

Summary
Altered metabolism is a hallmark of cancers, including shifting oxidative phosphorylation to glycolysis and up‐regulating glutaminolysis to divert carbon sources into biosynthetic pathways that promote proliferation and survival. Therefore, metabolic inhibitors represent promising anti‐cancer drugs. However, T cells must rapidly divide and survive in harsh microenvironments to mediate anti‐cancer effects. Metabolic profiles of cancer cells and activated T lymphocytes are similar, raising the risk of metabolic inhibitors impairing the immune system. Immune checkpoint blockade provides an example of how metabolism can be differentially impacted to impair cancer cells but support T cells. Implications for research with metabolic inhibitors are discussed.
Keywords: cancer, T cells, tumour immunology
Abbreviations
- 2DG
2‐deoxyglucose
- AMPK
AMP‐activated protein kinase
- CSCs
cancer stem‐like cells
- CTLA‐4
cytotoxic T lymphocyte‐associated antigen‐4
- CTLs
CD8+ cytotoxic T lymphocytes
- HIF
hypoxia‐inducible factor
- IFN‐γ
interferon‐γ
- IL
interleukin
- LDH
lactate dehydrogenase
- mTOR
mammalian target of rapamyacin
- PD‐1
programmed death protein‐1
- PD‐L1
programmed death protein ligand 1
- PI3K
phosphoinositide 3‐kinase
- TCA
tricarboxylic acid
- TCR
T‐cell receptor
- Th
CD4+ T helper
- TILs
tumour‐infiltrating lymphocytes
- Treg cells
regulatory T cells
Introduction
Cancer cells are characteristically different from their healthy counterparts, with one hallmark being altered metabolic programming.1 Factors such as hypoxia, oncogenic mutations, and altered signalling induce up‐regulation of anabolic processes and suppression of catabolic pathways in cancer cells.2 However, cancer cells are not the only proliferating cells in a tumour. Tumour‐infiltrating lymphocytes (TILs) are equally dependent on metabolic reprogramming to divert metabolites into biosynthetic pathways to promote survival and proliferation, and ultimately to mount a response against the tumour.3 The metabolic interplay between both cell types can contribute to the functional exhaustion of TILs.4 However, TILs remain a positive prognostic factor, suggesting that lymphocytes can reduce tumour growth even while functioning suboptimally.5, 6 Early successes of immunotherapies for treating aggressive malignancies such as metastatic melanomas, demonstrate that tumoricidal effects of TILs can be improved by reducing immunosuppressive effects of cancer cells.7 Importantly, the action of immunotherapies and metabolic targeting agents is not restricted to immunological and cancer cells, respectively; the hallmarks of metabolic alteration and immune evasion are probably more interdependent than previously anticipated. Therefore, novel therapies designed to disrupt the metabolic phenotype of cancer cells can potentially impair tumour‐specific immune responses that are a critical mechanism underlying conventional therapies and immunotherapies.
Cancer cell metabolism
Since the 1920s, when Otto Warburg demonstrated altered patterns of glucose breakdown in oxygenated regions of tumour tissues, the cellular metabolism of cancers has been the focus of extensive research.8 Further findings by Warburg et al. demonstrated increased glucose uptake and utilization compared with healthy cells and this knowledge led to advancements ranging from diagnostic imaging to metabolically targeted therapeutic agents.9, 10, 11 A host of metabolic adaptations allow cancer cells to support survival through rapid ATP generation and increased biosynthesis. Carbohydrate, amino acid, lipid and nucleic acid metabolism of cancer cells differs from that of quiescent cells.2 An extensively described phenomenon is the process by which cancer cells convert pyruvate to lactate in the presence of adequate oxygen, termed aerobic glycolysis or ‘The Warburg Effect’.9 This represents a shift from oxidative phosphorylation to glycolysis as the primary ATP‐producing pathway. Although initially thought to be a strategy to cope with hypoxia, it is now widely accepted that this shift to glycolytic metabolism serves primarily to divert carbon sources into biosynthetic pathways, producing nucleic acids, lipids and proteins for rapid cell division.3 Glutaminolysis, the process by which glutamine is imported into cells and converted to α‐ketoglutarate for entry into the tricarboxylic acid (TCA) cycle, is similarly up‐regulated in cancers. Aerobic glycolysis and glutaminolysis represent only two important metabolic changes that allow neoplastic growth.12 Cancer cells can use a variety of alternative energy‐producing pathways depending on nutrient availability, making it difficult to target cancers by deprivation of any one substrate or pathway.13
Tumour immunity
Cancers pose unique challenges to the immune system because they are derived from self and are, therefore, difficult to identify as a threat.14 Nevertheless, a small proportion of proteins are differentially expressed by cancer cells and the immune system can exploit these subtle differences to elicit tumour‐specific responses.15 However, differentiating healthy cells from neoplastic cells represents only one of many challenges posed to the immune system by cancers. Recruitment of immunoregulatory cells, down‐regulation of antigen presentation, and up‐regulation of inhibitory molecules represent other immunoevasive strategies of cancers.14 Tumour microenvironments are very hostile for T cells because of low oxygen tension, reduced pH, competition for nutrients and waste accumulation.4, 5, 16 The milieu of waste products is of particular concern because kynurenine, a by‐product of tryptophan catabolism, and lactic acid can suppress effector cells, while extracellular ATP can be metabolized by immunoregulatory subsets, with the net effect causing potent immunosuppression.17, 18, 19, 20 Additionally, cancer cells directly interact with TILs and modulate their activity by interfering with bioenergetic pathways, which limits their differentiation into effectors, consequently impairing their function.21, 22, 23 T lymphocytes have the unique ability to specifically target, destroy and establish memory for cancer cells through the antigen‐specific T‐cell receptor (TCR). Hence, T cells are imperative for selective destruction of cancer cells expressing endogenous antigens and establishment of long‐term anti‐cancer responses.24 Activation of naive T cells requires metabolic reprogramming and clonal expansion. The resulting effector T cells exhibit highly tailored bioenergetic requirements, suggesting that these lymphocytes may be the most affected by tumour‐induced metabolic challenges.22
Metabolic activation of T cells
Naive T cells have a metabolic phenotype similar to other quiescent cells of the body. They use fatty acid oxidation and low levels of glutaminolysis.24 Upon antigen stimulation, T cells increase nutrient uptake to fuel activation, proliferation and cytokine production. Similar to cancer cells, the increased uptake of substrates such as glucose, fuels biosynthetic pathways, whereas glutamine metabolism provides a carbon source for the TCA cycle.25
Activation and differentiation into effector CD8+ cytotoxic T lymphocytes (CTLs) requires a switch to glycolytic metabolism.26 The metabolic phenotype of CTLs is remarkably similar to cancer cells (Fig. 1), and serves the same purpose: to support macromolecule synthesis, rapid proliferation and heightened ATP demand.27 During antigen clearance, memory CD8+ cells are established, which requires another metabolic shift to fatty acid oxidation that supports long‐term survival.25
Figure 1.
