

Summary
All‐trans retinoic acid (atRA), the main biologically active metabolite of vitamin A, has been implicated in immunoregulation and anti‐cancer. A recent finding that vitamin A could decrease the risk of melanoma in humans indicates the beneficial role of atRA in melanoma. However, it remains unknown whether topical application of atRA could inhibit melanoma growth by influencing tumour immunity. We demonstrate topical application of tretinoin ointment (atRA as the active ingredient) effectively inhibited B16F10 melanoma growth. This is accompanied by markedly enhanced CD8+ T‐cell responses, as evidenced by significantly increased proportions of effector CD8+ T cells expressing granzyme B, tumour necrosis factor‐α, or interferon‐γ, and Ki67+ proliferating CD8+ T cells in atRA‐treated tumours compared with vaseline controls. Furthermore, topical atRA treatment promoted the differentiation of effector CD8+ T cells in draining lymph nodes (DLN) of tumour‐bearing mice. Interestingly, atRA did not affect tumoral CD4+ T‐cell response, and even inhibited the differentiation of interferon‐γ‐expressing T helper type 1 cells in DLN. Importantly, we demonstrated that the tumour‐inhibitory effect of atRA was partly dependent on CD8+ T cells, as CD8+ T‐cell depletion restored tumour volumes in atRA‐treated mice, which, however, was still significantly smaller than those in vaseline‐treated mice. Finally, we demonstrated that atRA up‐regulated MHCI expression in B16F10 cells, and DLN cells from tumour‐bearing mice had a significantly higher killing rate when culturing with atRA‐treated B16F10 cells. Hence, our study demonstrates that topical atRA treatment effectively inhibits melanoma growth partly by promoting the differentiation and the cytotoxic function of effector CD8+ T cells.
Keywords: all‐trans retinoic acid, CD8+ T cell, melanoma, MHCI
Abbreviations
- atRA
all‐trans retinoic acid
- CTL
cytotoxic T lymphocyte
- DLN
draining lymph node
- LDH
lactate dehydrogenase
- Mφ
macrophage
- MDSC
myeloid‐derived suppressor cell
- Treg
regulatory T cell
Introduction
Malignant melanoma is the most aggressive type of skin cancer, accounting for 90% of skin cancer mortality, and has been recognized as one of the most immunogenic malignancies with abundant infiltration of various immune cells.1 The types and activation status of immune cells have great impacts on the progression of melanoma and the responsiveness to immunotherapy.2, 3 Cytotoxic CD8+ T cells are well‐known to play a crucial anti‐tumour role in melanoma, primarily through the release of cytotoxic mediators such as granzymes and interferon‐γ (IFN‐γ) upon the recognition of MHCI molecule‐associated tumour antigens,4 whereas CD4+ T cells with different functional subsets play a complex role in melanoma, for example, IFN‐γ‐producing T helper type 1 (Th1) cells and Foxp3+ regulatory T (Treg) cells play anti‐tumour and pro‐tumour roles, respectively.5, 6 In addition, tumour infiltrating myeloid cells including macrophages and myeloid‐derived suppressor cells (MDSCs) could suppress anti‐tumour T‐cell immunity via various mechanisms, thereby leading to tumour immune escape and less responsiveness to immunotherapy.7, 8
Vitamin A and its main biologically active metabolite all‐trans retinoic acid (atRA) have been extensively studied to determine their role in immunoregulation and anti‐cancer.9, 10 AtRA is known to influence both CD4+ and CD8+ T cells by regulating the migration, differentiation and activation under different conditions.11 Accumulating evidence suggests a critical role of atRA in mucosal tolerance by imprinting the homing of CD4+ T cells to the gut and enhances the induction of regulator T cells while reciprocally inhibiting the development of Th17 cells in the intestinal lamina propria.12, 13 On the other hand, atRA was reported to promote effector T‐cell responses under some inflammatory conditions. For example, conditional ablation of atRA signalling in CD4+ T cells suppressed the inflammatory responses that mediate the rejection of allogeneic skin grafts,14 and genetic or pharmacological manipulation of the atRA–RARα axis influenced effector functions of Th1, Th17 cells and CD8+ T cells in response to infection.9, 15, 16, 17 Additionally, atRA has been shown to induce the differentiation of immature myeloid cells, which directly promoted T‐cell‐mediated tumour‐specific immune responses, leading to improved effect of cancer vaccines.18, 19 AtRA is also a well‐known anti‐cancer drug that is used clinically to treat leukaemia20 and pre‐clinically for the treatment of a number of types of cancer including hepatoma and breast cancer.21, 22 Most studies have been focused on the direct effect of atRA on the apoptosis and differentiation of tumour cells; however, little is known about the contribution of the immunoregulatory effect of atRA to its anti‐cancer role.
A recent study showed that taking vitamin A supplements may be able to decrease the risk of developing melanoma,23 suggesting a beneficial effect of atRA in melanoma. Most previous studies focused on the direct inhibitory effect of atRA on the biology of melanoma cells in vitro.24, 25, 26 There are only two studies focusing on the immunoregulatory role of atRA in melanoma, showing the opposing effect. Guo et al.27 demonstrated that genetic interruption of RARα, the primary functional receptor of atRA, in CD8+ T cells aggravated B16F10 tumour growth in CD4‐depleted mice. In contrast, Galvin et al.28 reported that treatment with a RARα inhibitor enhanced the protective efficacy of a DC vaccine against B16F10 tumours by suppressing the induction of Treg cells and promoting Th1 responses. It is noted that both experiments studied the role of endogenous atRA signalling in melanoma, and accumulating data indicate that atRA at physiological or pharmacological concentrations could have opposing effects on T‐cell immunity.29, 30, 31 We therefore aimed to investigate whether topical application of atRA was able to inhibit the established melanoma and the underlying immunological mechanism.
In this study, our results demonstrated that topical application of tretinoin ointment (with atRA as the active ingredient) effectively inhibited B16F10 melanoma growth in vivo, which was partly dependent on its ability to promote anti‐tumour CD8+ T‐cell immunity.
