

Summary
One of the most widespread and effective environmental factors is the infection with enteroviruses (EVs) which accelerate β cell destruction in type 1 diabetes (T1D). This study represented a comparison between diabetic EV+ and EV– children as well as correlation analysis between autoantibodies, T1D markers, cytokines, complement activation products and anti‐coxsackievirus (CV) immunoglobulin (Ig)G. EV RNA was detected in Egyptian children with T1D (26·2%) and healthy controls (0%). Detection of anti‐CV IgG in T1D‐EV+ resulted in 64% positivity. Within T1D‐EV+, previously diagnosed (PD) showed 74 versus 56% in newly diagnosed (ND) children. Comparisons between populations showed increased levels of haemoglobin A1c (HbA1c), C‐reactive protein (CRP), nitric oxide (NO), glutamic acid decarboxylase and insulin and islet cell autoantibodies [glutamic acid decarboxylase autoantibodies (GADA), insulin autoantibodies (IAA) and islet cell cytoplasmic autoantibodies (ICA), respectively], interferon (IFN)‐γ, tumour necrosis factor (TNF)‐α, interleukin (IL)‐1β, IL −10, IL −12, IL −17, C3d and sC5–9 in T1D‐EV+ versus T1D‐EV–. Conversely, both IL‐20 and transforming growth factor (TGF‐β) decreased in T1D‐EV+ versus EV–, while IL‐4, −6 and −13 did not show any changes. Correlation analysis showed dependency of accelerated autoimmunity and β cell destruction on increased IFN‐γ, IL‐12 and IL‐17 versus decreased IL‐4, −6 and −13. In conclusion, IFN‐γ, IL‐12 and IL‐17 played an essential role in exacerbating EV+‐T1D, while C3d, sC5b −9, IL‐10 and −20 displayed distinct patterns.
Keywords: complement, correlation, cytokines, enteroviruses, type 1 diabetes
Introduction
The global rapid spread of type 1 diabetes (T1D) disease cannot be explained solely by genetic predisposition but also to environmental factors, including viral infections 1. Enteroviruses (EVs) are found to play a major role in induction of the disease based on three possible mechanisms 2. The first is the invasion of pancreatic cells by viruses through cell surface receptors and replication followed by induction of innate immunity against the cells leading to destruction 3. The second is altered immunity as a result of viral infection leading to autoimmunity. The third is the induction of cross‐reactive immunity between EV and pancreatic β cell antigens as a result of antigen mimicry. EVs are the most likely viruses to be linked with the onset of T1D 4. The Enterovirus genus of the Picornaviridae family consists of small, non‐enveloped, positive, single‐strand RNA viruses, including polio viruses, coxsackieviruses (CVs) A and B and echoviruses 5. Transmission methods were identified to be faecal–oral or, less commonly, respiratory routes. Detection methods for EVs in serum, saliva or stool were dependent upon either reverse transcription–polymerase chain reaction (RT–PCR) or antibody assays 5.
Immunological and metabolic biomarkers were used widely in the diagnosis of T1D or β cell destruction 6. Immunological markers included islet cell cytoplasmic autoantibodies (ICA), glutamic acid decarboxylase autoantibodies (GADA), insulin autoantibodies (IAA) and pancreatic β cell‐specific zinc transporter (ZnT‐8) autoantibody. Cytokine production, including interleukin (IL)‐1β, tumour necrosis factor (TNF)‐α, interferon (IFN)‐γ, IL‐2 and IL‐17, were found to be highly characteristic for T1D patients compared to the controls, who were characterized by IL‐10 7, 8, 9, 10. However, IL‐10, together with IL‐6 and IL‐13, were found to be associated with the pathogenesis caused by EV infection 11. Haemoglobin A1c (HbA1c) is a direct indicator for glucose blood level in both of T1D and T2D, while C‐peptide correlates with insulin secretion. Both of them are used as markers for pancreatic β cell destruction 12.
Inflammatory responses, including elevated C‐reactive protein (CRP) and nitric oxide (NO), were found to be associated with T1D with or without EV infection, while anti‐inflammatory cytokines including IL‐4, IL‐10 and IL‐13 were found to be cytoprotective against β cell destruction 13. Regulatory T cells (Treg) by secretion of TGF‐β were also found to play an essentially protective role in prevention of T1D‐associated autoimmunity 14. The inflammatory reactions also promote complement activity, which could be measured by detecting the levels of C3d and sC5b–9 in serum 15.
The current study proposed to test the prevalence of EV in paediatric patients with T1D and compare between age‐ and sex‐matched T1D‐EV– and T1D‐EV+ patients. It also aimed to detect statistical correlations between anti‐CV immunoglobulins (IgGs), complement activity and cytokines.
Materials and methods
Study population and sample collection
The study population consisted of 382 children with T1D and 100 healthy controls. These children visited the Institute of Endocrinology and Diabetes, Cairo University, Cairo, Egypt during the period from October 2013 to September 2014 for consultancies. Children with T1D ranged from 2 to 16 [9·8 ± 2·9; mean ± standard deviation (s.d.)] years, while healthy controls ranged from 3 to 14 (9·1 ± 2·7; mean ± s.d.) years. Each of the diabetic and control groups consisted of an equal number of males and females. After written informed parental consents were obtained and the study was approved by the ethical committee, blood samples were collected. Venous blood (5 ml) was collected in serum tubes (Becton‐Dickinson, Franklin Lakes, NJ, USA) through venous puncture using a 21‐gauge needle. Serum samples were obtained by centrifugation at +4°C, 4500 g for 10 min and kept at −80°C until use. According to the criterion of the World Health Organization (WHO) and the American Diabetes Association, all participants were subjected to both random blood sugar and haemoglobin A1c (HbA1c) assays using commercial kits purchased from Spinreact, Girona, Spain and MyBiosource, San Diego, CA, USA, respectively. No participants (diabetic and controls) were obese or overweight. They were also free of infectious diseases, neoplastic, inflammatory and autoimmune disorders and allergies. Patients receiving immunomodulatory drugs were also excluded.
In‐vitro amplification
RNA extraction from serum as well as amplification reactions has been described previously 16, 17. Primers were selected to amplify sequences within the 5′ non‐coding region (5′NCR), which is a highly conserved zone among all EV serotypes. The external primers (EV/PCR1, 5′‐ATTGTCACCATAAGCAGCCA‐ 3′; EV/PCR2, 5′‐TCCTCCGGCCCCCTGAATGCG‐ 3′) generate a 154 base pairs (bp) fragment, whereas the internal primers (EV/PCR3, 5′‐ ACACGGACACCCAAAGTAGTCGGTTCC‐3′; EV/PCR4, 5′ TCCGGCCCCTGAATGCGGCTAATCC‐3′) generate a 114 bp PCR product.
Extracted samples (10 μl) were heated to 99°C for 5 min and placed immediately on ice. Salts, nucleotides, 0·3 μM of each primer and 100 U of reverse transcriptase (ThermoScientific, Waltham, MA, USA) were added in a 5‐μl final volume. The samples were incubated for 60 min at 42°C for the RT reaction. Five μl of the RT product was added to a final volume of 50 μl of the PCR reaction mix containing 5 μl of the PCR buffer (ThermoScientific) containing 1 μm of each primer (EV/PCR1 and EV/PCR2) and 2·5 U of the Taq DNA polymerase enzyme (ThermoScientific). After a denaturation step of 94°C for 4 min, 40 cycles of amplification at 92°C for 1·5 min, 55°C for 1·5 min and 72°C for 2 min were performed with a final extension of 72°C for 10 min. The nested PCR involved adding 2 µl of first‐round PCR product to a 48‐µl PCR mix containing 1 μM of each primer (EV/PCR3 and EV/PCR4) and 2·5 U of Taq DNA polymerase (ThermoScientific). The same cycling conditions used for the first nested PCR were also used in the second nested PCR. PCR products (10 μl) were analysed by electrophoresis on 1·5% agarose (Applichem GmbH, Darmstadt, Germany) gels containing 0·5 mg ethidium bromide/ml. All the control non‐diabetic samples were found negative to infection with EV. According to the diabetic positive and negative samples, patients were allocated into two groups of T1D‐EV+ and T1D‐EV–, respectively. For each group, newly diagnosed (ND) and previously diagnosed (PD) children were separated based on the duration (< 1 or > 1 year) of T1D diagnosis 18.