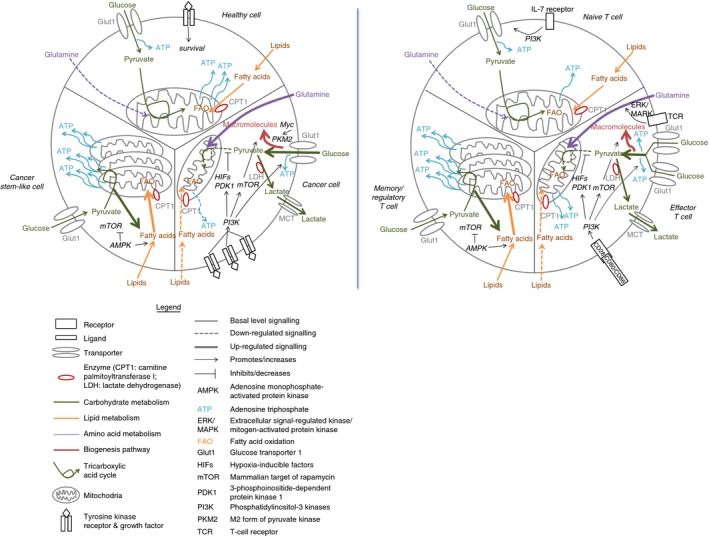
Overlapping metabolic profiles of cancer cells and T lymphocytes. The metabolism of cancer cells, including their healthy predecessor (left panel), compared with the metabolism of T cells (right panel). Different signals lead to the up‐regulation of glycolysis and glutaminolysis in cancer cells and T cells, but the activation of the PI3K/Akt signalling cascade is mechanistic in both scenarios. The result is increased expression of GLUT1, suppression of mitochondrial oxidation and fatty acid uptake, and increased biosynthesis. Cancer stem‐like cells and memory and regulatory T cells share a metabolic profile characterized by reliance upon mitochondrial fatty acid oxidation, an effect mediated largely by activated AMPK.
Similar to CTLs, CD4+ T cells up‐regulate glycolysis during activation.25 They then differentiate into a variety of effector subsets, depending on the cytokine profile in the microenvironment. Cytokine milieus in tumours are heavily influenced by type 2 macrophages, myeloid‐derived suppressor cells and regulatory T (Treg) cells, as well as cancer cells and cancer‐associated fibroblasts.28, 29 CD4+ T‐cell subsets can promote or suppress tumour growth, depending on their phenotype.29, 30 A more informative prognostic factor in cancer patients than total TILs is the ratio of CTLs : Treg cells, demonstrating the importance of T‐cell differentiation and effector subtypes.6 In general, CD4+ T helper type 1 (Th1) cells are associated with anti‐tumour responses through their ability to support activation of CTLs. CD4+ Th2 cells, and Treg cells are associated with tumour‐promoting immunosuppressive mechanisms.14
Each type of CD4+ T cell exhibits a characteristic metabolic phenotype, suggesting that metabolism is tightly linked to function.22, 31 All differentiated T‐cell populations have increased glycolytic rates compared with naive T cells, with Th2 cells having the most glycolysis and expression of GLUT1, the primary glucose transporter. Th1 cells are highly dependent on glutamine catabolism during differentiation; deprivation of this amino acid drives their fate towards Treg cells, even when cultured in Th1‐polarizing conditions.32 Treg cells have low glycolytic rates but high rates of lipid oxidation.22
Despite the reliance of T cells on glycolysis to meet energy demands, mitochondrial function is still important to T‐cell metabolism and loss of mitochondrial mass is a characteristic of functionally exhausted T cells.33 The fine line between tumour‐promoting and tumour‐inhibiting T‐cell subsets depends heavily on extracellular signals and metabolic adaptations. Because of the tight link between metabolism and function, disruptions to lymphocytic cellular energetics within tumours are a major source of immunosuppression.34
Glycolytic switch in cancer cells: proliferation
Underlying the glycolytic switch of cancer cells are oncogenic mutations that allow growth factor‐independent nutrient uptake and metabolic reprogramming towards anabolic pathways.2 Microenvironmental cues within tumours such as hypoxia, extracellular acidification and nutrient conditions further contribute to the abnormal metabolic phenotype.35 The shift to glycolysis in cancer cells is mediated by up‐regulation of phosphoinositide 3‐kinase (PI3K)‐Akt signalling, which activates the mammalian target of rapamyacin (mTOR) complex and downstream hypoxia‐inducible factor (HIF)‐regulated cellular reprogramming.2 HIF transcription factors exist transiently in cells under normoxic conditions and are targeted for degradation by the von Hippel–Lindau protein.36 During hypoxia, HIFs are stabilized and mediate transcription of genes promoting cellular survival. HIFs are over‐expressed in cancer cells due to hypoxia and PI3K‐Akt signalling, which is commonly up‐regulated due to mutations in tyrosine kinases or loss of function of the master regulator protein p53, and are responsible for regulating proliferation and glucose metabolism.3 The consequence of signalling through this pathway is increased glucose transporter expression, and mTOR activation, regulating nutrient availability and promoting biosynthetic pathways. HIF1‐activated genes also promote the glycolytic phenotype by up‐regulating glycolytic enzymes and expression of pyruvate dehydrogenase kinase isozyme‐1, which prevents pyruvate entry into the TCA cycle. Glycolytic intermediates accumulate due to MYC‐mediated expression of the fetal isozyme of pyruvate kinase, PKM2, which has lower catalytic activity, so allowing diversion into biosynthetic pathways such as the pentose phosphate pathway.8