Materials and methods
Mice and cell lines
Six‐ to eight‐week‐old female wild‐type C57BL/6 mice were purchased from the Chinese Academy of Sciences (Shanghai, China). All mice were kept and bred in a specific pathogen‐free environment. All animal experiments were conducted in accordance with protocols approved by the Animal Care and Use Committee at Shanghai Medical College, Fudan University. Murine melanoma B16F10 cell line was obtained from the American Type Culture Collection (Manassas, VA). Cells were grown in complete RPMI‐1640 media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Gibco, Grand Island, NY, USA).
Animal tumour model
B16F10 cells (5 × 105 in 100 μl PBS) were subcutaneously inoculated into the right flanks of C57BL/6 mice, and tumour sizes were measured every 2 days using calipers. The tretinoin ointment (0·15 g, PanGeo Pharma Inc., Montreal, Canada) or vaseline as control was locally rubbed on the tumour and the skin adjacent to the tumour every day when tumours were palpable. The tumour volume was calculated by the following formula: V = (larger diameter) × (smaller diameter)2/2.
CD8+ T‐cell depletion
Neutralizing anti‐mouse CD8α antibody (500 mg, clone YTS169.4; BioXCell, West Lebanon, NH, USA) was injected intraperitoneally into C57BL/6 mice once a week starting from the day when B16F10 cells were inoculated. The control group received the same amount of isotype control antibody (clone LTF2; BioXCell). The efficiency of CD8+ T‐cell depletion was determined by flow cytometric analysis of tumour, spleen, lymph nodes and blood (see Supplementary material, Fig. S1).
Preparation of single‐cell suspension from draining lymph nodes and tumours
Tumour‐bearing mice were killed and their surgically removed axillary lymph nodes (skin‐draining) were mechanically disrupted and filtered through a 200‐μm nylon mesh. Lymph node cells were stimulated with α‐CD3 (2 μg/ml, eBioscience, San Diego, CA, USA) for 3 days. The supernatants were harvested for determining the concentrations of IFN‐γ and tumour necrosis factor‐α (TNF‐α) using mouse ELISA kits (eBioscience), and cells for flow cytometry. Tumours were collected, cut into pieces and digested by Collagenase IV (1 mg/ml) and DNAase I (5 U/ml) for 20 min in 37°, and filtered through a 100‐μm nylon mesh. A single‐cell suspension was used for flow cytometry.
Flow cytometry
After blocking the Fc‐receptors with anti‐mouse CD16/32 (eBioscience), the following fluorochrome‐labelled anti‐mouse antibodies were used: CD45 (30‐F11), CD11b (M1/70), Gr‐1 (RB6‐8C5), F4/80 (BM8), CD4 (GK1.5), CD8 (APA5), CD3 (17A2), CD25 (M‐A251), NK1.1 (PK136), EpCAM (1B7), Foxp3 (FJK‐16s), CD31 (390), CD11c (N418), IFN‐γ (AN‐18), TNF‐α (MP6‐XT22), Granzyme B (NGZB), (all from BD, Franklin Lakes, NJ). For intracellular staining, cells were stimulated with cell stimulation cocktail (Cat#00‐4970; eBioscience) for 5 hr, and then cell surface staining of CD8 or CD4 was performed. Intracellular staining of IFN‐γ, TNF‐α, Granzyme B and Foxp3 were carried out after cells were fixed and permeabilized using the intracellular staining kit (Cat#560409 BD). Samples were acquired by FACS Cyan instrument (Beckman Coulter, Brea, CA, USA) and analysed by flowjo 7·6·1 software (FlowJo, LLC, Ashland, OR).
RNA isolation and quantitative real‐time PCR
Total RNA was extracted using TRIZOL (Ambion, Austin, TX), then cDNA was generated using a high‐capacity cDNA Reverse Transcription kit (Takara, Shiga, Japan). Quantitative real‐time PCR (qPCR) was performed using an SYBR green Gene Expression Assay (Takara). The data are represented as fold changes of the atRA‐treated group compared with the vaseline‐treated group. The primer sequences of all genes for PCR were used as followed: Gzmb: 5′‐TGTGCTGACTGCTGCTCACT‐3′ and 5′‐TCCTCTTGGCCTTACTCTTC‐3′, Prf1: 5′‐AACCTCCACTCCACCTTGAC‐3′ and 5′‐GTGCGTGCCATAGGAGGAGA‐3′, Ifng: 5′‐GGATGGTGACATGAAAATCCTGC‐3′ and 5′‐TGCTGATGGCCTGATTGTCTT‐3′, Cxcl9: 5′‐TCCTTTTGGGCATCATCTTCC‐3′ and 5′‐TTTGTAGTGGATCGTGCCTCG‐3′, Cxcl10: 5′‐CCAAGTGCTGCCGTCATTTTC‐3′ and 5′‐GGCTCGCAGGGATGATTTCAA‐3′, Cxcl1: 5′‐CTGGGATTCACCTCAAGAACATC‐3′ and 5′‐CAGGGTCAAGGCAAGCCTC‐3′, Cxcl2: CCAACCACCAGGCTACAGG‐3′ and 5′‐GCGTCACACTCAAGCTCTG‐3′, Ccl2: 5′‐GTTGGCTCAGCCAGATGCA‐3′ and 5′‐AGCCTACTCATTGGGATCATCTTG‐3′, β‐actin: 5′‐CCAGCCTTCCTTCTTGGGTATG‐3′ and 5′‐TGTGTTGGCATAGAGGTCTTTACG‐3′.
In vitro stimulation of cells
For in vitro experiments, B16F10 cells were treated with atRA (Sigma, St Louis, MO) at different concentrations (1 μm, 5 μm or 10 μm) or DMSO as control with or without 10 ng/ml IFN‐γ (Peprotech, Rocky Hill, NJ) for 48 hr. Cells were harvested for further flow cytometry.