Sequencing
Ten PCR products of EV+ samples were sequenced. Fifty to 100 µl PCR products were purified using the PCR products purification kit (Qiagen GmbH, Hilden, Germany), following the manufacturer's instructions. Cycle sequencing was performed on 1–7 μl of the purified products with BigDye® Terminator version 3.1 Cycle Sequencing kit (ThermoScientific) using 40 pmol of the same primers as in PCR, following the manufacturer's instructions. Cycle sequencing consisted of 25 cycles of 94°C for 10 s, 55°C for 5 s and 60°C for 4 min. Analysis of the products was carried out on an ABI prism 310 genetic analyser (Applied Biosystems, Waltham, MA, USA). Sequence data from both strands of the PCR products were aligned and compared using the Basic Local Alignment Search Tool (BLAST).
Assessment of serum C‐peptide, CRP and NO
The C‐peptide enzyme‐linked immunosorbent assay (ELISA) kit was purchased from DRG Diagnostics (Marburg, Germany) and the CRP ELISA kit was purchased from R&D Systems (Minneapolis, MN, USA), while NO was assayed using a colorimetric kit (BioVision, Milpitas, CA, USA). The procedures were performed according to the kit instructions provided.
Autoantibody determinations
GADA and ICA were determined using ELISA kits purchased from MyBiosource, and IAA was assessed using an ELISA kit purchased from AlphaDiagnostic International (San Antonio, CA, USA).
Determination of anti‐CV IgG
The ELISA classic CV IgG kit was purchased from Serion GmbH (Würzburg, Germany). Procedure was performed according to the kit instructions. EV+ patients who had positive (above cut‐off value) and negative (below cut‐off value) IgG readings were allocated into CV+ and CV– groups. Cut‐off value was calculated by mean of control + (2 × s.d. of control).
Cytokine assays
Sandwich ELISA kits for TNF‐α, IFN‐γ, IL‐1β, IL‐6, IL‐12, IL‐17, IL‐10, IL‐13, IL‐20 and TGF‐β were purchased from R&D Systems.
Complement activity
The complement activation product C3d was measured by ELISA, as described previously 19, where polyclonal rabbit anti‐human C3d antibodies (Dako, Glostrup, Denmark) were used as the coating antibody and murine monoclonal antibody to C3d (Quidel, San Diego, CA, USA) was used as detecting antibody. Horseradish peroxidase (HRP)‐conjugated goat anti‐mouse antibody (KPL, Gaithersburg, MD, USA) and O‐phenylenediamine (Sigma, St Loius, MO, USA) were used for colorimetric enzyme‐substrate reaction.
The soluble terminal complement complex (sC5b‐9) level was assessed by ELISA using a monoclonal antibody (Quidel) against a neoepitope 20, as described previously 21.
Statistical analysis
Data are presented as mean ± s.d. Statistical analysis was performed using spss version 20 for Windows (SPSS Inc., Chicago, IL, USA). The comparisons between healthy and T1D‐EV– children or between T1D‐EV– and T1D‐EV+ children were determined by one‐way analysis of variance (anova) least significant difference (LSD) t‐test. A simple linear correlation analysis was processed by Pearson's method to measure the degree of dependency between variables using the MedCalc statistical program (MedCalcOstend, Belgium). Statistical significance was assumed at P < 0·05.
Results
Frequency of EV RNA and anti‐CV IgGs
A higher frequency of EV RNA was found in children with T1D (26·2%, 100 of 382) and none in healthy control children (0%, 0 of 100). Positive samples (two of nine samples) were identified by appearance of bands at 114 bp size (Fig. 1). Detection of anti‐CV IgG revealed 2% positivity in control, 0% in the T1D‐EV– (EV RNA‐negative) and 64% in T1D‐EV+ (EV RNA‐positive) groups (Table 1). The T1D‐EV– group showed no change (P > 0·05) in IgG levels compared to control, while T1D‐EV+ was significantly higher (P < 0·001) than T1D‐EV–. Comparison between ND and PD patients for anti‐CV IgG in the EV+ group indicated 56 and 74% in ND and PD, respectively. Sequence analysis of PCR positive samples indicated 97% similarity with human CVB4 isolate CB4_Cph15 polyprotein gene (Accession no. KC558570).
Figure 1.
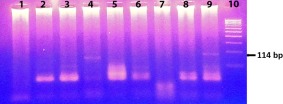
Nested reverse transcription–polymerase chain reaction (RT–PCR) products of nine samples from type 1 diabetes (T1D) children. The test revealed two positive samples [lanes 4 and 9 show enterovirus (EV)‐specific fragment of 114 base pairs (bp)] for EV infection out of nine samples. Lane 10 corresponds to DNA ladder. [Colour figure can be viewed at wileyonlinelibrary.com].
Table 1.
Number and percentages of positive serum samples in reverse transcription–polymerase chain reaction (RT–PCR) [detection of enterovirus (EV)] and enzyme‐linked immunosorbent assay (ELISA) immunoglobulin (IgG) [detection of coxsackievirus (CV)]
| Group | No. of children | Screening RT–PCR (EV) | Group | No. of children | ELISA IgG (CV) | Group | No. of children | ELISA IgG (CV) | Duration of T1D |
|---|---|---|---|---|---|---|---|---|---|
| NO. of positive samples (%) | Mean ± s.d. (%) ** | Mean ± s.d. (%) | |||||||
| Control | 100 | 0 (0%) | Control | 100 | 0·34 ± 0·2 (2%) | Control | 100 | 0·34 ± 0·2 (2%) | |
| T1D | 382 | 100 (26·2%) | T1D‐EV– | 100 | 0·33 ± 0·1 (0%) | T1D‐EV– (ND) * | 50 | 0·32 ± 0·1 (0%) | <1 year |
| T1D‐EV– (PD) † | 50 | 0·34 ± 0·1 (0%) | >1 year | ||||||
| T1D‐EV+ | 100 | 0·96 ± 0·45 (64%) ‡ | T1D‐EV+ (ND) | 50 | 0·83 ± 0·4 (56%) § | <1 year | |||
| T1D‐EV+ (PD) | 50 | 1·12 ± 0·4 (74%) ¶ | >1 year |
*Newly diagnosed. †Previously diagnosed. ‡Significant (P < 0·001) compared to type 1 diabetes (T1D)‐EV–. §Significant (P < 0·001) compared to T1D‐enterovirus (EV)– (newly diagnosed). ¶Significant (P < 0·001) compared to T1D‐EV– (previously diagnosed). **Cut‐off value = 0·74.
