed by immunofluorescence confocal microscopy. As shown in Fig. 6, TIIA inhibited NETs formation, along with NETs‐associated MPO and NE release.
Figure 6.

Tanshinone IIA (TIIA) inhibits neutrophil extracellular traps (NETs) formation and myeloperoxidase (MPO) and neutrophil elastase (NE) release in vitro. Phorbol 12‐myristate 13‐acetate (PMA)‐induced (NETs) formation was visualized by staining with anti‐MPO (red, a), anti‐NE (green, b) and 4',6‐diamidino‐2‐phenylindole (DAPI) (blue) and observed by immunofluorescence confocal microscopy. The data were obtained from three independent experiments. [Colour figure can be viewed at wileyonlinelibrary.com].
Discussion
RA is the most common inflammatory joint disease. The aetiopathogenesis of this disease has been classically explained by a T cell‐driven process. However, recent studies have highlighted the significant contribution of neutrophils to RA physiopathology 1, 2. Neutrophils are the major infiltrating inflammatory cells and serve as a first‐line defence against invading microorganisms or tissue injury 22, 23. If neutrophils infiltrate tissues in large numbers and secrete high levels of cytokines and chemokines, these processes may overwhelm the anti‐protease and anti‐oxidant protective mechanisms in tissues, which can lead to indiscriminate tissue damage, further stimulating neutrophils infiltration, thus forming a vicious cycle. A large number of activated neutrophils are found in RA synovial fluid and pannus, which play pivotal roles in tissue damage within RA joints. Therefore, neutrophils serve as important targets for RA therapy 27.
TNF‐α and IL‐6 are essential for neutrophil activation and migration 7, 8. The signalling of neutrophils to leave the circulation depends upon the up‐regulation of ICAM‐1 on the surfaces of endothelial cells 28. The major inducer of ICAM‐1 on endothelial cells is TNF‐α 29, 30. Thus, down‐regulating TNF‐α expression will prevent the first important step in neutrophil emigration and activation. IL‐6 mediates similar effects to those of TNF‐α in the local synovial environment, with the added role of directly driving the acute‐phase response 31. Neutralizing IL‐6 can inhibit the adhesion of neutrophils to co‐cultured endothelial cells 32. Our study showed that TIIA reduced TNF‐α and IL‐6 expression and secretion significantly.
Neutrophils are characterized by a very short lifespan. In the absence of activating stimuli, neutrophils stay in circulation for 6–18 h before undergoing constitutive apoptosis 33. Apoptosis is crucial for neutrophil turnover and for the resolution of inflammation. Apoptotic neutrophils are recognized and ingested by macrophages, leading to the production of anti‐inflammatory mediators. If there is an impairment in neutrophil clearance, apoptotic neutrophils undergo secondary necrosis. Ingestion of necrotic cellular debris by macrophages induces the production of proinflammatory cytokines, thus amplifying the inflammatory scenario 34. In patients with early RA, synovial neutrophils show significantly lower levels of apoptosis compared with patients with other persistent forms of arthritis 35. Therefore, to resolve inflammation, it is essential to promote neutrophil apoptosis in a timely manner in inflamed tissues. Our study also showed that TIIA treatment promoted neutrophil apoptosis by enhancing caspase 3 activity.
NETosis is a distinct death process of neutrophils. NETs externalize various autoantigens and granule enzymes 36, 37, which perpetuate a vicious cycle leading to the generation of specific autoantibodies and inflammatory responses. In the current study, we found that TIIA inhibited NETs formation significantly, and NETs‐associated MPO and NE release.
In conclusion, our study showed that TIIA can ameliorate inflammation in a murine model of RA by targeting neutrophils in three ways. First, TIIA reduced TNF‐α and IL‐6 expression and secretion, thus inhibiting neutrophil activation and infiltration. Secondly, TIIA promoted neutrophil apoptosis, thus decreasing the numbers of activated neutrophils in the inflamed joints. Thirdly, TIIA inhibited NETs formation and NETs‐associated MPO and NE release, thus reducing tissue damage and inflammation. These results indicate that TIIA may act as a potential therapeutic for RA.