Cytotoxic T lymphocyte assay
Cytotoxicity was determined by lactate dehydrogenase (LDH) release from B16F10 cells into the culture medium, as LDH is a cytoplasmic enzyme released by dying cells. draining lymph nodes (DLN) cells were prepared from atRA‐treated B16F10 tumours, and then incubated with B16F10 cell lysates prepared by a repeated freeze–thaw process in the presence of interleukin‐2 (Peprotech, 10 ng/ml) for 5 days. B16F10 (target cells) cells treated with or without atRA were seeded in 96‐well plates as target cells for DLN cells (effector cells) at the effector : target (E : T) ratio of 50 : 1 and 100 : 1 for 5 hr, and then supernatants were collected for the measurement of LDH cytotoxicity activity according to the manufacturer's instructions of the LDH Cytotoxicity Assay Kit (Beyotime, Shanghai, China). DLN cells or B16F10 cells alone were set up as controls for spontaneous LDH release. Maximum LDH release of target cells was determined by lysing B16F10 cells for 1 hr with the lysis buffer provided by the assay kit. The killing rates were calculated using the following formula: killing rates = [(experimental release − target spontaneously release − effector spontaneously release group)/(target maximum release − target spontaneously release)] × 100.
Statistical analysis
The comparisons between two groups were performed by two‐tailed Student's t‐tests. Multiple‐group comparisons were performed by two‐way analysis of variance. The statistical analysis was performed with graphpad prism 6 (GraphPad Software, Inc., San Diego, CA). Significant difference was defined as P < 0·05.
Results
Topical treatment with atRA suppresses B16F10 tumour growth
We first attempted to examine whether topical treatment with atRA was able to influence the murine melanoma growth. Briefly, tretinoin ointment with atRA as the active ingredient or vaseline as control was locally rubbed on the tumour and the skin adjacent to the tumour every day when tumours were palpable on day 4 after subcutaneous inoculation of B16F10 melanoma cells, and mice were killed on day 14 (Fig. 1a). We found that atRA treatment significantly slowed down B16F10 tumour growth (Fig. 1b) and reduced the volume and weight of B16F10 tumours (Fig. 1b–d), demonstrating a therapeutic effect of topical treatment with atRA on murine melanoma.
Figure 1.
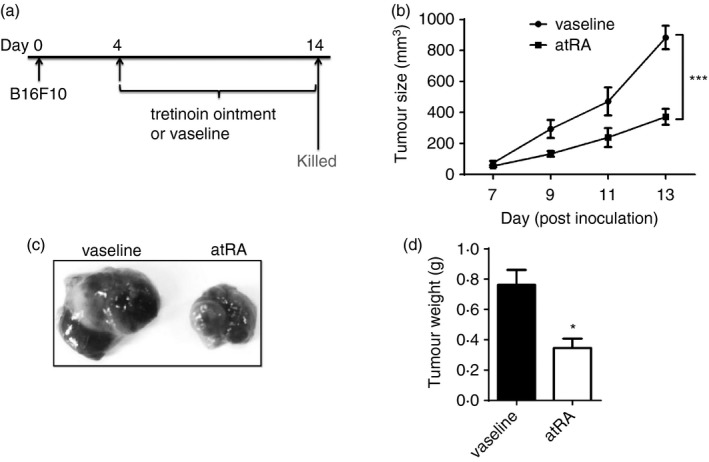
Topical all‐trans retinoic acid (atRA) treatment suppresses the growth of murine melanoma. (a) Experimental protocol for the development of a mouse model of melanoma. B16F10 cells (5 × 105 in 100 μl PBS) were subcutaneously inoculated into the right flanks of 6‐ to 8‐week‐old female C57BL/6 wild‐type mice, and tretinoin ointment (0·15 g), or vaseline used as control, was locally rubbed on the tumour and the skin adjacent to the tumour every day on day 4 when tumours were palpable. (b) Tumour growth was measured at the indicated time‐points. Statistical significance calculated by analysis of variance (c) Representative images of melanoma. (d) the tumour weight. Columns and error bars represent mean ± SEM (n = 5 to n = 7 per group). *P < 0·05, ***P < 0·001. Similar results were obtained from three independent experiments.
Topical treatment with atRA causes a pronounced CD8+ T‐cell response in the tumour microenvironment
Given the immunogenic property of melanoma and the immunoregulatory role of atRA, we next investigated whether topical treatment with atRA influenced the type and the activation of immune cells in tumour sites. Significantly increased frequency of CD45+ leucocytes was observed in atRA‐treated B16F10 tumours compared with those treated with vaseline by flow cytometric analysis of enzymatically dissociated cell suspensions (Fig. 2a). Further analysis revealed that atRA treatment significantly increased the frequency of tumour‐infiltrating CD8+ T cells, but not CD4+ T cells, in atRA‐treated mice compared with control mice (Fig. 2b). Moreover, there were significant decreases in the frequencies of tumour‐infiltrating MDSCs (CD11b+ Gr‐1+) and macrophages (CD11b+ F4/80+ Gr‐1−) in atRA‐treated mice (Fig. 2c). Importantly, atRA treatment significantly increased the ratios of tumour‐infiltrating CD8+ T cells to MDSCs or macrophages (Fig. 2d), suggesting an enhanced anti‐tumour immune response in the tumour microenvironment.
Figure 2.