Serum levels of HbA1c, C‐peptide and autoantibodies
HbA1c was significantly higher (P < 0·001) in T1D‐EV– than controls. In comparison to the former group, HbA1c was significantly higher (P < 0·001) in T1D‐EV+ (Fig. 2a). C‐peptide was significantly lower (P < 0·001) in T1D‐EV– than controls, while the T1D‐EV+ group was non‐significantly (P > 0·05) lower than T1D‐EV– (Fig. 2b). Similar to the pattern of HbA1c serum levels in three different groups, all the autoantibodies (GADA, IAA and ICA) were significantly higher (P < 0·001) in T1D‐EV– than controls, but showed a further increase (P < 0·001) in T1D‐EV+ compared to T1D‐EV– (Fig. 3a–c). Comparison of autoantibody levels between EV+‐CV– and EV+‐CV+ revealed higher frequency of autoantibodies in CV+ than CV–, while comparison between ND and PD patients who were CV– or CV+ showed generally higher significance in ND than PD (Fig. 3d–f). This was more prominent in GADA and ICA than IAA. All autoantibodies were significantly higher in CV+ ND than CV– ND. Only ICA levels were significantly (P < 0·001) higher in PD (CV+) than PD (CV–). EV+ patients were separated into IgG– (< 0·74) and IgG+ who were, in turn, allocated according to the strength of IgG into + (0·74–1·0), ++ (1·0–1·4) and +++ (> 1·4). Comparison of autoantibody levels between the latter groups clearly revealed the dependence of IAA on IgG but less prominently in ICA compared to GADA (Fig. 3g–i).
Figure 2.
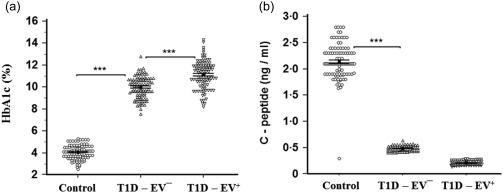
Determination of both haemoglobin A1c (HbA1c) (a) and C‐peptide (b) levels in human serum samples of control, enterovirus (EV)– and EV+ groups. HbA1c was significantly higher (P < 0·001) in EV– than controls and in EV+ than EV– (P < 0·001). C‐peptide was significantly lower (P < 0·001) in EV– than controls, while EV+ did not show any changes (P > 0·05) compared to EV–. Means ± standard deviations are shown.
Figure 3.
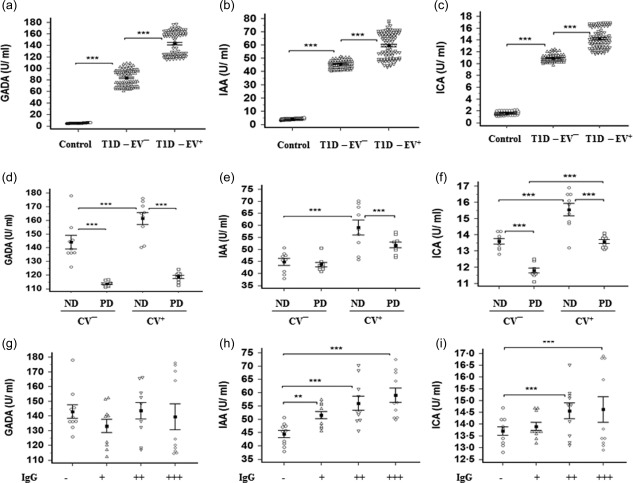
Levels of glutamic acid decarboxylase autoantibodies (GADA) (a), insulin autoantibodies (IAA) (b) and islet cell cytoplasmic autoantibodies (ICA) (c) in human serum samples of control, enterovirus (EV)– and EV+ groups. Autoantibodies were significantly higher in EV– than controls (P < 0·001) and in EV+ than EV– (P < 0·001). Levels of GADA (d), IAA (e) and ICA (f) were also compared between newly diagnosed (ND) and previously diagnosed (PD) patients who were EV+‐CV– or EV+‐CV+ showed generally higher significance (P < 0·001) in ND than PD. The same levels were compared between EV+‐immunoglobulin (Ig)G−, IgG+ (+), IgG+ (++) and IgG+ (+++) showed dependence of IAA and ICA on IgG strength. Means ± standard deviations are shown.
Serum levels of cytokines, TGF‐β and NO
Inflammatory cytokines (IFN‐γ, TNF‐α and IL‐1β) showed a similar pattern to that of autoantibodies. NO showed also the same pattern as an inflammatory mediator (Fig. 4). Levels of T helper type 2 (Th2) cytokines were investigated where only IL‐4, IL‐6 and IL‐13 were significantly higher in T1D‐EV– (P < 0·001) than control and no change (P > 0·05) in T1‐EV+ compared to T1D‐EV–. Again, IL‐10 showed the same pattern to that of autoantibodies and inflammatory cytokines with a clearly higher increase (P < 0·001) in T1D‐EV+ versus T1D‐EV– (Fig. 5). IL‐12 and IL‐17 showed the same pattern as inflammatory cytokines and NO, while IL‐20 and TGF‐β showed different patterns between groups (Fig. 6). The TGF‐β level was significantly higher in T1D‐EV– than controls and in T1D‐EV+ compared to T1D‐EV– (P < 0·001). IL‐20 was significantly higher in T1D‐EV– than controls and lower in T1D‐EV+ than T1D‐EV– (P < 0·001).
Figure 4.
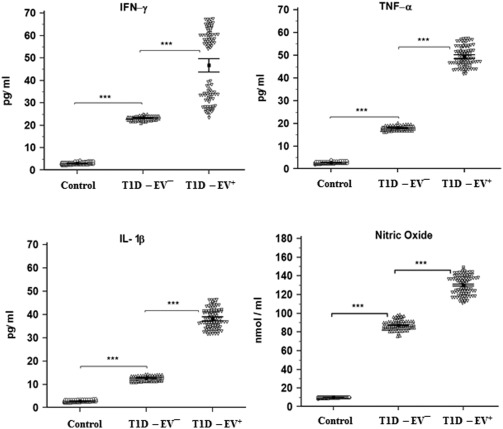
Detection of interferon (IFN)‐γ, tumour necrosis factor (TNF)‐α, interleukin (IL)‐1β and nitric oxide (NO) in human serum samples of control, enterovirus (EV)– and EV+ groups. Inflammatory cytokines and NO were significantly higher in EV– than control (P < 0·001) and in EV+ than EV– (P < 0·001). Means ± standard deviations are shown.
Figure 5.
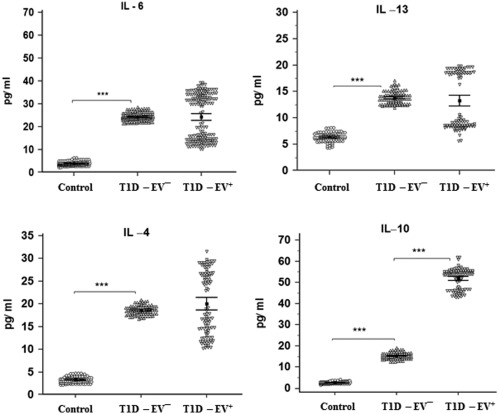
Detection of interleukin (IL)‐4, IL‐6, IL‐10 and IL‐13 in human serum samples of control, enterovirus (EV)– and EV+ groups. IL‐4, IL‐6 and IL‐13 were significantly higher in EV– than controls (P < 0·001) and no changes in EV+ versus EV– (P > 0·05). IL‐10 was significantly higher in EV– than controls (P < 0·001) and in EV+ than EV– (P < 0·001). Means ± standard deviations are shown.
Figure 6.
