

Summary
Graft‐versus‐host disease (GVHD) remains a major problem after allogeneic haematopoietic stem cell transplantation, a curative therapy for haematological malignancies. Previous studies have demonstrated a role for the adenosine triphosphate (ATP)‐gated P2X7 receptor channel in allogeneic mouse models of GVHD. In this study, injection of human peripheral blood mononuclear cells (PBMCs) into immunodeficient non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice established a humanized mouse model of GVHD. This model was used to study the effect of P2X7 blockade in this disease. From five weeks post‐PBMC injection, humanized mice exhibited clinical signs and histopathology characteristic of GVHD. The P2X7 antagonist, Brilliant Blue G (BBG), blocked ATP‐induced cation uptake into both murine and human cells in vitro. Injection of BBG (50 mg/kg) into NSG mice did not affect engraftment of human leucocytes (predominantly T cells), or the clinical score and survival of mice. In contrast, BBG injection reduced circulating human interferon (IFN)‐γ significantly, which was produced by human CD4+ and CD8+ T cells. BBG also reduced human T cell infiltration and apoptosis in target organs of GVHD. In conclusion, the P2X7 antagonist BBG reduced circulating IFN‐γ in a humanized mouse model of GVHD supporting a potential role for P2X7 to alter the pathology of this disease in humans.
Keywords: bone marrow transplantation, Brilliant Blue G, graft‐versus‐host disease, humanized mice, lymphocyte, P2X7 receptor, purinergic signalling
Introduction
Allogeneic haematopoietic stem cell transplantation (HSCT) is a current curative therapy for a range of haematological malignancies, including leukaemia and lymphoma 1. A common complication following transplantation is the development of graft‐versus‐host disease (GVHD), which arises in approximately half of HSCT recipients worldwide 2. GVHD is mediated by transplanted donor immune cells attacking ‘foreign’ host tissue, resulting in inflammatory damage to healthy host tissue. The initial host tissue damage by preconditioning regimens used to treat cancer causes a proinflammatory environment which leads, in turn, to activation of donor CD4+ T cells through host antigen‐presenting cells 3. CD4+ T cells then release additional proinflammatory cytokines, including interferon (IFN)‐γ and interleukin (IL)‐17, directly causing damage to tissue, as well as activating cytotoxic CD8+ T cells which further exacerbate disease 4, 5. Current therapies aim to prevent GVHD by preventing these T cell responses through general immunosuppression, leaving a patient vulnerable to subsequent infection or relapse, demonstrating a vital need for novel therapeutics 6.
The purinergic system comprises a complex network of extracellular signalling molecules and plasma membrane receptors, and is involved in a range of physiological processes including inflammation and immunity 7. Adenosine triphosphate (ATP) is a common signalling molecule in this system, acting as both an autocrine activation molecule and a danger‐associated molecular pattern when released by damaged or dying cells 8. In these contexts, extracellular ATP predominantly mediates its effects through activation of the P2X7 receptor 8. P2X7 is an ATP‐gated cation channel, whereby activation by ATP allows the flux of Ca2+, K+ and other cations, including fluorescent dyes 9. P2X7 is present on numerous immune cell types, including antigen‐presenting cells and T cells 10. Activation of the P2X7 receptor can result in a variety of downstream signalling events, including cytokine release, reactive oxygen species formation and cell proliferation 11.
Extracellular ATP and P2X7 have emerging roles in GVHD. In allogeneic mouse models, extracellular ATP accumulates at sites of inflammatory damage, while P2X7 is up‐regulated on dendritic cells (DCs) in lymphoid tissues and livers of mice with severe GVHD 12. Notably, pharmacological blockade or genetic deletion of P2X7 reduces disease severity and improves survival in allogeneic mouse models of GVHD 12, 13, 14. Studies investigating the role of P2X7 in human GVHD are limited, but it has been shown that P2X7 expression is higher in colon biopsies from HSCT patients 12. Additionally, the presence of a loss‐of‐function P2RX7 single nucleotide polymorphism (E496A) in either the donor or recipient cells correlates with reduced survival after allogeneic HSCT 15.
Therefore, the current study aimed to investigate the effect of the P2X7 antagonist, Brilliant Blue G (BBG), in a preclinical humanized mouse model of GVHD. BBG prevented ATP‐induced dye uptake into splenic DCs from non‐obese diabetic‐severe combined immunodeficiency‐ (NOD‐SCID)‐IL‐2Rγnull (NSG) mice and peripheral blood T cells from humans. NSG mice engrafted with human (h) peripheral blood mononuclear cells (PBMCs) demonstrated increased P2X7 expression in the spleen and small intestine. Injection of BBG did not alter the engraftment of human leucocytes or the clinical signs of GVHD in these mice. In contrast, injection of BBG caused a significant reduction in serum hIFN‐γ concentrations and reduced human T cell infiltration and apoptosis in target organs, suggesting a potential role for P2X7 in GVHD pathogenesis in humans.
Methods
Antibodies for flow cytometry
Fluorescein isothiocyanate (FITC)‐conjugated mouse anti‐hCD4 (clone: RPA‐T4), mouse anti‐hCD8 (clone: RPA‐T8) and mouse anti‐hCD45 (clone: HI30) monoclonal antibodies (mAb); R‐phycoerythrin (PE)‐conjugated mouse anti‐hCD3 (clone: UCHT1) and mouse anti‐hCD8 (clone: RPA‐T8) mAb; peridinin chlorophyll protein (PerCP‐Cy5·5)‐conjugated mouse anti‐hCD4 (clone: L200) and rat anti‐mCD45 (clone: 30‐F11) mAb; allophycocyanin (APC)‐conjugated mouse anti‐hCD3 (clone: UCHT1) and mouse anti‐hCD19 (clone: HIB19) mAb were obtained from BD Pharmingen (San Jose, CA, USA). PE‐conjugated hamster anti‐murine (m) CD11c mAb (clone: N418) was from BioLegend (San Diego, CA, USA).