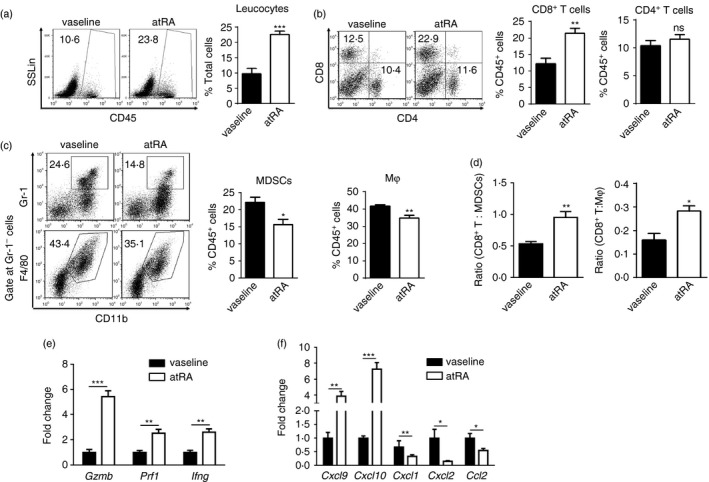
Topical all‐trans retinoic acid (atRA) treatment leads to significantly increased tumour‐infiltrating CD8+ T cells and decreased myeloid cells. Enzymatically disassociated single‐cell suspensions were prepared from atRA‐ or vaseline‐treated tumuors. (a–c) Representative flow cytometry data and averaged percentages of CD45+ leucocytes in total cells (a), CD4+ T cells and CD8+ T cells (b), as well as myeloid‐derived suppressor cells (MDSCs) (CD11b+ Gr‐1+) and macrophages (Mφ, CD11b+ F4/80+ Gr‐1−) in CD45+ leucocytes (c). (d) Ratios of CD8+ T cells to MDSCs or macrophages (Mφ) in tumours. (e, f) Quantitative RT‐PCR analysis of gene expression of anti‐tumour effector molecules incuding Gzmb, Prf1, Ifng (e) and chemokines including Cxcl9, Cxcl10, Cxcl1, Cxcl2, Ccl2 (f) in melanoma of mice treated with atRA or vaseline. Columns and error bars represent mean ± SEM (n = 5 to n = 7 per group). *P < 0·05, **P < 0·01, ***P < 0·001, NS = no significance. Similar results were obtained from three independent experiments.
Furthermore, atRA treatment significantly increased gene expression of several important anti‐tumour effector molecules including granzyme B, perforin and IFN‐γ, which are primarily expressed by activated CD8+ T cells. There was a significant increase in gene expression of chemokines that are important for T‐cell migration, including CXCL9 and CXCL10, whereas there was a significant decrease in those that are important for tumour recruitment of MDSCs, including CXCL1, CXCL2 and CCL2, in atRA‐treated B16F10 tumours compared with those treated with vaseline (Fig. 2e, f). This is consistent with the changes in the composition of immune cells upon atRA treatment. Taken together, these results demonstrate that topical treatment with atRA changes the balance between pro‐ and anti‐tumour immune responses in the tumour microenvironment, skewing toward anti‐tumour CD8+ T‐cell immunity.
Topical treatment with atRA promotes the activation and proliferation of anti‐tumour CD8+ T cells
We next investigated whether topical treatment with atRA activated effector CD8+ T cells in tumour sites. The portions of CD8+ T cells expressing granzyme B, TNF‐α or IFN‐γ, which are closely related to the cytotoxic activities, significantly increased in atRA‐treated B16F10 tumour compared with controls (Fig. 3a). Moreover, atRA treatment significantly enhanced the proliferation of CD8+ T cells, as evidenced by much higher frequency of Ki67+ proliferating CD8+ T cells in atRA‐treated B16F10 tumours (Fig. 3b). In contrast, comparable frequencies of IFN‐γ‐expressing CD4+ T cells and Ki67+ proliferating CD4+ T cells were detected in B16F10 tumours treated with atRA or vaseline (Fig. 3c). These results demonstrate that atRA treatment favourably promotes a robust effector CD8+ T‐cell response in the tumour microenvironment.
Figure 3.
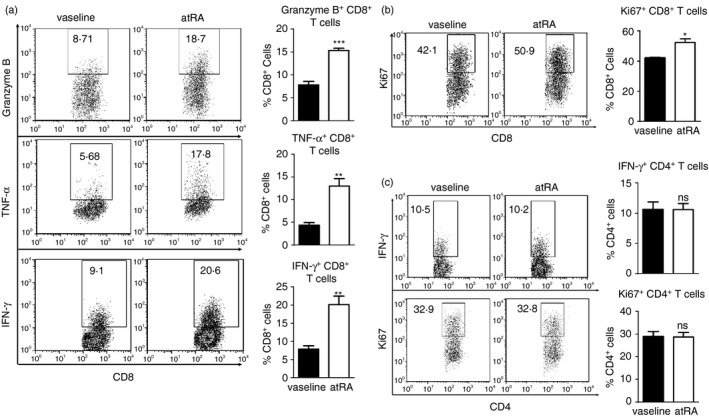
Topical all‐trans retinoic acid (atRA) treatment enhances effector CD8+ T‐cell response at tumour sites. (a, b) Representative flow cytometry data and averaged percentages of Granzyme B+, TNF‐α + and IFN‐γ + (a) as well as Ki67+ (b) cells in CD8+ T cells. (c) Representative flow cytometry data and averaged percentages of IFN‐γ + and Ki67+ cells in CD4+ T cells. Columns and error bars represent mean ± SEM (n = 5 to n = 7 per group). *P < 0·05, **P < 0·01, ***p < 0·001, NS = no significance. Similar results were obtained from three independent experiments.
Topical treatment with atRA promotes the differentiation of effector CD8+ T cells in DLN of B16F10 tumour‐bearing mice
AtRA is well‐known as a differentiation factor in developmental biology, and accumulating data demonstrate the role of atRA in determining T‐cell differentiation under homeostatic or inflammatory conditions.9 We then asked whether topical treatment with atRA influenced T‐cell differentiation in B16F10 tumour‐bearing mice. To this end, DLN were collected from tumour‐bearing mice on day 7 when the tumour size was similar between atRA‐treated and vaseline‐treated mice to avoid the influence of the size of tumour on T‐cell immunity. Greatly elevated concentrations of TNF‐α and IFN‐γ were detected in the cultures of DLN cells of atRA‐treated mice compared with those of vaseline‐treated mice upon α‐CD3 stimulation (Fig. 4a). Furthermore, there were significant increases in the portions of CD8+ T expressing granzyme B, TNF‐α or IFN‐γ in α‐CD3‐stimulated DLN cells of atRA‐treated mice (Fig. 4b). In contrast, significantly decreased portion of IFN‐γ‐expressing CD4+ T cells was detected in α‐CD3‐stimulated DLN cells from atRA‐treated mice (Fig. 4c). Taken together, these results demonstrate that topical treatment with atRA promotes the differentiation of effector CD8+ T cells in DLN of B16F10 melanoma‐bearing mice.
Figure 4.