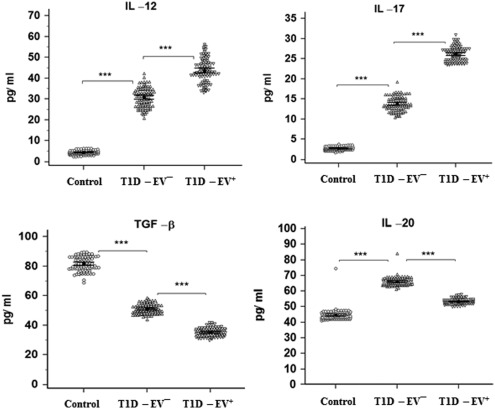
Detection of interleukin (IL)‐12, IL‐17, IL‐20 and transforming growth factor (TGF)‐β in human serum samples of control, enterovirus (EV)– and EV+ groups. IL‐12 and IL‐17 were significantly higher in EV– than controls (P < 0·001) and in EV+ than EV– (P < 0·001). Conversely, TGF‐β was significantly higher (P < 0·001) in EV– than controls and in EV+ than EV– (P < 0·001). IL‐20 was significantly higher (P < 0·001) in EV– than controls but lower (P < 0·001) in EV+ than EV–. Means ± standard deviations are shown.
Serum levels of CRP and complement activity
Similar to inflammatory cytokines and NO, CRP showed the same pattern between the groups as a marker for inflammation. While no changes (P > 0·05) were observed between control and T1D‐EV–, complement activity was higher in T1D‐EV+ than T1D‐EV– (Fig. 7). C3d was significantly higher compared to sC5b–9 (P < 0·001 and P < 0·01, respectively).
Figure 7.
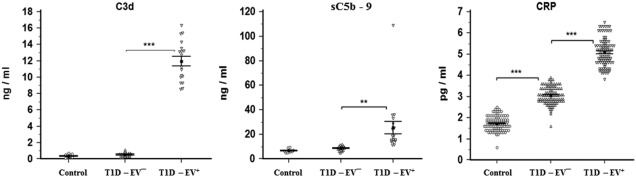
Detection of complement activity and C‐reactive protein (CRP) serum levels in controls, EV– and EV+ groups. Both C3d and sC5b–9 were significantly higher in EV+ than EV– (P < 0·001 and P < 0·01, respectively). CRP was significantly higher in EV– than controls (P < 0·001) and in EV+ than EV– (P < 0·001). Means ± standard deviations are shown.
Correlations of autoantibodies, cytokines and TGF‐β with HbA1c, C‐ peptide, NO, CRP, C3d and sC5b–9 in diabetic groups
For T1D‐EV–, positive correlation was found between ICA and sC5b–9 (0·45; P < 0·05), while negative correlations appeared between C3d and IL‐13 (–0·6; P < 0·05) or sC5b–9 with IL‐17 (–0·5; P < 0·05; Table 2). In addition, HbA1c correlated negatively with IL‐20 (–0.3; P < 0·01). In T1D‐EV+, HbA1c, NO and CRP showed highly significant and positive correlations with autoantibodies, IFN‐γ and IL‐12 levels but negative correlations with IL‐4, IL‐6 and IL‐13 (Table 3). IL‐10 showed both positive and negative correlations with C‐peptide (–0·2; P < 0·05) and NO (–0·24; P < 0·05), respectively. Negative correlations also appeared between IL‐20 and CRP (–0.2; P < 0·05) or IL‐17 and NO (–0.23; P < 0·05), while positive correlations appeared between NO and CRP with anti‐CV IgG (0·25; P < 0·05). Levels of C3d and sC5b–9 did not show any significant correlations in T1D‐EV+.
Table 2.
Correlations with haemoglobin A1c (HbA1c), C‐peptide, nitric oxide (NO), C‐reactive protein (CRP), C3d and sC5b–9 in the type 1 diabetes (T1D)‐enterovirus (EV)– groups
| HbA1c | C‐ peptide | NO | CRP | C3d | sC5b–9 | |
|---|---|---|---|---|---|---|
| r (P‐value) | ||||||
| GADA | 0·02 (0·849) | 0·04 (0·695) | 0·2 (0·05) | −0·2 (0·09) | −0·2 (0·4) | −0·07 (0·783) |
| IAA | −0·14 (0·175) | 0·16 (0·114) | 0·1 (0·336) | −0·07 (0·05) | −0·37 (0·121) | 0·115 (0·65) |
| ICA | 0·10 (0·318) | 0·13 (0·181) | −0·08 (0·382) | 0·007 (0·93) | −0·23 (0·359) | 0·453 (0·04) * |
| IFN‐γ | −0·12 (0·254) | −0·05 (0·6) | −0·07 (0·485) | −0·06 (0·54) | 0·06 (0·81) | −0·2 (0·422) |
| TNF‐α | 0·11 (0·269) | −0·11 (0·27) | 0·12 (0·233) | 0·077 (0·44) | −0·006 (0·98) | −0·15 (0·546) |
| IL‐1β | −0·02 (0·881) | −0·08 (0·43) | −0·13 (0·2) | 0·122 (0·225) | 0·11 (0·657) | 0·3 (0·2) |
| IL‐12 | 0·11 (0·287) | 0·04 (0·7) | −0·03 (0·76) | 0·154 (0·126) | 0·017 (0·94) | −0·173 (0·5) |
| IL‐4 | 0·06 (0·535) | −0·16 (0·11) | −0·04 (0·68) | −0·026 (0·8) | −0·48 (0·05) | 0·27 (0·27) |
| IL‐6 | 0·02 (0·808) | 0·02 (0·876) | −0·04 (0·6) | 0·05 (0·6) | 0·063 (0·8) | −0·13 (0·6) |
| IL‐13 | −0·14 (0·160) | −0·17 (0·1) | −0·09 (0·366) | −0·12 (0·22) | −0·6 (0·01) * | 0·1 (0·7) |
| IL‐20 | −0·33 (0·001) ** | −0·10 (0·326) | −0·04 (0·7) | −0·06 (0·55) | −0·04 (0·86) | 0·23 (0·35) |
| IL‐10 | −0·20 (0·05) | −0·08 (0·408) | −0·05 (0·6) | 0·06 (0·55) | 0·28 (0·25) | 0·28 (0·25) |
| IL‐17 | 0·14 (0·150) | 0·10 (0·303) | 0·1 (0·24) | 0·035 (0·72) | 0·136 (0·58) | −0·5 (0·03) * |
| TGF‐β | −0·04 (0·7) | −0·015 (0·87) | −0·07 (0·46) | 0·07 (0·44) | −0·3 (0·21) | 0·08 (0·7) |
GADA = glutamic acid decarboxylase; IAA = insulin autoantibodies; ICA = islet cell cytoplasmic autoantibodies; IFN = interferon; TNF = tumour necrosis factor; IL = interleukin; TGF = transforming growth factor. *P < 0·05; **P < 0·01.
Table 3.