During aerobic glycolysis, lactate dehydrogenase (LDH) catalyses the conversion of pyruvate to lactate. Melanoma cells are particularly glycolytic, and rely heavily on LDH‐mediated lactate generation.13 HIF‐mediated up‐regulation of H+/lactate transporters such as monocarboxylate transporters are essential to prevent intracellular acidification, which causes growth arrest in cancer cells.35 Lactate extrusion may not merely be a mechanism of waste clearance, but maintains glycolytic rate and promotes tumour progression and metastasis.13 The resulting acidic extracellular microenvironment promotes epithelial‐to‐mesenchymal transition in cancer cells, contributes to drug resistance, and impairs immunological cell signalling.37 The dependence of cancer cells on fermentation and extrusion of lactate suggests an additional reason for aerobic glycolysis: rapid, albeit inefficient, ATP generation supports demand for membrane transporter activity to regulate nutrient flux and pH homeostasis.38
Despite the majority of cancer cells relying on aerobic glycolysis, their metabolic phenotypes are heterogeneous. Cancer stem‐like cells (CSCs) have become a focus of research because of their ability to self‐renew, repopulate tumours and mediate resistance to conventional therapies.39 A unique feature of CSCs that makes them resistant to common therapeutic interventions is probably their metabolic distinction from bulk tumour cells. However, whether or not CSCs rely primarily on oxidative phosphorylation or glycolysis is unclear and appears to vary by tumour type.40 Regardless, CSCs share a common metabolic feature: reliance on mitochondrial function. Consequently, pharmaceutical agents that inhibit mitochondrial biogenesis have become attractive options for targeting CSC metabolism and combating drug resistance in melanomas.41
Glycolytic switch in T cells: activation
T cells reprogramme to gain the same survival and growth advantages as cancers. The metabolic phenotypes are so similar that lymphocytes have been recommended as models to understand the molecular mechanisms of carcinogenesis.27 The mediators of metabolic reprogramming in T cells include PI3K‐Akt signalling, mTOR, AMP‐activated protein kinase (AMPK) and HIF1‐α transcriptional programming.24
Naive CD8+ T cells maintain basal levels of glucose uptake due to signalling through the interleukin‐7 (IL‐7) receptor.25 During activation, antigen recognition at the TCR promotes glutamine uptake through the extracellular signal‐regulated kinase/mitogen‐activated protein kinase pathway. Co‐stimulation through CD28 promotes enhanced glucose uptake through the PI3K‐Akt pathway.42 There are many similarities in the pathways used to activate T cells and those used by glycolytic cancer cells, illustrating the ubiquity of these metabolic mediators in rapidly proliferating cells (Fig. 1). For example, mTOR activity is essential for the increased metabolic rate and effector phenotype of CTLs.43 Downstream of mTOR, HIF1‐α stabilization up‐regulates the expression of glycolytic genes, including GLUT1, which is required by CD4+ T cells for activation, expansion and survival.22, 44 Aerobic glycolysis promotes differentiation of CD4+ cells into a Th1 phenotype through an epigenetic mechanism mediated by LDH‐A, which raises concentrations of acetyl‐coenzyme A that promotes histone acetylation and, ultimately, transcription of interferon‐γ (IFN‐γ).45 Compared with naive CD4+ T cells, those with an effector memory phenotype have greater spare respiratory capacity that can be accessed immediately upon exposure to hypoxia to sustain bioenergetic requirements, allowing glycolysis to be diverted to maintaining mitochondrial membrane potential.46 This allows CD4+ memory cells to maintain effector functions, at least transiently, in tumours.
Treg cells and memory CD8+ cells rely on the suppression of mTOR to adopt an oxidative phenotype. Reliance of Treg cells on fatty acid oxidation is believed to be partially due to suppression of mTOR by AMPK.22, 32 The importance of this metabolic shift is demonstrated in mice with a deletion of tuberous sclerosis complex‐2, the negative regulator of mTOR, which are unable to form memory T cells.43 Upon re‐stimulation with antigens and co‐stimulation via CD28, memory T cells reprogramme to a glycolytic phenotype quicker than after initial exposure to antigens.47 This metabolic priming of memory T cells is likely imprinted at the epigenetic level.47, 48, 49
Tumour acidity
Tumours secreting large amounts of lactate have greater metastatic potential and confer worse prognoses for patients.31 This effect may be due to impaired anti‐tumour immunity and altered cancer cell metabolism. Extracellular lactate reduces T‐cell proliferation and function while promoting a shift to oxidative phosphorylation in cancer cells.50, 51 Under acidic conditions cytokine production by activated T cells is abolished due to inhibition of glycolysis.52 Tumour‐derived lactate also contributes to T‐cell polarization toward a Treg phenotype.31 The acidic microenvironment of tumours has consequently become a target for intervention with lactate dehydrogenase‐A and monocarboxylate transporter inhibitors or oral bicarbonate supplementation. In a melanoma model, oral bicarbonate reduced tumour growth when administered with a programmed cell death protein‐1 (PD‐1) ‐specific antibody, and improved survival when combined with adoptive T‐cell transfer.52 These results suggest that extracellular pH in tumours is a mechanism whereby cancer cell metabolism can mediate lymphocyte dysfunction.