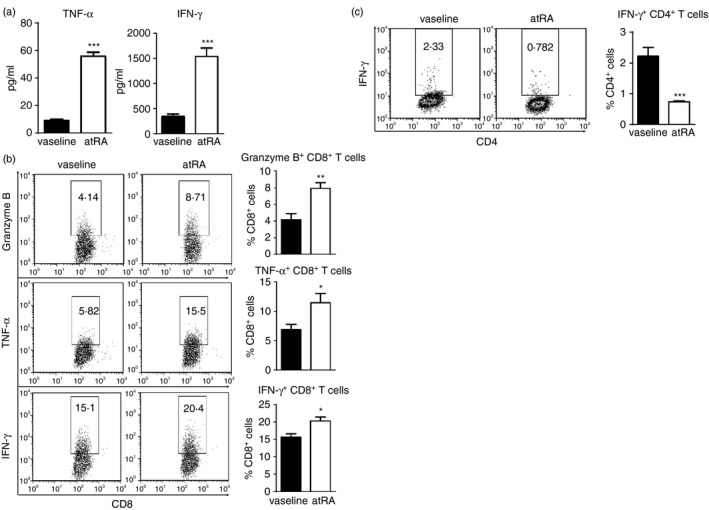
Topical all‐trans retinoic acid (atRA) treatment enhances the differentiation of effector CD8+ T cells in draining lymph nodes (DLN) of melanoma‐bearing mice. Axillary lymph nodes were collected from tumour‐bearing mice on day 7 when similar tumour size was detected in atRA‐ and vaseline‐treated mice to avoid the influence of the tumour size on T‐cell immunity. DLN cells were cultured with α‐CD3 for 3 days. (a) The concentrations of tumour necrosis factor‐α (TNF‐α) and interferon‐γ (IFN‐γ) in the cultures were determined by ELISA. (b) Representative flow cytometry data and averaged percentages of Granzyme B+, TNF‐α + and IFN‐γ + cells in CD8+ T cells. (c) Representative flow cytometry data and averaged percentages of IFN‐γ + cells in CD4+ T cells. Columns and error bars represent mean ± SEM (n = 5 to n = 7 per group). *P < 0·05, **P < 0·01, ***P < 0·001. Similar results were obtained from three independent experiments.
Depletion of CD8+ T cells greatly impairs the melanoma‐inhibitory effect of atRA
We then investigated the role of CD8+ T cells in the melanoma‐inhibitory effect of atRA by depleting CD8+ T cells in atRA‐treated melanoma‐bearing mice. CD8+ T‐cell depletion significantly impaired the tumour‐inhibiting effect of atRA, as evidenced by significant increases in the tumour volume and weight in mice treated with neutralizing anti‐CD8 antibody compared with those with isotype antibody (Fig. 5a–c). However, atRA treatment still significantly decreased the tumour volume and weight of CD8+ T‐cell‐depleted mice (Fig. 5a–c), suggesting that both CD8+ T‐cell‐dependent and T‐cell‐independent mechanisms mediate the melanoma‐inhibitory effect of atRA. Consistently, depletion of CD8+ T cells drastically reduced gene expression of granzyme, perforin and IFN‐γ (Fig. 5d), underscoring that CD8+ T cells are the primary source of these molecules in our model. Interestingly, significantly decreased frequency of CD4+ T cells was detected in tumours of atRA‐treated mice with neutralizing anti‐CD8 antibody compared with those with isotype antibody (Fig. 5e). Significantly increased MDSCs and macrophages were detected in tumours of atRA‐treated mice with CD8+ T‐cell depletion (Fig. 5e). CD8+ T‐cell depletion had no effect on gene expression of CXCL1, CXCL2 and CCL2 in tumours of atRA‐treated mice (Fig. 5d). Taken together, these results demonstrate that the therapeutic effect of atRA is partly dependent on CD8+ T cells in our model.
Figure 5.
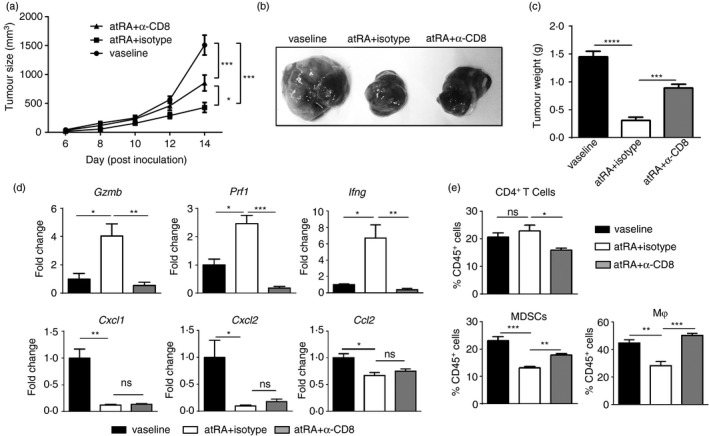
CD8+ T cells partly mediate the melanoma‐inhibitory effect of all‐trans retinoic acid (atRA). For CD8+ T‐cell depletion, 500 μg neutralizing anti‐mouse CD8α antibody (clone YTS169.4, BioXCell) was intraperitoneally injected into C57BL/6 female mice once a week starting from the day when B16F10 cells were inoculated into mice (a) Tumour growth was measured at the indicated time‐points. Statistical significance calculated by analysis of variance n = 5 to n = 7. (b) Representative images of melanoma. (c) The tumour weight. (d) Quantitative RT‐PCR analysis of gene expression of Gzmb, Prf1, Ifng, Cxcl1, Cxcl2, Ccl2. (e) Averaged percentages of CD4+ T cells, myeloid‐derived suppressor cells (MDSCs) and macrophages (Mφ) in enzymatically dissociated melanoma cell suspensions and was gated at CD45+ leucocytes. Columns and error bars represent mean ± SEM (n = 5 to n = 7 per group). *P < 0·05, **P < 0·01, ***P < 0·001, NS = no significance. Similar results were obtained from three independent experiments.