Correlations with haemoglobin A1c (HbA1c), C‐reactive peptide (CRP), nitric oxide (NO), CRP, C3d and sC5b–9 in type 1 diabetes (T1D)‐enterovirus (EV)+
| HbA1c | C‐ peptide | NO | CRP | C3d | sC5b–9 | |
|---|---|---|---|---|---|---|
| r (P‐value) | ||||||
| GADA | 0·37 (0·0001) *** | −0·14 (0·170) | 0·8 (<0·0001) *** | 0·6 (<0·0001) *** | −0·006 (0·98) | 0·18 (0·47) |
| IAA | 0.34 (0.0005) *** | −0.03 (0.736) | 0.8 (<0.0001) *** | 0.6 (<0.0001) *** | 0.17 (0.48) | 0.35 (0.15) |
| ICA | 0.36 (0.0002) *** | −0.16 (0.122) | 0.75 (<0.0001) *** | 0.45 (<0.0001) *** | −0.14 (0.57) | 0.15 (0.54) |
| IFN‐γ | 0.34 (0.0005) *** | −0.15 (0.146) | 0.8 (<0.0001) *** | 0.6 (<0.0001) *** | 0.27 (0.26) | 0.17 (0.48) |
| TNF‐α | 0.08 (0.423) | 0.04 (0.681) | −0.04 (0.7) | −0.01 (0.9) | 0.002 (0.9) | 0.27 (0.27) |
| IL‐1β | −0.07 (0.501) | 0.14 (0.153) | −0.15 (0.13) | −0.09 (0.35) | −0.06 (0.8) | 0.09 (0.7) |
| IL‐12 | 0.29 (0.003) ** | −0.15 (0.126) | 0.66 (<0.0001) *** | 0.53 (<0.0001) *** | 0.4 (0.1) | 0.1 (0.64) |
| IL‐4 | −0·33 (0·001) ** | 0·09 (0·388) | −0·77 (<0·0001) *** | −0·6 (<0·0001) *** | 0·1 (0·7) | −0·14 (0·57) |
| IL‐6 | −0·33 (0·0009) *** | 0·09 (0·382) | −0·84 (<0·0001) *** | −0·6 (<0·0001) *** | −0·27 (0·26) | −0·02 (0·9) |
| IL‐13 | −0·36 (0·0002) *** | 0·11 (0·283) | −0·84 (<0·0001) *** | −0·6 (<0·0001) *** | −0·33 (0·17) | −0·0009 (1) |
| IL‐20 | −0·11 (0·272) | 0·05 (0·627) | −0·2 (0·07) | −0·2 (0·02) * | −0·06 (0·8) | 0·07 (0·7) |
| IL‐10 | −0·03 (0·769) | 0·22 (0·032) * | −0·24 (0·02) * | −0·14 (0·15) | 0·06 (0·8) | −0·34 (0·15) |
| IL‐17 | −0·21 (0·038) | 0·02 (0·824) | −0·23 (0·02) * | −0·16 (0·09) | −0·08 (0·7) | −0·05 (0·8) |
| TGF‐β | 0·37 (0·12) | −0·05 (0·6) | 0·13 (0·2) | 0·1 (0·26) | 0·2 (0·36) | 0·37 (0·12) |
| Anti‐CV IgGs | −0·14 (0·245) | 0·10 (0·389) | 0·25 (0·03) * | 0·25 (0·04) * | −0·05 (0·8) | 0·21 (0·38) |
GADA = glutamic acid decarboxylase; IAA = insulin autoantibodies; ICA = islet cell cytoplasmic autoantibodies; IFN = interferon; TNF = tumour necrosis factor; IL = interleukin; TGF = transforming growth factor. *P < 0·05; ** P < 0·01; ***P < 0·01.
Correlations of HbA1c and C‐peptide with NO, CRP, C3d and sC5b–9 in diabetic groups
While T1D‐EV– did not show any significant correlations (Table 4), T1D‐EV+ showed positive correlations between HbA1c and each of NO (0·36; P < 0·001) and CRP (0·2; P < 0·05) or C‐peptide with C3d (0·45; P < 0·05).
Table 4.
Correlations between haemoglobin A1c (HbA1c) & C‐peptide with nitric oxide (NO), C‐reactive protein (CRP), C3d and sC5b–9 in type 1 diabetes (T1D)‐enterovirus (EV)– and EV+ groups
| NO | CRP | C3d | sC5b–9 | ||
|---|---|---|---|---|---|
| r (P‐value) | |||||
| HbA1c | T1D‐EV– | 0·13 (0·18) | −0·07 (0·47) | 0·03 (0·9) | −0·18 (0·46) |
| T1D‐EV+ | 0·36 (0·0002) *** | 0·2 (0·036) * | 0·02 (0·9) | −0·4 (0·12) | |
| C‐ peptide | T1D‐EV– | −0·045 (0·65) | 0·07 (0·5) | 0·3 (0·18) | 0·015 (0·95) |
| T1D‐EV+ | −0·2 (0·1) | 0·07 (0·4) | 0·45 (0·04) * | 0·1 (0·64) | |
*P < 0·05; ***P < 0·01.
Correlations of autoantibodies with cytokines in diabetic groups
In T1D‐EV–, GADA correlated positively with IL‐6 (0.4; P < 0.001), TNF‐α, IL‐13 and IL‐17 (0·2; P < 0·05) and negatively with IL‐10 (–0·3; P < 0·01; Table 5). IAA correlated negatively with both IL‐4 and IL‐12 (–0·2; P< 0·05) but ICA correlated positively with IL‐4 (0·4; P< 0·001) and IL‐13 (0·2; P< 0·05). In T1D‐EV+, all autoantibodies correlated positively with both IFN‐γ and IL‐12 and negatively with IL‐4, IL‐6, IL‐13 and IL‐17 (P < 0·001). GADA and IAA but not ICA correlated negatively with IL‐20 (P < 0·05), while all correlated negatively with IL‐10 (P < 0·05 for GADA and ICA but P < 0·01 for IAA).
Table 5.
Correlations between autoantibodies and cytokines in type 1 diabetes (T1D)‐enterovirus (EV)– and EV+ groups
| T1D‐EV– | T1D‐EV+ | |||||
|---|---|---|---|---|---|---|
| GADA | IAA | ICA | GADA | IAA | ICA | |
| r (P‐value) | r (P‐value) | |||||
| IFN‐γ | 0·1 (0·2) | 0·01 (0·89) | 0·1 (0·44) | 0·9 (<0·0001) *** | 0·9 (<0·0001) *** | 0·8 (<0·0001) *** |
| TNF‐α | 0·2 (0·02) * | 0·01 (0·78) | 0·01 (0·69) | 0·0 (0·65) | 0·0 (0·83) | 0·0 (0·62) |
| IL‐1β | 0·01 (0·83) | 0·01 (0·72) | 0·01 (0·92) | −0·1 (0·44) | −0·1(0·38) | −0·1 (0·25) |
| IL‐12 | −0·1 (0·24) | −0·2 (0·02) * | 0·1 (0·45) | 0·7 (<0·0001) *** | 0·7 (<0·0001) *** | 0·6 (<0·0001) *** |
| IL‐4 | 0·1 (0·21) | −0·2 (0·03) * | 0·4 (<0·0001) *** | −0·8 (<0·0001) *** | −0·8 (<0·0001) *** | −0·7 (<0·0001) *** |
| IL‐6 | 0·4 (<0·0001) *** | 0·2 (0·07) | 0·01 (0·82) | −0·9 (<0·0001) *** | −0·8 (<0·0001) *** | −0·7 (<0·0001) *** |
| IL‐13 | 0·2 (0·02) * | 0·01 (0·73) | 0·2 (0·04) * | −0·9 (<0·0001) *** | −0·9 (<0·0001) *** | −0·8 (<0·0001) *** |
| IL‐20 | −0·1 (0·15) | 0·01 (0·76) | 0·01 (0·84) | −0·2 (0·04) * | −0·3 (0·012) * | −0·2 (0·06) |
| IL‐10 | −0·3 (0·004) ** | −0·2 (0·12) | 0·1 (0·47) | −0·2 (0·048) * | −0·3 (0·007) ** | −0·2 (0·04) * |
| IL‐17 | 0·2 (0·01) * | 0·01 (0·1) | −0·1 (0·32) | −0·3 (0·0004) *** | −0·3 (0·006) ** | −0·3 (0·009) ** |
| TGF‐β | −0·15 (0·12) | −0·18 (0·06) | −0·17 (0·08) | 0·05 (0·63) | −0·0005 (0·1) | 0·05 (0·65) |
GADA = glutamic acid decarboxylase; IAA = insulin autoantibodies; ICA = islet cell cytoplasmic autoantibodies; IFN = interferon; TNF = tumour necrosis factor; IL = interleukin; TGF = transforming growth factor. *P < 0.05; **P < 0.01; ***P < 0.01.