Nutrient restriction
Altered nutrient availability in tumours affects metabolic reprogramming of T cells, resulting in impaired effector functions and differentiation towards suppressive phenotypes. TILs are exposed to low extracellular glucose and glutamine due to high nutrient uptake by cancer cells. Intratumoural glucose is approximately ten times lower than in blood or spleen.26 The mere presence of cancer cells in co‐culture reduces glucose uptake by Th1 cells.26 TCR‐stimulated cells fail to proliferate in the absence of glucose, and have impaired effector functions.26, 48 Reductions in extracellular glucose do not impair ATP generation, suggesting that these cells are able to use alternative energy‐generating pathways such as glutaminolysis.53 In response to inadequate extracellular glucose, AMPK is activated in T cells and promotes glutamine metabolism to maintain cellular energetics and survival.21 However, the tumoricidal potential of T cells is remarkably reduced under low glucose conditions.21 One mechanism is mediated by the glucose metabolite phosphoenolpyruvate, which maintains cytosolic Ca2+ concentrations by inhibiting endoplasmic reticulum Ca2+ channels.26 Cytosolic Ca2+ concentration serves as a metabolic threshold, allowing activation of the family of transcription factors collectively named nuclear factor of activated T cells. Consequently, glucose deprivation results in a dose‐dependent decrease in IFN‐γ, mediated at the translational level by decreased mTOR activity.21 Specifically, translation of IFN‐γ from mRNA can be blocked by glyceraldehyde 3‐phosphate dehydrogenase when it is not engaged in glycolysis, highlighting the importance of glucose metabolism for maintenance of T‐cell function.54 Interferon‐γ is essential for CTL‐mediated cell‐cycle arrest and growth inhibition of murine B16 melanomas.55 Furthermore, glucose deprivation increases secretion of transforming growth factor‐β by Th cells, confirming a switch from an immunostimulatory to immunosuppressive microenvironment.26
Although nutrient restriction is not metabolically favourable for CTLs, T cells that rely on fatty acid oxidation thrive. TCR‐stimulated T cells in glutamine‐poor and glucose‐poor conditions preferentially differentiate into Treg cells, probably because their oxidative phenotype is metabolically suited to survive in this environment.31 α‐Ketoglutarate, the breakdown product of glutaminolysis is required for incorporation into the TCA cycle to fuel proliferation, and is necessary for expression of Tbet, a transcription factor required for Th1 differentiation.32 Under low glucose and glutamine conditions, differentiation of Treg cells and their proliferation predominate over Th1 cells, even in the presence of Th1‐polarizing cytokines.32 Specifically, the defining transcription factor of Treg cells, known as Foxp3, has been shown to reduce the expression of Myc, leading to reduced glycolysis and increased oxidative phosphorylation.56 This suggests that an environment with adequate nutrients is optimal for the differentiation of effector T cells.
Inhibitory checkpoints
Immunological checkpoints suppress immune responses following antigen clearance to avoid pathological autoimmunity. Cancer cells aberrantly express ligands for receptors on lymphocytes, and their interaction impairs T‐cell effector functions by inhibiting signalling downstream of the TCR.57 Cytotoxic T lymphocyte‐associated antigen‐4 (CTLA‐4) is expressed on T cells following activation, where it negatively regulates effector functions to maintain immunological homeostasis.14, 57 CTLA‐4 competes for the same ligands as CD28, the co‐stimulatory receptor, creating a negative feedback loop that ensures CTL activity is controlled and temporary.14 Ligation of CTLA‐4 inhibits glycolysis, preventing activation and differentiation of naive CTLs.50 PD‐1 is a T‐cell co‐inhibitory receptor that interacts with its ligands PD‐L1 and PD‐L2. PD‐L1 is normally expressed by self‐tissues to regulate peripheral tolerance, and is often expressed by cancer cells to impair T cells.57 The expression of both PD‐1 and PD‐L1 are highly dependent on microenvironmental conditions. Extracellular adenosine, a by‐product of altered tumour metabolism, induces expression of both CTLA‐4 and PD‐1 on T cells.16 Furthermore, tissue hypoxia and the presence of immunoregulatory cells impose metabolic stresses on T cells, resulting in increased expression of PD‐1.5, 58
Immune checkpoint blockade differentially affects metabolism of cancer and T cells
Antibodies that block ligation of CTLA‐4 or PD‐1 have remarkable clinical effectiveness in melanomas, where they reverse immunosuppression and promote T‐cell‐mediated cytotoxicity.57, 59 Although tumours with a higher mutational load seem to be more responsive to anti‐PD‐1 therapy, there remains a subset of melanoma patients who are non‐responsive despite similar mutational load, suggesting that PD‐1 blockade relies on more than just antigenicity of tumours.60 Recent evidence suggests that checkpoint inhibition may directly modify metabolism of T cells and cancer cells.7 Three checkpoint inhibitors are approved for the treatment of malignant melanomas in humans: Ipilimumab, a CTLA‐4‐specific monoclonal antibody, and pembrolizumab and nivolumab, which are PD‐1‐specific monoclonal antibodies.57, 61, 62
Programmed death protein ligand 1 is expressed on cancer cells and tumour‐associated macrophages.63 Expression on melanoma cells can be constitutive or induced by IFN‐γ secreted by TILs, and the degree of PD‐L1 expression in melanomas correlates with tumour growth.4, 64 Ligation of PD‐1 alters the metabolic phenotype of activated T cells, impairing mechanisms of energy generation and macromolecule synthesis needed for effector functions.65 By inhibiting glycolysis, and up‐regulating fatty acid oxidation through increased expression of carnitine palmitoyltransferase‐I, ligation of PD‐1 reduces cytokine secretion by activated CTLs.5, 65
Signalling through PD‐L1 also has direct metabolic effects on cancer cells. In response to PD‐L1 blockade, glucose uptake and lactate extrusion are decreased, suggesting that pathological expression of PD‐L1 by cancer cells not only impairs T‐cell metabolism, but benefits cancer cell metabolism.7 PD‐1 blockade inhibits melanoma xenograft growth in immunocompromised mice, an effect attributed to suppression of downstream mTOR signalling.66 Therefore, anti‐PD1/PD‐L1 therapy appears to be able to restore the metabolic balance in favour of T cells, providing an example of how to differentially impact cancer and T cells with shared metabolic requirements (Fig. 2).
Figure 2.
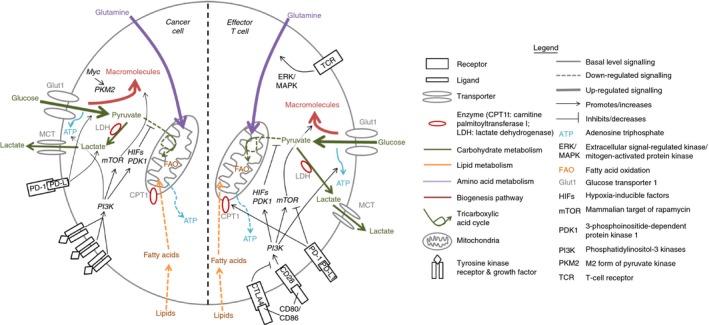
How immunological checkpoints differentially impact the metabolic profiles of cancer versus T cells. The effect of imposing immunological checkpoints on the metabolism of cancer cells (left side of illustration) compared with the metabolism of T cells (right side). Ligation of programmed death ligand‐1 (PD‐L1) on cancer cells promotes glucose uptake and production of lactate, which promotes survival. In contrast, ligation of programmed death receptor‐1 (PD‐1) on T cells inhibits glycolysis and up‐regulates fatty acid oxidation leading to impaired energy generation and macromolecule synthesis, which compromises proliferation and effector functions such as cytokine production. Therefore, inhibition of the PD‐1/PD‐L1 immune checkpoint would preferentially inhibit cancer cells, while promoting T‐cell functions. T cells have an additional inhibitory receptor, cytotoxic T lymphocyte‐associated protein 4 (CTLA‐4), which competes with CD28 for binding to the ligands CD80 and CD86 on mature antigen‐presenting cells, resulting in a reduction in PI3K/Akt signalling. This causes decreased expression of GLUT1, increased mitochondrial oxidation and fatty acid uptake, and decreased biosynthesis. This is why there is also interest in blocking CTLA4 on tumour‐infiltrating T cells.