Disclosure
All authors declare that there are no financial conflicts of interest associated with this work.
Acknowledgements
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. A. X. had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study conception and design: S. Z., G. H., K.Y. and A. X. Acquisition of data: S. Z., G. H., K. Y., Q. Z. and K. Yu. Analysis and interpretation of data: S. Z., G. H, K. Y., H. S., G. L. and Q. Xu. This paper presents independent research supported by the National Natural Science Foundation of China Grant 81430099, Projects of International Cooperation and Exchanges Grant 2014DFA32950 and Beijing Municipal Commission of Education grant 521/0101312 to A. X.
References
- 1. Cascão R, Rosário HS, Souto‐Carneiro MM, Fonseca JE. Neutrophils in rheumatoid arthritis: more than simple final effectors. Autoimmun Rev 2010; 9:531–5. [DOI] [PubMed] [Google Scholar]
- 2. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol 2014; 10:593–601. [DOI] [PubMed] [Google Scholar]
- 3. Raza K, Scheel‐Toellner D, Lee CY et al Synovial fluid leukocyte apoptosis is inhibited in patients with very early rheumatoid arthritis. Arthritis Res Ther 2006; 8:R120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eyles JL, Roberts AW, Metcalf D, Wicks IP. Granulocyte colony‐stimulating factor and neutrophils–forgotten mediators of inflammatory disease. Nat Clin Pract Rheumatol 2006; 2:500–10. [DOI] [PubMed] [Google Scholar]
- 5. Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM‐1 regulates neutrophil adhesion and transcellular migration of TNF‐alpha‐activated vascular endothelium under flow. Blood 2005; 106:584–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Woodfin A, Voisin MB, Nourshargh S. Recent developments and complexities in neutrophil transmigration. Curr Opin Hematol 2010; 17:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis – shaping the immunological landscape. Nat Rev Rheumatol 2016; 12:63–8. [DOI] [PubMed] [Google Scholar]
- 8. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol 2007; 7:429–42. [DOI] [PubMed] [Google Scholar]
- 9. Smolen JS, Aletaha D. Rheumatoid arthritis therapy reappraisal: strategies, opportunities and challenges. Nat Rev Rheumatol 2015; 11:276–89. [DOI] [PubMed] [Google Scholar]
- 10. Murphy G, Nagase H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: destruction or repair? Nat Clin Pract Rheumatol 2008; 4:128–35. [DOI] [PubMed] [Google Scholar]
- 11. Kaplan MJ. Role of neutrophils in systemic autoimmune diseases. Arthritis Res Ther 2013; 15:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sohn DH, Rhodes C, Onuma K et al Local Joint inflammation and histone citrullination in a murine model of the transition from preclinical autoimmunity to inflammatory arthritis. Arthritis Rheumatol 2015; 67:2877–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khandpur R, Carmona‐Rivera C, Vivekanandan‐Giri A et al NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 2013; 5:178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu M, Cao FL, Zhang YF et al Tanshinone IIA therapeutically reduces LPS‐induced acute lung injury by inhibiting inflammation and apoptosis in mice. Acta Pharmacol Sin 2015; 36:179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fan GW, Gao XM, Wang H et al The anti‐inflammatory activities of Tanshinone IIA, an active component of TCM, are mediated by estrogen receptor activation and inhibition of iNOS. J Steroid Biochem Mol Biol 2009; 113:275–80. [DOI] [PubMed] [Google Scholar]
- 16. Tang C, Xue HL, Bai CL, Fu R. Regulation of adhesion molecules expression in TNF‐α‐stimulated brain microvascular endothelial cells by tanshinone IIA: involvement of NF‐κB and ROS generation. Phytother Res 2011; 25:376–80. [DOI] [PubMed] [Google Scholar]