Correlations between autoantibodies and cytokines with anti‐CV IgGs in T1D‐EV+
Anti‐CV IgGs correlated positively with GADA (0·34; P < 0·01), IAA and ICA (0·37; P < 0·001) and both IFN‐γ and IL‐12 (0·3; P < 0·01) but negatively with IL‐4, IL‐6 and IL‐13 (–0·3; P < 0·01; Table 6).
Table 6.
Correlations between autoantibodies & cytokines with anti‐ coxsackievirus (CV) immunoglobulins (IgGs) in the type 1 diabetes (T1D)‐EV+ group
| Anti‐CV IgGs r (P‐value) | |
|---|---|
| GADA | 0·34 (0·003) ** |
| IAA | 0·37 (0·0009) *** |
| ICA | 0·37 (0·0009) *** |
| IFN‐ γ | 0·3 (0·001) ** |
| TNF‐α | −0·07 (0·56) |
| IL‐1β | 0·13 (0·25) |
| IL‐12 | 0·3 (0·006) ** |
| IL‐4 | −0·3 (0·008) ** |
| IL‐6 | −0·3 (0·007) ** |
| IL‐13 | −0·3 (0·005) ** |
| IL‐20 | −0·02 (0·86) |
| IL‐10 | −0·05 (0·7) |
| IL‐17 | −0·02 (0·86) |
| TGF‐β | 0·05 (0·68) |
GADA = glutamic acid decarboxylase; IAA = insulin autoantibodies; ICA = islet cell cytoplasmic autoantibodies; IFN = interferon; TNF = tumour necrosis factor; IL = interleukin; TGF = transforming growth factor. **P < 0·01; ***P < 0·01.
Discussion
The prevalence of EV infection among T1D children revealed 26·2% positive cases based on EV RNA detection in serum. This finding indicated a probability of infection with other environmental viruses which could be a cause of T1D. Another possibility is the involvement of genetic risk, which contributed to autoimmunity to pancreatic β cell antigens. The study highlighted the necessity of investigation of other viral infections among T1D patients 22. Indeed, Rubella virus, rotavirus, cytomegalovirus and mumps virus were found to be associated previously with children with T1D 23, 24, 25. The study tested only 10 PCR products (EV+) for sequencing, where all showed high homology with CVB4. The sequencing results could not be direct evidence or an indicator for the involvement of CVB4 in T1D, especially considering that the ELISA test for anti‐CV IgGs gave 64% positivity. This, in turn, might also imply the involvement of other EVs such as echoviruses in the disease 26. In this study, the T1D‐EV+ population was selected based on the results of nested PCR for detection of viral RNA in T1D patients. The viral RNA detection in serum represented a diagnostic method for active infection, which might be sufficient for induction of autoimmunity before development of anti‐viral IgG 27. The other possibility is viral infection after ongoing autoimmunity, which could lead to T1D acceleration 28. Indeed, the sequence of events, whether the viral infections occurred earlier and autoimmunity occurred later or vice versa, cannot be predicted because all the RNA‐negative children were IgG‐negative.
Increased IgG levels could be an indicator for recurrent infections with same virus in children as a result of drinking water contaminated with EVs 29. In addition, IgG levels among EV+‐PD (more than 1 year) who might have been exposed to recurrent infections were significantly higher than EV+‐ND (less than 1 year). If we speculate that EV+‐CV– and EV+‐CV+ represent new and recurrent infections, respectively, the recurrent infections down‐regulate the production of autoantibodies. This is because the autoantibodies in ND children were significantly higher than PD. In the case that the development of anti‐viral IgG occurs after a long time of first infection, this is often associated with down‐regulated production of autoantibodies. However, separation of EV+ according to IgG strength into positive, +, ++ and +++ revealed a dependence of IAA and ICA on IgG strength. If we speculate the dependence of IgG strength on acute and recurrent infections, the latter would increase IAA and ICA, which supports the hypothesis of accelerated ongoing autoimmunity and overt diabetes after viral infection. The correlation analysis between IgG and autoantibodies supports this hypothesis further.
As most cases were anti‐CV IgG‐positive, it could be considered that CV invaded β cells through specific receptors, replicated and led to autoimmunity and cell destruction 30. This cell destruction was widespread as a result of highly exposed autoantigens and activation of bystander T cells 5. The increased expression of major histocompatibility complex (MHC‐I) proteins on the cell surface with viral antigens could also activate CD8+ T cells 31. Innate immunity also played an important role through activation of Toll‐like receptors (TLRs) on macrophages and neutrophils which, in turn, secreted inflammatory cytokines 32. These cytokines could recruit and activate natural killer cells (NKs), which played an important role in killing and destroying β cells 33. Recurrent infections could invade more β cells and increase the activated CD8+ T cells and levels of autoantibodies.
HbA1c showed an observable increase in diabetic groups, either infected or not. However, diabetic EV+ showed a further increase over EV–. Thus, EV infection exacerbated β cell destruction and decreased secretion of insulin. This was also confirmed by measured C‐peptide which, conversely, decreased in diabetic groups. The effective role of EV in inhibition of insulin secretion was assigned to depletion of its granule stores and hence inhibition of insulin secretion 34. In addition, increased production of autoantibodies to multiple cell antigens, including GAD, IA and IC in diabetic groups, with observable exacerbation in EV+ compared to EV–, implicated the potential role of EV in autoimmunity induction 35. Increased levels of proinflammatory cytokines (IFN‐γ, TNF‐α and IL‐1β) and NO coincided with and interpreted further the observed levels of HbA1c, C‐peptide and autoantibodies. These cytokines had been known to be associated with viral infections, while NO was also known to induce damage to the cell 36, 37. The direction of immune response to Th1 phenotype by the production of IFN‐γ was observable in T1D‐associated autoimmunity. However, IL‐10, which is a Th2 cytokine and known to be secreted by Tregs, was also elevated in diabetic groups, with an observable increase in EV+. Indeed, this finding was also observed in autoimmune T1D patients in comparison to non‐diabetic controls and even in diabetic EV+ versus diabetic EV– children 7, 11, 38. IL‐4, IL‐6 and IL‐13 appeared higher in diabetic groups, but no difference between EV+ and EV– was observed. Thus, EV, rather than IL‐4, IL‐6 and IL‐13, motivated production of IFN‐γ. Increased levels of IL‐4 and IL‐6 in hyperglycaemic T1D children were found sustained even after 2 h of euglycaemia/correction 39. It could be considered that the increased levels of IFN‐γ due to viral infection dampened the levels of Th2 cytokines (IL‐4, IL‐6 and IL‐13) but not IL‐10, showing the latter as a distinct cytokine. IL‐12 and IL‐17 behaved similarly to inflammatory cytokines. Thus, increased levels of IFN‐γ, IL‐12 and IL‐17 in viral infection revealed a predominance of Th1 and Th17 versus Th2 immune responses. Both IFN‐γ and IL‐12 were reported to be responsible for activation of natural killer (NK) cells which are, in turn, responsible for pancreatic β cell apoptosis 40. Thus, NKs with help from IFN‐γ and IL‐12 initiated β cell lysis before activation of CD8+ T cells and production of anti‐viral antibodies. Generally, IFN‐γ, IL‐10, IL‐12 and IL‐17 were found to play an important role in induction of autoimmunity and disease progression with EV infection 38. In addition, elevated levels of IL‐12 and IL‐17 were found to be associated with low frequencies of Tregs 9. TGF‐β decreased in diabetic groups with observably exacerbated decrease in EV+ versus EV– children. Human serum TGF‐β levels were also decreased in diabetic children with either types 1 or 2 14. Indeed, the levels of this growth factor in serum were indicators for the infiltration and activity of Tregs which decreased during autoimmunity 41. IL‐20 levels were found to be up‐regulated in diabetic EV– children in comparison to healthy controls, but down‐regulated in EV+ versus EV– children. This indicated an association of this cytokine with T1D‐EV– but down‐regulated in T1D‐EV+. IL‐20 belongs to the IL‐10 family of cytokines and is linked to other autoimmune diseases, such as rheumatoid arthritis 42.