Metabolic targeting agents
Therapies aimed at restoring normal cellular energetics attempt to exploit the dependence of cancer cells on glucose and glutamine.13, 67 However, metabolic disruptors can potentially impair T cells that may be critical for therapeutic success due to metabolic similarities with cancer cells (Fig. 1). Energy disruptors aim to reduce glycolysis and disturb mitochondrial biogenesis to leverage the metabolic differences between cancer cells and quiescent host cells. Drugs proposed for treating cancers include 2‐deoxyglucose (2DG), a glycolysis inhibitor, metformin, a mitochondrial respiratory complex inhibitor and a host of mitochondrial biogenesis inhibitors developed to selectively target CSCs.68, 69, 70 Since there is potential for these agents to interfere with T‐cell metabolism, the long‐term effects of their use as therapeutic agents should be evaluated in immunocompetent animal models, with special consideration given to combinations with immunotherapies.
2‐Deoxyglucose is structurally similar to glucose but inhibits hexokinase activity, shutting down glycolysis.71, 72 This leads to metabolic disruptions in cancer cells, including depletion of ATP, impairment of biosynthetic pathways, and reactive oxygen species‐mediated growth arrest. Notably, 2DG could be safely administered to human patients with gliomas that received radiotherapy in a phase I/II clinical trial.73 A well‐tolerated dose of 2DG was also found in a phase I trial that enrolled patients with castration‐resistant prostate cancers and other advanced malignancies.74 However, relatively low serum concentrations of 2DG were achieved; levels that cancer cells could resist by induction of autophagy in pre‐clinical studies.75 Disconcertingly, 2DG not only affects cancer cells but similarly impairs the metabolism of human T cells, and results in a greater reduction of cytokine production than glucose deprivation alone.76
Due to a heavy reliance of many cancer cells on lactate generation to support their growth, the inhibition of LDH shows promise as a therapeutic intervention. Indeed, the growth of melanomas can be arrested by oxamate and dichloroacetate‐mediated inhibition of LDH.13 Cancer stem‐like cells can be particularly reliant on LDH, as demonstrated in the context of non‐small cell lung cancer.77 Also, blocking lactate extrusion in colorectal cancer cells impairs the glycolytic phenotype and increases the cytotoxicity of 5‐Fluorouracil treatment.78 Importantly, LDH inhibition can also rescue proliferating T cells from the immunosuppressive effects of l‐lactate, making this an ideal target to differentially impact cancer cells and the immune system.17
Metformin is commonly used to treat type II diabetes due to its effect of reducing hepatic glucose production. A link between metformin use and reduced risk of cancer in individuals with type II diabetes was established from retrospective epidemiological studies.79 The protective effect of metformin has been confirmed in randomized control trials examining the incidence of various cancers including lung and prostate cancers in people with diabetes taking metformin.80 Metformin has been proposed as a treatment for melanomas due to the limitations of current therapies.79 Beneficial effects of metformin have been observed in melanoma cells in vitro, but in animal models the results have been inconclusive, perhaps due to differences in dosing.81, 82 The two main targets of metformin are the mitochondrial respiratory complex I and AMPK. Both are essential mediators of cancer cell and T‐cell metabolism, suggesting that metformin may be a potent regulator of anti‐cancer immunity as well as a metabolic disruptor of cancer cells.
Inhibition of complex I
Metformin targets complex I of the mitochondrial respiratory chain, effectively depleting cellular ATP levels.83 Although most cancers are not reliant on this oxidative phosphorylation, and simply up‐regulate glycolysis and glutaminolysis to generate ATP, certain cancer cells such as CSCs, are unable to compensate.71, 72 For this reason metformin is thought to preferentially target CSCs. Inhibition of complex I also has implications on anti‐tumour immunity. Experimental small interfering RNA‐mediated knockdown of complex I results in reduced IL‐2 and IL‐4 secretion by activated T cells.84 As IL‐4 is associated with differentiation of CD4+ T cells into Th2 cells, blocking IL‐4 through complex I inhibition may represent a preferential and beneficial effect against tumour‐promoting T‐cell subsets. However, complex I blockade also has pro‐tumorigenic effects, leading to elevated lactate production and vascular endothelial growth factor expression in cancer cells, causing increased tumour growth in mice.13 Due to potentially offsetting effects of its down‐regulation, the role of complex I in lymphocyte and cancer cell metabolism requires greater understanding before being targeted for therapeutic intervention.
Activation of AMPK
Metformin also activates AMPK, which is a protein complex at the interface of metabolism and cellular function, regulating intracellular energy status by suppressing anabolic pathways and promoting catabolic pathways in times of cellular stress.85, 86 Because of its pleiotropic effects, AMPK can regulate tumour‐suppressing and tumour‐promoting processes, in a manner that is poorly understood.85 AMPK's ability to counteract the Warburg effect by down‐regulating glycolytic gene expression suggests that metformin's AMPK‐activating effects may have anti‐proliferative effects in cancer cells.87 This has been supported by models of prostate, colon, lung and breast cancers.79 However, AMPK activation may be pro‐tumorigenic by mediating adaptation to metabolic stresses, contributing to increased survival of cancer cells.88, 89, 90 Similarly, metformin's AMPK‐activating ability may have pleiotropic and conflicting effects on T cells. Metformin can protect CTLs from apoptosis and promote effector functions, causing regression of murine B16 melanomas.91 However, AMPK is active during metabolic reprogramming of effector T cells to memory cells and during differentiation to Treg cells by promoting the oxidative phenotype of these cells. AMPK activation suppresses glycolysis and promotes more efficient means of generating energy such as lipid oxidation, which is required by Treg cells and memory T cells, but suppresses effector T cells.22 This suggests that AMPK activation by metformin may alter the composition of TILs, favouring those with less cytotoxic functions. Indeed, metformin treatment improved memory T‐cell responses in mice, as demonstrated by improved secondary responses to leukaemia cells.91 Also, metformin pushes the Treg/Th17 balance toward immunosuppressive Treg cells in autoimmune diseases.92, 93 AICAR, a direct AMPK activator, similarly promotes expansion of Treg cells and reduces Th17 cells by promoting fatty acid oxidation.94 The implications of metformin treatment on anti‐tumour immunity have yet to be conclusively resolved.