AtRA directly enhances the cytotoxic activity of effector CD8+ T cells by up‐regulating MHCI expression in melanoma cells
We next attempted to determine the possible mechanism by which atRA promoted anti‐tumour CD8+ T‐cell responses. Since CD8+ T‐cell‐mediated cytotoxicity is MHC class I restricted, we evaluated whether atRA treatment altered the expression of MHCI on B16F10 cells. AtRA treatment significantly up‐regulated MHCI expression in CD45− CD31− cells, the majority of which were B16F10 cells, from enzymatically dissociated B16F10 melanoma tumours (Fig. 6a, b). Consistently, in vitro treatment of B16F10 cells with atRA up‐regulated MHCI expression in a dose‐dependent manner (Fig. 6c). AtRA at only 10 μm could lead to cytotoxicity as evidenced by significantly increased apoptotic B16F10 cells (see Supplementary material, Fig. S2). Interferon‐γ is well‐known to increase MHCI expression. We therefore examined whether atRA treatment could amplify the promoting‐effect of IFN‐γ on B16F10 cells. As expected, IFN‐γ significantly increased MHCI expression on B16F10 cells (Fig. 6c). The addition of atRA could further increase the MHCI expression on B16F10 cells (Fig. 6c). To determine whether up‐regulated MHCI expression on B16F10 cells enhanced the ability of CD8+ T cells to kill these cells, the cytotoxicity assay were performed by co‐culturing DLN cells of the tumour‐bearing mice and B16F10 cells treated with atRA or DMSO at different ratios. We demonstrated that DLN cells co‐cultured with atRA‐treated B16F10 cells had significantly higher killing rates than those with DMSO‐treated B16F10 cells (Fig. 6d). These results show that atRA directly up‐regulates MHCI expression on B16F10 cells, thereby rendering them sensitive to CD8+ T‐cell‐mediated killing.
Figure 6.
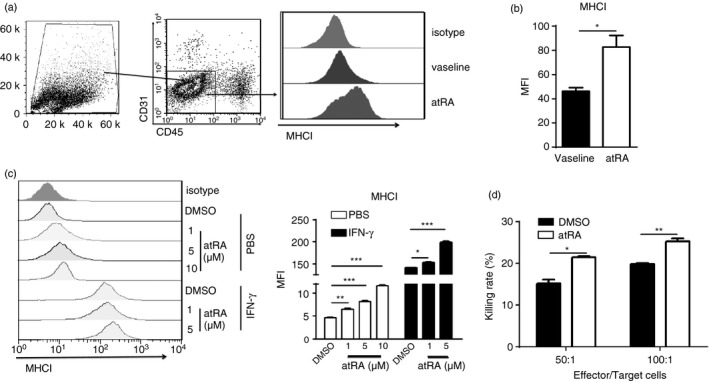
All‐trans retinoic acid (atRA) directly up‐regulates MHCI expression in melanoma cells and enhances the cytotoxic activity of cytotoxic T lymphocytes. (a, b) The gating strategy of CD45− CD31− cells in enzymatically disassociated single‐cell suspensions prepared from tumours, and representative histograms of MHCI expression (a) and flow cytometry plots of geometric mean fluorescence intensity (MFI) of MHCI expression (b) in CD45− CD31− cells of tumours from atRA‐ or vaseline‐treated mice. Columns and error bars represent mean ± SEM (n = 5 to n = 7 per group). Similar results were obtained from three independent experiments. (c) Representative histogram of MHCI expression and average MFI of MHCI on B16F10 cells after in vitro treatment with atRA at different concentrations with or without interferon‐γ (IFN‐γ; 10 ng/ml) for 48 hr. (d) The cytotoxicity assay was performed by co‐culturing draining lymph node cells (effector cells) from the tumour‐bearing mice and B16F10 cells (target cells) treated with atRA or DMSO at different ratio, shown are killing rates determined by lactate dehydrogenase release assay. DMSO was used as control in (c) and (d). Columns and error bars represent three replicate wells from a representative of three independent experiments in (c) and (d). *P < 0·05, **P < 0·01, ***P < 0·001.
Discussion
We here demonstrate that topical application of atRA effectively inhibits B16F10 tumour growth, which is accompanied by pronounced CD8+ T‐cell responses in tumour sites and DLN. Furthermore, we reveal that the therapeutic effect of atRA is partly dependent on CD8+ T cells, as depletion of CD8+ T cells partly restored B16F10 tumour growth in atRA‐treated mice. Moreover, our in vitro study reveals that atRA is able to up‐regulate MHCI expression in B16F10 cells in a dose‐dependent manner, which in turn enhances the cytotoxic activity of CD8+ T cells, while atRA at higher concentrations also enhances the apoptosis of B16F10 cells.
The beneficial role of atRA in melanoma is implicated by a recent finding that vitamin A supplementation could decrease the risk of melanoma in humans. We here demonstrated the therapeutic effect of topical application of atRA on murine B16F10 melanoma. Considering the profound effects of atRA on immune cells, particularly T cells, and the relatively high sensitivity of melanoma to immunotherapy, the melanoma‐inhibitory effect of atRA observed in our study was probably due to its immunoregulatory functions. We found that topical treatment with atRA markedly increased the frequency of leucocytes in B16F10 tumour, which was primarily attributed to increased tumour‐infiltrating CD8+ T cells. Furthermore, atRA treatment promoted the activation of effector CD8+ T cells, as evidenced by marked increases in the proportions of CD8+ T cells that expressed effector molecules including granzyme B, TNF‐α or IFN‐γ and proliferating CD8+ T cells in tumours. Interestingly, atRA treatment had no effect on the activation state and proliferation of tumour‐infiltrating CD4+ T cells. Additionally, topical atRA treatment on established B16F10 tumours did not affect the frequency of tumour‐infiltrating Treg cells (see Supplementary material, Fig. S3a), although inhibition of endogenous atRA‐RARα signalling was shown to suppress tumour‐infiltrating CD4+ CD25+ Foxp3+ Treg cells.32 This could be due to the different effects of atRA on CD4+ T cells in physiological or pharmacological concentrations. Notably, in addition to elevated CD8+ T‐cell response in tumour sites, topical treatment with atRA could enhance effector CD8+ T‐cell differentiation, as significantly increased effector CD8+ T cells expressing granzyme B, TNF‐α or IFN‐γ were detected in DLN of tumour‐bearing mice on day 7 after B16F10 cell inoculation. This is supported by previous studies showing that administration of atRA provides protection against infection by promoting the differentiation of effector CD8+ T cells.15 In contrast, there was significantly decreased IFN‐γ‐expressing CD4+ T cells in DLN of atRA‐treated mice, which is consistent with the previous findings that exogenous atRA inhibited IFN‐γ production during the early phase of Th1 differentiation.33 Collectively, these results indicate that topical treatment with exogenous atRA promotes a robust CD8+ T‐cell response, which could contribute to the inhibition of B16F10 melanoma.