- 17. Robertson AL, Holmes GR, Bojarczuk AN et al A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti‐inflammatory mechanism. Sci Transl Med 2014; 6:225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferrell WR, Lockhart JC, Kelso EB et al Essential role for proteinase‐activated receptor‐2 in arthritis. J Clin Invest 2003; 111:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Durai M, Gupta RS, Moudgil KD. The T cells specific for the carboxyl‐terminal determinants of self (rat) heat‐shock protein 65 escape tolerance induction and are involved in regulation of autoimmune arthritis. J Immunol 2004; 172:2795–802. [DOI] [PubMed] [Google Scholar]
- 20. Antoine F, Simard JC, Girard D. Curcumin inhibits agent‐induced human neutrophil functions in vitro and lipopolysaccharide‐induced neutrophilic infiltration in vivo . Int Immunopharmacol 2013; 17:1101–7. [DOI] [PubMed] [Google Scholar]
- 21. Holub M, Cheng CW, Mott S, Wintermeyer P, van Rooijen N, Gregory SH. Neutrophils sequestered in the liver suppress the proinflammatory response of Kupffer cells to systemic bacterial infection. J Immunol 2009; 183:3309–16. [DOI] [PubMed] [Google Scholar]
- 22. Carlson M, Raab Y, Sevéus L, Xu S, Hällgren R, Venge P. Human neutrophil lipocalin is a unique marker of neutrophil inflammation in ulcerative colitis and proctitis. Gut 2002; 50:501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006; 6:173–82. [DOI] [PubMed] [Google Scholar]
- 24. El Kebir D, Gjorstrup P, Filep JG. Resolvin E1 promotes phagocytosis‐induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci USA 2012; 109:14983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life‐or‐death switch. Nat Rev Cancer 2002; 2:647–56. [DOI] [PubMed] [Google Scholar]
- 26. Juraver‐Geslin HA, Durand BC. Early development of the neural plate: new roles for apoptosis and for one of its main effectors caspase‐3. Genesis 2015; 53:203–24. [DOI] [PubMed] [Google Scholar]
- 27. Wittkowski H, Foell D, af Klint E et al Effects of intra‐articular corticosteroids and anti‐TNF therapy on neutrophil activation in rheumatoid arthritis. Ann Rheum Dis 2007; 66:1020–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013; 13:159–75. [DOI] [PubMed] [Google Scholar]
- 29. Edwards SW, Hallett MB. Seeing the wood for the trees: the forgotten role of neutrophils in rheumatoid arthritis. Immunol Today 1997; 18:320–4. [DOI] [PubMed] [Google Scholar]
- 30. Paleolog EM, Hunt M, Elliott MJ, Feldmann M, Maini RN, Woody JN. Deactivation of vascular endothelium by monoclonal anti‐tumor necrosis factor alpha antibody in rheumatoid arthritis. Arthritis Rheum 1996; 39:1082–91. [DOI] [PubMed] [Google Scholar]
- 31. Tanaka T, Narazaki M, Kishimoto T. IL‐6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 2014; 6:a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lally F, Smith E, Filer A et al A novel mechanism of neutrophil recruitment in a coculture model of the rheumatoid synovium. Arthritis Rheum 2005; 52:3460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bostan M, Brasoveanu LI, Livescu A, Manda G, Neagu M, Iordachescu D. Effects of synovial fluid on the respiratory burst of granulocytes in rheumatoid arthritis. J Cell Mol Med 2001; 5:188–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol 2004; 36:372–8. [DOI] [PubMed] [Google Scholar]
- 35. Andersson AK, Li C, Brennan FM. Recent developments in the immunobiology of rheumatoid arthritis. Arthritis Res Ther 2008; 10:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nzeusseu Toukap A, Delporte C, Noyon C et al Myeloperoxidase and its products in synovial fluid of patients with treated or untreated rheumatoid arthritis. Free Radic Res 2014; 48:461–5. [DOI] [PubMed] [Google Scholar]
- 37. Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 2006; 6:541–50. [DOI] [PubMed] [Google Scholar]