Future directions
The plethora of mechanisms that cancers use to escape treatments argues in favour of implementing combination therapies. Indeed, simultaneous use of multiple metabolic inhibitors, such as metformin with 2DG, has been proposed.95 However, these kinds of studies need to account for the potential to compromise tumour‐specific immunity. From this perspective, combining metabolic disruptors with immune checkpoint blockade may be particularly appealing. Both treatment modalities are most commonly used as adjuvant therapies with chemotherapeutic agents or small molecule inhibitors, where they can contribute to direct and immune‐mediated cancer cell death. By operating through distinct and complementary mechanisms, immune checkpoint blockade combined with metabolic disruptors has the potential to impair cancer cell metabolism and increase tumour‐specific immunity, skewing the balance in favour of tumour clearance. Indeed, pre‐clinical research has demonstrated the benefit of using metformin to counteract hypoxia in the tumour microenvironment, resulting in potentiation of PD‐1 blockade.96 Also, in a retrospective analysis of users of chronic medications while receiving Ipilimumab, those on metformin or proton pump inhibitors were more likely to respond to Ipilimumab treatment, providing support for intentionally combining these therapies.97 Importantly, the combination of these chronic medications with immune checkpoint inhibitors does not appear to increase the risk of immune‐related adverse reactions.97 Future therapeutic regimens could combine these agents to attack cancers by multiple mechanisms. However, to reiterate, great caution must be practiced when testing metabolic inhibitors to ensure that deleterious effects are focused on cancer cells, not T lymphocytes. A particularly promising alternative to metabolic inhibitors is respiratory hyperoxia, a strategy in which supraphysiological oxygen levels in the blood reduce hypoxia‐driven extracellular accumulation of highly immunosuppressive adenosine in tumours.98 Excitingly, this modulates the immunological milieu of the tumour microenvironment in a way that potentiates the recruitment and function of T and natural killer cells. Supplemental oxygen represents a safe, simple and non‐invasive strategy that could interface well with a variety of immunotherapies, especially cancer vaccines and adoptive T‐cell therapy.99
Conclusions
There is compelling evidence for a link between metabolism and T‐cell functions. Indeed, expression profiling identified 55 immunometabolic genes that are essential mediators of metabolic pathways that contribute to lymphocyte functions.53 Testing of metabolic inhibitors should emphasize the use of immunocompetent animal models, with a focus on comprehensively assessing impacts on tumours and the immune system. Immunocompromised hosts facilitate testing drugs on patient‐derived tumour xenografts. However, exclusive use of these models to study metabolic disruptors should be avoided. Ideal animal modelling may be facilitated by humanized mice in which tumours from patients could be tested in the context of matched immune systems. Mice that encode multiple human cytokines and growth factors enable engraftment of innate components of the human immune system along with matched tumour grafts.100 Although major components of the adaptive immune system can develop in these mice, there is room for improvement. Including agents that optimize immunometabolism in cancer treatments has the potential to prevent or reverse immunosuppression in patients, reduce doses of cytotoxic drugs, and promote long‐term anti‐cancer responses that persist after drug withdrawal.
Disclosures
The authors have no competing interests.
Acknowledgements
All three authors conducted literature searches and wrote the paper. Funding for this work was provided by the Terry Fox Research Institute under Project #1041 . K.E.A. received an MSc scholarship from the Ontario Veterinary College, University of Guelph.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–74. [DOI] [PubMed] [Google Scholar]
- 2. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11:85–95. [DOI] [PubMed] [Google Scholar]
- 3. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiang Y, Li Y, Zhu B. T‐cell exhaustion in the tumor microenvironment. Cell Death Dis 2015; 6:e1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y, Ertl HC. Starved and asphyxiated: how can CD8+ T cells within a tumor microenvironment prevent tumor progression. Front Immunol 2016; 7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour‐infiltrating lymphocytes in cancer: a systematic review with meta‐analysis. Br J Cancer 2011; 105:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD et al Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015; 162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ferreira LM, Hebrant A, Dumont JE. Metabolic reprogramming of the tumor. Oncogene 2012; 31:3999–4011. [DOI] [PubMed] [Google Scholar]
- 9. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol 1927; 8:519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pauwels EK, Sturm EJ, Bombardieri E, Cleton FJ, Stokkel MP. Positron‐emission tomography with [18F]fluorodeoxyglucose. Part I. Biochemical uptake mechanism and its implication for clinical studies. J Cancer Res Clin Oncol 2000; 126:549–59. [DOI] [PubMed] [Google Scholar]
- 11. Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med 2013; 45:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rodrigues MF, Obre E, de Melo FH, Santos GC Jr, Galina A, Jasiulionis MG et al Enhanced OXPHOS, glutaminolysis and β‐oxidation constitute the metastatic phenotype of melanoma cells. Biochem J 2016; 473:703–15. [DOI] [PubMed] [Google Scholar]
- 13. Chaube B, Malvi P, Singh SV, Mohammad N, Meena AS, Bhat MK. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015; 6:37281–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weinberg RA. The Biology of Cancer, 2nd edn. New York, USA: Garland Science, 2014. [Google Scholar]
- 15. Frosig TM, Lyngaa R, Met O, Larsen SK, Donia M, Svane IM et al Broadening the repertoire of melanoma‐associated T‐cell epitopes. Cancer Immunol Immunother 2015; 64:609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol 2016; 7:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M et al Inhibitory effect of tumor cell‐derived lactic acid on human T cells. Blood 2007; 109:3812–9. [DOI] [PubMed] [Google Scholar]
- 18. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Can Res 2012; 72:5435–40. [DOI] [PubMed] [Google Scholar]
- 19. Belladonna ML, Puccetti P, Orabona C, Fallarino F, Vacca C, Volpi C et al Immunosuppression via tryptophan catabolism: the role of kynurenine pathway enzymes. Transplantation 2007; 84(1 Suppl):S17–20. [DOI] [PubMed] [Google Scholar]
- 20. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J et al A2A receptor signaling promotes peripheral tolerance by inducing T‐cell anergy and the generation of adaptive regulatory T cells. Blood 2008; 111:251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia‐Vazquez G, Yurchenko E et al The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo . Immunity 2015; 42:41–54. [DOI] [PubMed] [Google Scholar]