We further confirmed the requirement of CD8+ T cells for the melanoma‐inhibiting effect of atRA by showing that depletion of CD8+ T cells restored B16F10 tumour growth in atRA‐treated mice. However, atRA treatment still had the inhibitory effect on B16F10 tumour growth in the absence of CD8+ T cells, suggesting the existence of a CD8+ T‐cell‐independent mechanism. Natural killer (NK) cells were reported to be in synergy with CD8+ T cells to inhibit established B16F10 tumour.34 However, we found that topical treatment with atRA had no effect on the frequency of NK cells and IFN‐γ‐expressing NK cells in B16F10 tumours regardless of the presence of CD8+ T cells (see Supplementary material, Fig. S3b), which excluded the possibility that NK cells mediate the therapeutic effect of atRA on established B16F10 tumour. AtRA can induce apoptosis of many types of cancer cells including melanoma at high non‐physiological concentrations.25, 35 Consistently, we found that atRA at higher concentration (10 μm) could significantly induce the apoptosis of B16F10 cells, suggesting that the direct pro‐apoptotic effect of atRA on melanoma cells may also contribute to the inhibition of B16F10 tumour growth in vivo.
Early studies have demonstrated that MHCI is a direct transcriptional target of atRA.36, 37 A recent study also demonstrated that the therapeutic effect of atRA on murine colon cancer is dependent on its ability to directly up‐regulate MHCI expression in tumour epithelial cells, thereby rendering them susceptible to CD8+ T‐cell‐mediated killing.38 We also found that atRA treatment could up‐regulate MHCI expression in B16F10 cells that are not transformed from epithelial cells in a dose‐dependent manner. Furthermore, atRA could further amplify the ability of IFN‐γ to up‐regulate MHCI expression in B16F10 cells. More importantly, we demonstrated that DLN cells from tumour‐bearing mice exhibited a significantly higher killing ability when cultured with atRA‐treated B16F10 cells. These results indicate that topical treatment with atRA could up‐regulate MHCI expression in B16F10 cells, which in turn makes them a better target of CD8+ cytotoxic T‐cell killing.
In summary, our study provides the evidence that topical treatment with atRA effectively inhibits murine melanoma growth, and indicates that the melanoma‐inhibitory effect of atRA is partly dependent on its ability to promote anti‐tumour CD8+ T‐cell immunity. Given the fact that systemic administration of atRA has a risk to cause retinoid toxicity, and topical atRA has long been clinically used, our results suggest that topical application atRA may be an effective and safe therapeutic strategy for combination treatment of melanoma.
Disclosures
None of the authors has a conflicting financial interest.
Supporting information
Figure S1. The efficiency of CD8+ T‐cell depletion.
Figure S2. All‐trans retinoic acid at 10 μm induced apoptosis of B16F10 cells.
Figure S3. Topical all‐trans retinoic acid treatment has no effect on tumour‐infiltrating regulatory T cells and natural killer cells.
Acknowledgements
This work is supported by National Natural Science Foundation of China Grant 81471555, 91642112 (to R.H.) and The National Key Research and Development Program of China 2016YFC1305103 (to R.H.)
References
- 1. Berwick M, Buller DB, Cust A, Gallagher R, Lee TK, Meyskens F et al Melanoma epidemiology and prevention. Cancer Treat Res 2016; 167:17–49. [DOI] [PubMed] [Google Scholar]
- 2. Lee AF, Sieling PA, Lee DJ. Immune correlates of melanoma survival in adoptive cell therapy. Oncoimmunology 2013; 2:e22889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leontovich AA, Dronca RS, Nevala WK, Thompson MA, Kottschade LA, Ivanov LV et al Effect of the lymphocyte‐to‐monocyte ratio on the clinical outcome of chemotherapy administration in advanced melanoma patients. Melanoma Res 2016; 27:32–42. [DOI] [PubMed] [Google Scholar]
- 4. Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M et al TIL therapy broadens the tumor‐reactive CD8+ T cell compartment in melanoma patients. Oncoimmunology 2012; 1:409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Viguier M, Lemaitre F, Verola O, Cho MS, Gorochov G, Dubertret L et al Foxp3 expressing CD4+CD25high regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol 2004; 173:1444–53. [DOI] [PubMed] [Google Scholar]
- 6. Hung K, Hayashi R, Lafond‐Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J Exp Med 1998; 188:2357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meyer C, Cagnon L, Costa‐Nunes CM, Baumgaertner P, Montandon N, Leyvraz L et al Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother 2014; 63:247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diaz‐Montero CM, Finke J, Montero AJ. Myeloid‐derived suppressor cells in cancer: therapeutic, predictive, and prognostic implications. Semin Oncol 2014; 41:174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raverdeau M, Mills KH. Modulation of T cell and innate immune responses by retinoic acid. J Immunol 2014; 192:2953–8. [DOI] [PubMed] [Google Scholar]
- 10. Chen MC, Hsu SL, Lin H, Yang TY. Retinoic acid and cancer treatment. Biomedicine (Taipei) 2014; 4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity 2011; 35:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coombes JL, Siddiqui KR, Arancibia‐Carcamo CV, Hall J, Sun CM, Belkaid Y et al A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF‐β and retinoic acid‐dependent mechanism. J Exp Med 2007; 204:1757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17‐producing T cell responses. Nat Immunol 2007; 8:1086–94. [DOI] [PubMed] [Google Scholar]