The levels of C3d and sC5b–9 indicated increased complement activity in diabetic EV+ versus diabetic EV– children. Moreover, their levels in diabetic EV– children did not show any significant differences compared to healthy controls. Thus, the profile of these complement activation products (especially C3d) was distinct for diabetic EV+. However, complement activation was described preciously to play a role in the disease pathogenesis, irrespective of viral infections 43. The involvement of activated complement components in inflammatory reactions after EV infection was also found 44. Furthermore, capsid proteins of CVB3 were found to interact with C3 and activate the alternative pathway 45. The levels of CRP showed a similar profile to inflammatory cytokines (IFN‐γ, TNF‐α and IL‐1β, IL‐12 and IL‐17), as well as IL‐10 and NO. CRP was found to polarize human monocytes towards the proinflammatory M1 phenotype and was involved in complement activation 46, 47.
Statistical correlations in diabetic EV– showed fewer significantly positive and negative associations. Statistical analysis showed the dependence of complement activity on ICA in diabetic EV– 48. Conversely, complement activation was independent of IL‐13 and IL‐17. Furthermore, IL‐20 showed an inhibitory effect on pancreatic β cell destruction. However, this was not apparent in EV infection.
A strong dependence was found of inflammation and β cell destruction in EV infection on the levels of anti‐viral antibodies, autoantibodies and Th1, but not Th2, cytokines 13, 49. In addition, a positive correlation between the levels of C‐peptide and C3d revealed the dependence of β cell destruction on complement activity. This can be considered as evidence for a complementary role in T1D‐EV+ disease.
Statistical correlation revealed also that the increased levels of both IL‐4 and IL‐6 were responsible for increased levels of ICA and GADA, respectively, in diabetic EV–. Conversely, the decreased levels of these cytokines together with IL‐13 and less potently IL‐17 were responsible for increased GADA, IAA and ICA in diabetic EV+. Moreover, IFN‐γ and IL‐12 were responsible for exposure of more β cell antigens to the immune system as a result of EV entry to the cell (breaking tolerance) leading to autoimmunity 50.
In conclusion, T1D‐EV+ showed increased levels of inflammatory cytokines and NO when compared to T1D‐EV–. Viral infection increased Th1 and decreased Th2 cytokines with remarkable down‐regulation for Tregs. IL‐10, IL‐20 and complement activation products showed distinct profiles for viral infection, highlighting their roles in diagnosis of T1D‐EV+. The recurrent viral infections accelerate the ongoing autoimmunity and overt T1D.
Disclosures
None.
Author contributions
A. A. M. designed the experimental study, M. H. H. diagnosed the disease, R. G. K. performed the experiments, M. A. L. wrote the paper.
Acknowledgement
The authors are grateful for Professor Dr Michael Kirschfink, Department of Immunochemistry, Institute of Immunology, Heidelberg University for providing C3d and sC5b–9 detection antibodies.
References
- 1. Onkamo P, Vaananen S, Karvonen M et al Worldwide increase in incidence of type I diabetes – the analysis of the data on published incidence trends. Diabetologia 1999; 42:1395–403. [DOI] [PubMed] [Google Scholar]
- 2. Jun HS, Yoon JW. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev 2003; 19:8–31. [DOI] [PubMed] [Google Scholar]
- 3. Zanone MM, Favaro E, Ferioli E et al Human pancreatic islet endothelial cells express coxsackievirus and adenovirus receptor and are activated by coxsackie B virus infection. Faseb J 2007; 21:3308–17. [DOI] [PubMed] [Google Scholar]
- 4. Varela‐Calvino R, Peakman M. Enteroviruses and type 1 diabetes. Diabetes Metab Res Rev 2003; 19:431–41. [DOI] [PubMed] [Google Scholar]
- 5. Bergamin CS, Dib SA. Enterovirus and type 1 diabetes: what is the matter? World J Diabetes 2015; 6:828–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lebastchi J, Herold KC. Immunologic and metabolic biomarkers of β‐cell destruction in the diagnosis of type 1 diabetes. Cold Spring Harb Perspect Med 2012; 2:a007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arif S, Tree TI, Astill TP et al Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest 2004; 113:451–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arif S, Moore F, Marks K et al Peripheral and islet interleukin‐17 pathway activation characterizes human autoimmune diabetes and promotes cytokine‐mediated b‐cell death. Diabetes 2011; 60:2112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryba‐Stanisławowska M, Rybarczyk‐Kapturska K, Myśliwiec M et al Elevated levels of serum IL‐12 and IL‐18 are associated with lower frequencies of CD4(+)CD25 (high)FOXP3 (+) regulatory t cells in young patients with type 1 diabetes. Inflammation 2014; 37:1513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dogan Y, Akarsu S, Ustundag B et al Serum IL‐1beta, IL‐2, and IL‐6 in insulin‐dependent diabetic children. Mediators Inflamm 2006; 2006:59206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Z, Li R, Xie Z et al IL‐6, IL‐10 and IL‐13 are associated with pathogenesis in children with Enterovirus 71 infection. Int J Clin Exp Med 2014; 7:2718–23. [PMC free article] [PubMed] [Google Scholar]
- 12. Kaneko K, Satake C, Yamamoto J et al A case of idiopathic type 1 diabetes with subsequent recovery of endogenous insulin secretion despite initial diagnosis of fulminant type 1 diabetes. Endocr J 2017; 64:369–74. [DOI] [PubMed] [Google Scholar]
- 13. Russell MA, Morgan NG. The impact of anti‐inflammatory cytokines on the pancreatic β‐cell. Islets 2014; 6:e950547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roohi A, Tabrizi M, Abbasi F et al Serum IL‐17, IL‐23, and TGF‐β levels in type 1 and type 2 diabetic patients and age‐matched healthy controls. Biomed Res Int 2014; 2014:718946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thordardottir S, Vikingsdottir T, Bjarnadottir H et al Activation of complement following total hip replacement. Scand J Immunol 2016; 83:219–24. [DOI] [PubMed] [Google Scholar]
- 16. Zoll GJ, Melchers WJ, Kopecka H et al General primer‐mediated polymerase chain reaction for detection of enteroviruses: application for diagnostic routine and persistent infections. J Clin Microbiol 1992; 30:160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarmiento L, Cabrera‐Rode E, Lekuleni L et al Occurrence of enterovirus RNA in serum of children with newly diagnosed type 1 diabetes and islet cell autoantibody‐positive subjects in a population with a low incidence of type 1 diabetes. Autoimmunity 2007; 40:540–5. [DOI] [PubMed] [Google Scholar]
- 18. Maha MM, Ali MA, Abdel‐Rehim SE et al The role of coxsackieviruses infection in the children of insulin dependent diabetes mellitus. J Egypt Public Health Assoc 2003; 78:305–18. [PubMed] [Google Scholar]