- 22. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF et al Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 2011; 186:3299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang R, Solt LA. Metabolism of murine T 17 cells: impact on cell fate and function. Eur J Immunol 2016; 46:807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity 2015; 43:435–49. [DOI] [PubMed] [Google Scholar]
- 25. Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol 2014; 32:609–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R et al Phosphoenolpyruvate is a metabolic checkpoint of anti‐tumor T cell responses. Cell 2015; 162:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Macintyre AN, Rathmell JC. Activated lymphocytes as a metabolic model for carcinogenesis. Cancer Metab. 2013; 1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ho PC, Liu PS. Metabolic communication in tumors: a new layer of immunoregulation for immune evasion. J Immunother Cancer 2016; 4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W et al Tumor‐induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta 2014; 1845:182–201. [DOI] [PubMed] [Google Scholar]
- 30. Whiteside TL. Induced regulatory T cells in inhibitory microenvironments created by cancer. Expert Opin Biol Ther 2014; 14:1411–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Molon B, Cali B, Viola A. T cells and cancer: how metabolism shapes immunity. Front Immunol 2016; 7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L et al Glutamine‐dependent α‐ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal 2015; 8:ra97. [DOI] [PubMed] [Google Scholar]
- 33. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC et al The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 2016; 45:374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delgoffe GM, Powell JD. Feeding an army: the metabolism of T cells in activation, anergy, and exhaustion. Mol Immunol 2015; 68:492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parks SK, Cormerais Y, Marchiq I, Pouyssegur J. Hypoxia optimises tumour growth by controlling nutrient import and acidic metabolite export. Mol Aspects Med 2016; 47–48:3–14. [DOI] [PubMed] [Google Scholar]
- 36. Semenza GL. HIF‐1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013; 123:3664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhattacharya B, Mohd Omar MF, Soong R. The Warburg effect and drug resistance. Br J Pharmacol 2016; 173:970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Epstein T, Xu L, Gillies RJ, Gatenby RA. Separation of metabolic supply and demand: aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Cancer Metab. 2014; 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vlashi E, Pajonk F. The metabolic state of cancer stem cells‐a valid target for cancer therapy? Free Radic Biol Med 2015; 79:264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer 2016; 114:1305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S et al Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest 2016; 126:1834–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR et al The CD28 signaling pathway regulates glucose metabolism. Immunity 2002; 16:769–77. [DOI] [PubMed] [Google Scholar]
- 43. Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J et al mTORC1 and mTORC2 selectively regulate CD8+ T cell differentiation. J Clin Invest 2015; 125:2090–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D et al The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 2014; 20:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016; 354:481–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dimeloe S, Mehling M, Frick C, Loeliger J, Bantug GR, Sauder U et al The immune‐metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J Immunol 2016;196:106–14. [DOI] [PubMed] [Google Scholar]
- 47. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G et al Rapid effector function of memory CD8+ T cells requires an immediate‐early glycolytic switch. Nat Immunol 2013; 14:1064–72. [DOI] [PubMed] [Google Scholar]
- 48. Finlay DK. Starved human T lymphocytes keep fighting. Eur J Immunol 2015; 45:2480–3. [DOI] [PubMed] [Google Scholar]
- 49. Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab 2012; 16:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol 2015; 36:257–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peppicelli S, Toti A, Giannoni E, Bianchini F, Margheri F, Del Rosso M et al Metformin is also effective on lactic acidosis‐exposed melanoma cells switched to oxidative phosphorylation. Cell Cycle 2016; 15:1908–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pilon‐Thomas S, Kodumudi KN, El‐Kenawi AE, Russell S, Weber AM, Luddy K et al Neutralization of tumor acidity improves antitumor responses to immunotherapeutic interventions. Cancer Res 2015; 76:1381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Han F, Li G, Dai S, Huang J. Genome‐wide metabolic model to improve understanding of CD4+ T cell metabolism, immunometabolism and application in drug design. Mol BioSyst 2016; 12:431–43. [DOI] [PubMed] [Google Scholar]
- 54. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D et al Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013; 153:1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matsushita H, Hosoi A, Ueha S, Abe J, Fujieda N, Tomura M et al Cytotoxic T lymphocytes block tumor growth both by lytic activity and IFNγ‐dependent cell‐cycle arrest. Cancer Immunol Res 2015; 3:26–36. [DOI] [PubMed] [Google Scholar]
- 56. Angelin A, deGil‐ ‐Gómez L , Dahiya S, Jiao J, Guo L, Levine MH et al Foxp3 reprograms T cell metabolism to function in low‐glucose, high‐lactate environments. Cell Metab 2017; 25:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol 2015; 33:1974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang Y, Ertl HC. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+ T cells within tumors. Oncotarget 2016; 7:23282–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Valko‐Rokytovska M, Bruchata K, Simkova J, Milkovicova M, Kostecka Z. Current trends in the treatment of malignant melanoma. Neoplasma 2016; 63:333–41. [DOI] [PubMed] [Google Scholar]
- 60. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu‐Lieskovan S et al Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 2016; 165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen YS, Shen CR. Immune checkpoint blockade therapy: the 2014 Tang prize in biopharmaceutical science. Biomed J 2015; 38:5–8. [DOI] [PubMed] [Google Scholar]
- 62. Swatler J, Kozlowska E. Immune checkpoint targeted cancer immunotherapies. Postepy Hig Med Dosw 2016; 70:25–42. [DOI] [PubMed] [Google Scholar]