- 14. Pino‐Lagos K, Guo Y, Brown C, Alexander MP, Elgueta R, Bennett KA et al A retinoic acid‐dependent checkpoint in the development of CD4+ T cell‐mediated immunity. J Exp Med 2011; 208:1767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan X, Sande JL, Pufnock JS, Blattman JN, Greenberg PD. Retinoic acid as a vaccine adjuvant enhances CD8+ T cell response and mucosal protection from viral challenge. J Virol 2011; 85:8316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, Osma‐Garcia I et al Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015; 42:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cantorna MT, Nashold FE, Hayes CE. In vitamin A deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J Immunol 1994; 152:1515–22. [PubMed] [Google Scholar]
- 18. Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R et al All‐trans‐retinoic acid eliminates immature myeloid cells from tumor‐bearing mice and improves the effect of vaccination. Cancer Res 2003; 63:4441–9. [PubMed] [Google Scholar]
- 19. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all‐trans retinoic acid effect on tumor‐associated myeloid‐derived suppressor cells. Cancer Res 2007; 67:11021–8. [DOI] [PubMed] [Google Scholar]
- 20. Okuno M, Kojima S, Matsushima‐Nishiwaki R, Tsurumi H, Muto Y, Friedman SL et al Retinoids in cancer chemoprevention. Curr Cancer Drug Targets 2004; 4:285–98. [DOI] [PubMed] [Google Scholar]
- 21. Hsu SL, Lin HM, Chou CK. Suppression of the tumorigenicity of human hepatoma hep3B cells by long‐term retinoic acid treatment. Cancer Lett 1996; 99:79–85. [DOI] [PubMed] [Google Scholar]
- 22. Arisi MF, Starker RA, Addya S, Huang Y, Fernandez SV. All trans‐retinoic acid (ATRA) induces re‐differentiation of early transformed breast epithelial cells. Int J Oncol 2014; 44:1831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Asgari MM, Brasky TM, White E. Association of vitamin A and carotenoid intake with melanoma risk in a large prospective cohort. J Invest Dermatol 2012; 132:1573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sengupta S, Ray S, Chattopadhyay N, Biswas N, Chatterjee A. Effect of retinoic acid on integrin receptors of B16F10 melanoma cells. J Exp Clin Cancer Res 2000; 19:81–7. [PubMed] [Google Scholar]
- 25. Siddikuzzaman, Grace VM. Anti‐metastatic study of liposome‐encapsulated all trans retinoic acid (ATRA) in B16F10 melanoma cells‐implanted C57BL/6 mice. Cancer Invest 2014; 32:507–17. [DOI] [PubMed] [Google Scholar]
- 26. Wang Z, Cao Y, D'Urso CM, Ferrone S. Differential susceptibility of cultured human melanoma cell lines to enhancement by retinoic acid of intercellular adhesion molecule 1 expression. Cancer Res 1992; 52:4766–72. [PubMed] [Google Scholar]
- 27. Guo Y, Pino‐Lagos K, Ahonen CA, Bennett KA, Wang J, Napoli JL et al A retinoic acid‐rich tumor microenvironment provides clonal survival cues for tumor‐specific CD8+ T cells. Cancer Res 2012; 72:5230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Galvin KC, Dyck L, Marshall NA, Stefanska AM, Walsh KP, Moran B et al Blocking retinoic acid receptor‐alpha enhances the efficacy of a dendritic cell vaccine against tumours by suppressing the induction of regulatory T cells. Cancer Immunol Immunother 2013; 62:1273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown CC, Noelle RJ. Seeing through the dark: new insights into the immune regulatory functions of vitamin A. Eur J Immunol 2015; 45:1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schuster GU, Kenyon NJ, Stephensen CB. Vitamin A deficiency decreases and high dietary vitamin A increases disease severity in the mouse model of asthma. J Immunol 2008; 180:1834–42. [DOI] [PubMed] [Google Scholar]
- 31. Wu J, Zhang Y, Liu Q, Zhong W, Xia Z. All‐trans retinoic acid attenuates airway inflammation by inhibiting Th2 and Th17 response in experimental allergic asthma. BMC Immunol 2013; 14:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Penkert RR, Surman SL, Jones BG, Sealy RE, Vogel P, Neale G et al Vitamin A deficient mice exhibit increased viral antigens and enhanced cytokine/chemokine production in nasal tissues following respiratory virus infection despite the presence of FoxP3+ T cells. Int Immunol 2016; 28:139–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iwata M, Eshima Y, Kagechika H. Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. Int Immunol 2003; 15:1017–25. [DOI] [PubMed] [Google Scholar]
- 34. Xu D, Gu P, Pan PY, Li Q, Sato AI, Chen SH. NK and CD8+ T cell‐mediated eradication of poorly immunogenic B16‐F10 melanoma by the combined action of IL‐12 gene therapy and 4‐1BB costimulation. Int J Cancer 2004; 109:499–506. [DOI] [PubMed] [Google Scholar]
- 35. Liu WJ, Zhang T, Guo QL, Liu CY, Bai YQ. Effect of ATRA on the expression of HOXA5 gene in K562 cells and its relationship with cell cycle and apoptosis. Mol Med Rep 2016; 13:4221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jansa P, Forejt J. A novel type of retinoic acid response element in the second intron of the mouse H2Kb gene is activated by the RAR/RXR heterodimer. Nucleic Acids Res 1996; 24:694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marks MS, Hallenbeck PL, Nagata T, Segars JH, Appella E, Nikodem VM et al H‐2RIIBP (RXR β) heterodimerization provides a mechanism for combinatorial diversity in the regulation of retinoic acid and thyroid hormone responsive genes. EMBO J 1992; 11:1419–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker‐Flynn NE et al Normalizing microbiota‐induced retinoic acid deficiency stimulates protective CD8+ T cell‐mediated immunity in colorectal cancer. Immunity 2016; 45:641–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The efficiency of CD8+ T‐cell depletion.
Figure S2. All‐trans retinoic acid at 10 μm induced apoptosis of B16F10 cells.
Figure S3. Topical all‐trans retinoic acid treatment has no effect on tumour‐infiltrating regulatory T cells and natural killer cells.