- 19. Thordardottir S, Vikingsdottir T, Bjarnadottir H et al Reciprocal changes in complement activity and immune‐complex levels during plasma infusion in a C2‐deficient SLE patient. Lupus 1993; 2:161–5. [DOI] [PubMed] [Google Scholar]
- 20. Mollnes TE, Lea T, Froland SS et al Quantification of the terminal complement complex in human plasma by an enzyme‐linked immunosorbent assay based on monoclonal antibodies against a neoantigen of the complex. Scand J Immunol 1985; 22:197–202. [DOI] [PubMed] [Google Scholar]
- 21. Messias‐Reason IJ, Hayashi SY, Nisihara RM et al Complement activation in infective endocarditis: correlation with extracardiac manifestations and prognosis. Clin Exp Immunol 2002; 127:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Komulainen J, Knip M, Sabbah E et al Autoimmune and clinical characteristics of type I diabetes in children with different genetic risk loads defined by HLA‐DQB1 alleles. Childhood Diabetes in Finland Study Group. Clin Sci (Lond) 1998; 94:263–9. [DOI] [PubMed] [Google Scholar]
- 23. Szopa TM, Titchener PA, Portwood ND et al Diabetes mellitus due to viruses – some recent developments. Diabetologia 1993; 36:687–95. [DOI] [PubMed] [Google Scholar]
- 24. Goto A, Takahashi Y, Kishimoto M et al A case of fulminant type 1 diabetes associated with significant elevation of mumps titers. Endocr J 2008; 55:561–4. [DOI] [PubMed] [Google Scholar]
- 25. Blomqvist M, Juhela S, Erkkila S et al Rotavirus infections and development of diabetes‐associated autoantibodies during the first 2 years of life. Clin Exp Immunol 2002; 128:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klemola P, Kaijalainen S, Ylipaasto P et al Diabetogenic effects of the most prevalent enteroviruses in Finnish sewage. Ann NY Acad Sci 2008; 1150:210–2. [DOI] [PubMed] [Google Scholar]
- 27. Lönnrot M, Salminen K, Knip M et al Enterovirus RNA in serum is a risk factor for beta‐cell autoimmunity and clinical type 1 diabetes: a prospective study. Childhood Diabetes in Finland (DiMe) Study Group. J Med Virol 2000; 61:214–20. [PubMed] [Google Scholar]
- 28. Serreze DV, Ottendorfer EW, Ellis TM et al Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T‐cells in pancreatic islets. Diabetes 2000; 49:708–11. [DOI] [PubMed] [Google Scholar]
- 29. Ali MA, El‐Esnawy NA, Shoaeb AR et al RT–PCR and cell culture infectivity assay to detect enteroviruses during drinking water treatment processes. J Egypt Public Health Assoc 1999; 74:651–61. [PubMed] [Google Scholar]
- 30. Oikarinen M, Tauriainen S, Honkanen T et al Analysis of pancreas tissue in a child positive for islet cell antibodies. Diabetologia 2008; 51:1796–802. [DOI] [PubMed] [Google Scholar]
- 31. Foulis AK. Pancreatic pathology in type 1 diabetes in human. Novartis Found Symp 2008; 292:2–13. [PubMed] [Google Scholar]
- 32. Takeda K, Kaisho T, Akira S. Toll‐like receptors. Annu Rev Immunol 2003; 21:335–76. [DOI] [PubMed] [Google Scholar]
- 33. Dotta F, Censini S, van Halteren AG et al Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent‐onset type 1 diabetic patients. Proc Natl Acad Sci USA 2007; 104:5115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petzold A, Solimena M, Knoch KP. Mechanisms of beta cell dysfunction associated with viral infection. Curr Diab Rep 2015; 15:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta‐analysis of observational molecular studies. BMJ 2011; 342:d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ylipaasto P, Smura T, Gopalac haryulu P et al Enterovirus‐induced gene expression profile is critical for human pancreatic islet destruction. Diabetologia 2012; 55:3273–83. [DOI] [PubMed] [Google Scholar]
- 37. Sharma V, Kalim S, Srivastava MK et al Oxidative stress and coxsackievirus infections as mediators of beta cell damage: a review. Sci Res Essays 2009; 4:42–58. [Google Scholar]
- 38. Yeung WC, Al‐Shabeeb A, Pang CN et al Children with islet autoimmunity and enterovirus infection demonstrate a distinct cytokine profile. Diabetes 2012; 61:1500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosa JS, Flores RL, Oliver SR et al Sustained IL‐1alpha, IL‐4, and IL‐6 elevations following correction of hyperglycemia in children with type 1 diabetes mellitus. Pediatr Diabetes 2008; 9:9–16. [DOI] [PubMed] [Google Scholar]
- 40. Chrul S, Polakowska E, Mycko M et al The effect of IL‐2, IL‐12 and IL15 on the function of natural killer cells in children suffering from type 1 diabetes mellitus. Pediatr Endocrinol Diabetes Metab 2013; 19:91–5. [PubMed] [Google Scholar]
- 41. Tonkin DR, Haskins K. Regulatory T cells enter the pancreas during suppression of type 1 diabetes and inhibit effector T cells and macrophages in a TGF‐beta‐dependent manner. Eur J Immunol 2009; 39:1313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Šenolt L, Leszczynski P, Dokoupilová E et al Efficacy and safety of anti‐interleukin‐20 monoclonal antibody in patients with rheumatoid arthritis: a randomized phase IIa trial. Arthritis Rheumatol 2015; 67:1438–48. [DOI] [PubMed] [Google Scholar]
- 43. Rowe P, Wasserfall C, Croker B et al Increased complement activation in human type 1 diabetes pancreata. Diabetes Care 2013; 36:3815–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qiu S, Liu N, Jia L et al A new treatment for neurogenic inflammation caused by EV71 with CR2‐targeted complement inhibitor. Virol J 2012; 9:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anderson DR, Carthy CM, Wilson JE et al Complement component 3 interactions with coxsackievirus B3 capsid proteins: innate immunity and the rapid formation of splenic antiviral germinal centers. J Virol 1997; 71:8841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Devaraj S, Jialal I. C‐reactive protein polarizes human macrophages to an M1 phenotype and inhibits transformation to the M2 phenotype. Arterioscler Thromb Vasc Biol 2011; 31:1397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zimmermann O, Bienek‐Ziolkowski M, Wolf B et al Myocardial inflammation and non‐ischaemic heart failure: is there a role for C‐reactive protein? Basic Res Cardiol 2009; 104:591–9. [DOI] [PubMed] [Google Scholar]
- 48. Okada S, Ichiki K, Sato K et al Complement activation pathways associated with islet cell surface antibody (ICSA) derived from child patients with insulin‐dependent diabetes mellitus (IDDM). Acta Med Okayama 1991; 45:185–6. [DOI] [PubMed] [Google Scholar]
- 49. Bason C, Lorini R, Lunardi C et al In type 1 diabetes a subset of anti‐coxsackievirus B4 antibodies recognize autoantigens and induce apoptosis of pancreatic beta cells. PLOS ONE 2013; 8:e57729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hill SL, Rose NR. The transition from viral to autoimmune myocarditis. Autoimmunity 2001; 34:169–76. [DOI] [PubMed] [Google Scholar]