- 63. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A et al Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA‐4, and PD‐L1 in mice with brain tumors. Clin Cancer Res 2014; 20:5290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but not constitutive expression of PD‐L1 in human melanoma cells is dependent on activation of NF‐κB. PLoS ONE 2015; 10:e0123410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN et al PD‐1 alters T‐cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 2015; 6:6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E et al Melanoma cell‐intrinsic PD‐1 receptor functions promote tumor growth. Cell 2015; 162:1242–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gostner JM, Becker K, Uberall F, Fuchs D. The potential of targeting indoleamine 2,3‐dioxygenase for cancer treatment. Expert Opin Ther Targets 2015; 19:605–15. [DOI] [PubMed] [Google Scholar]
- 68. Wang J, Wen X, Liu J, Sun H. Mitochondrial biogenesis inhibitors for anticancer therapy: a review of recent patents. Recent Pat Anticancer Drug Discov 2016; 11:332–41. [DOI] [PubMed] [Google Scholar]
- 69. Liu H, Hu YP, Savaraj N, Priebe W, Lampidis TJ. Hypersensitization of tumor cells to glycolytic inhibitors. Biochemistry 2001; 40:5542–7. [DOI] [PubMed] [Google Scholar]
- 70. Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B et al Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase‐dependent manner. Cell Metab 2010; 11:390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bost F, Decoux‐Poullot AG, Tanti JF, Clavel S. Energy disruptors: rising stars in anticancer therapy? Oncogenesis 2016; 5:e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochem Biophys Acta 2002; 1555:14–20. [DOI] [PubMed] [Google Scholar]
- 73. Mohanti BK, Rath GK, Anantha N, Kannan V, Das BS, Chandramouli BA et al Improving cancer radiotherapy with 2‐deoxy‐d‐glucose: phase I/II clinical trials on human cerebral gliomas. Int J Radiat Oncol Biol Phys 1996; 35:103–11. [DOI] [PubMed] [Google Scholar]
- 74. Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K et al Targeting tumor metabolism with 2‐Deoxyglucose in patients with castrate‐resistant prostate cancer and advanced malignancies. Prostate 2010; 70:1388–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. DiPaola RS, Dvorzhinski D, Thalasila A, Garikapaty V, Doram D, May M et al Therapeutic starvation and autophagy in prostate cancer: a new paradigm for targeting metabolism in cancer therapy. Prostate 2008; 68:1743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Renner K, Geiselhoringer AL, Fante M, Bruss C, Farber S, Schonhammer G et al Metabolic plasticity of human T cells: preserved cytokine production under glucose deprivation or mitochondrial restriction, but 2‐deoxy‐glucose affects effector functions. Eur J Immunol 2015; 45:2504–16. [DOI] [PubMed] [Google Scholar]
- 77. Xie H, Hanai J, Ren JG, Kats L, Burgess K, Bhargava P et al Targeting lactate dehydrogenase A inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor‐initiating cells. Cell Metab 2014; 19:795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Amorim R, Pinheiro C, Miranda‐Goncalves V, Pereira H, Moyer MP, Preto A et al Monocarboxylate transport inhibition potentiates the cytotoxic effect of 5‐fluorouracil in colorectal cancer cells. Cancer Lett 2015; 365:68–78. [DOI] [PubMed] [Google Scholar]
- 79. Cerezo M, Tomic T, Ballotti R, Rocchi S. Is it time to test biguanide metformin in the treatment of melanoma? Pigment Cell Melanoma Res 2015; 28:8–20. [DOI] [PubMed] [Google Scholar]
- 80. Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta‐analysis. PLoS ONE 2012; 7:e33411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A et al Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis 2011; 2:e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Martin MJ, Hayward R, Viros A, Marais R. Metformin accelerates the growth of BRAF V600E‐driven melanoma by upregulating VEGF‐A. Cancer Discov 2012; 2:344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti‐diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 2000; 348:607–14. [PMC free article] [PubMed] [Google Scholar]
- 84. Kaminski MM, Sauer SW, Klemke CD, Suss D, Okun JG, Krammer PH et al Mitochondrial reactive oxygen species control T cell activation by regulating IL‐2 and IL‐4 expression: mechanism of ciprofloxacin‐mediated immunosuppression. J Immunol 2010; 184:4827–41. [DOI] [PubMed] [Google Scholar]
- 85. Zadra G, Batista JL, Loda M. Dissecting the dual role of AMPK in cancer: from experimental to human studies. Molecular cancer research: MCR 2015; 13:1059–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk‐Melody J et al Role of AMP‐activated protein kinase in mechanism of metformin action. Journal of Clinical Investigation 2001; 108:1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z et al AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo . Cell Metab 2013; 17:113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ng TL, Leprivier G, Robertson MD, Chow C, Martin MJ, Laderoute KR et al The AMPK stress response pathway mediates Anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ 2012; 19:501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kato K, Ogura T, Kishimoto A, Minegishi Y, Nakajima N, Miyazaki M et al Critical roles of AMP‐activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene 2002; 21:6082–90. [DOI] [PubMed] [Google Scholar]
- 90. Park HU, Suy S, Danner M, Dailey V, Zhang Y, Li H et al AMP‐activated protein kinase promotes human prostate cancer cell growth and survival. Mol Cancer Ther 2009; 8:733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune‐mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A. 2015; 112:1809–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Son HJ, Lee J, Lee SY, Kim EK, Park MJ, Kim KW et al Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediators Inflamm 2014; 2014:973986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lee SY, Lee SH, Yang EJ, Kim EK, Kim JK, Shin DY et al Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/Treg balance. PLoS ONE 2015; 10:e0135858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gualdoni GA, Mayer KA, Goschl L, Boucheron N, Ellmeier W, Zlabinger GJ. The AMP analog AICAR modulates the Treg/Th17 axis through enhancement of fatty acid oxidation. FASEB J 2016; 30:3800–9. [DOI] [PubMed] [Google Scholar]
- 95. Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P et al Targeting cancer cell metabolism: the combination of metformin and 2‐deoxyglucose induces p53‐dependent apoptosis in prostate cancer cells. Can Res 2010; 70:2465–75. [DOI] [PubMed] [Google Scholar]
- 96. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD‐1 blockade is potentiated by metformin‐induced reduction of tumor hypoxia. Cancer Immunol Res 2017; 5:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Failing JJ, Finnes HD, Kottschade LA, Allred JB, Markovic SN. Effects of commonly used chronic medications on the outcomes of ipilimumab therapy in patients with metastatic melanoma. Melanoma Res 2016; 26:609–15. [DOI] [PubMed] [Google Scholar]
- 98. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK et al A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA 2006; 103:13132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R et al Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med 2015; 7:277ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato‐lymphoid system mice. Haematologica 2016; 101:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]