Cells
Human multiple myeloma RPMI‐8226 cells were obtained from the European Collection of Cell Cultures (Wiltshire, UK). Murine macrophage RAW264.7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were maintained in RPMI‐1640 medium containing 2 mM GlutaMAX (Thermo Fisher Scientific, Waltham, MA, USA) and 10% heat‐inactivated fetal bovine serum (FBS) (Bovogen Biologicals, East Keller, Australia) at 37°C/5% CO2. Cell lines were checked every 2 months for Mycoplasma spp. infections, using a MycoAlertTM Mycoplasma Detection Kit (Lonza, Basel, Switzerland), as per the manufacturer's instructions. Cells were routinely negative for Mycoplasma spp.
Mice
Mouse experiments were approved by the Animal Ethics Committee, University of Wollongong (Wollongong, NSW, Australia). C57BL/6 mice were obtained from Australian BioResources (Moss Vale, NSW, Australia). P2X7 knock‐out (KO) mice 16, back‐crossed onto a C57BL/6 background 17, were bred at the University of Wollongong. Deletion of the P2RX7 gene was confirmed periodically as described previously 16. Female NSG mice, obtained originally from The Jackson Laboratory (Bar Harbor, ME, USA), were bred at the Westmead Animal Research Facility (Westmead, NSW, Australia). NSG mice were housed in filter top cages in Tecniplast (Buggugiate, Italy) isolation cabinets, and provided ad libitum with autoclaved food and water.
Isolation of human PBMCs
Experiments with human blood were approved by the Human Ethics Committee, University of Wollongong. Blood was collected by venepuncture into VACUETTE® lithium heparin tubes (Greiner Bio‐One, Frickenhausen, Germany). Whole blood was diluted with an equal volume of sterile phosphate‐buffered saline (PBS) (Thermo Fisher Scientific), underlaid with Ficoll‐Paque PLUS (GE Healthcare, Uppsala, Sweden) and centrifuged (560 g for 30 min). hPBMCs were collected and washed with two volumes of PBS by centrifugation (430 g for 5 min) and resuspended in PBS.
Humanized mouse model of GVHD
NSG mice were injected i.p. with 10 × 106 hPBMCs in 100 μl sterile PBS (hPBMC group) or with 100 μl sterile PBS alone (control group) (day 0). At 3 weeks post‐hPBMC injection, mice were checked for engraftment by immunophenotyping of tail vein blood. Mice were monitored up to weeks 8 or 10 for signs of GVHD using a scoring system (involving a score out of 2, for each of the following five criteria: weight loss, hunching, decreased activity, fur ruffling and skin involvement), giving a total clinical score out of 10. Mice were euthanized at 8 or 10 weeks post‐injection of hPBMCs, or earlier if exhibiting a clinical score of ≥ 8 or a weight loss of ≥ 10%, according to the approved animal ethics protocol. For BBG blockade, mice were injected with 10 × 106 hPBMCs as above, and with either 200 μl sterile saline (saline group) or 200 μl sterile saline containing BBG (50 mg/kg) (BBG group) (Sigma‐Aldrich, St Louis, MO, USA) on days 0 (1 h post‐hPBMC injection), 2, 4, 6 and 8.
Immunophenotyping by flow cytometry
Tail vein blood (50 μl) was collected into 200 μl of citrate solution (Sigma‐Aldrich), diluted with PBS and centrifuged (500 g for 5 min). Spleens from euthanized mice were homogenized and filtered through 70 μm nylon filters (Falcon Biosciences, New York, NY, USA) and centrifuged (300 g for 5 min). Blood and spleen cells were incubated with ammonium chloride potassium lysis buffer (150 mM NH4Cl, 1 mM KHCO3, 0·1 mM Na2CO3, pH 7·3) for 5 min and washed in PBS (300 g for 5 min). Cells were washed in PBS containing 2% FBS (300 g for 3 min) and incubated for 30 min with fluorochrome conjugated mAb, including respective isotype controls. Cells were washed with PBS (300 g for 3 min) and data were collected using a BD Biosciences LSRII Flow Cytometer (using band‐pass filters 515/20 for FITC, 575/26 for PE, 675/40 for PerCP‐Cy5.5 and 660/20 for APC). The relative percentages of cells were analysed using FlowJo software version 8.7.1 (TreeStar Inc., Ashland, OR, USA) (Supporting information, Figs S1 and S2).
Histological analysis
Tissues from euthanized mice were incubated overnight in neutral buffered (10%) formalin (Sigma‐Aldrich). Fixed tissues were removed, coated in paraffin, sectioned (5 μm) and stained with haematoxylin and eosin (POCD, Artarmon, Australia). Histological changes were assessed using a Leica (Wetzlar, Germany) DMIL inverted light microscope at ×4 objective and images captured using a Motic (Causeway Bay, Hong Kong) Moticam 2 microscope camera, using Motic Images Plus software version 2.0.
Isolation of RNA
Tissues removed from euthanized mice were stored in RNAlater (Sigma‐Aldrich) at −20°C until required. RNA was isolated using TRIzol reagent (Thermo Fisher Scientific), as per the manufacturer's instructions. Isolated RNA was converted immediately to complementary DNA (cDNA) using the qScript cDNA Synthesis Kit (Quanta Biosciences, Beverly, MA, USA), as per the manufacturer's instructions, and stored at −80°C. cDNA was checked by polymerase chain reaction (PCR) amplification of the housekeeping gene glyceraldehyde 3‐phosphate dehydrogenase (Invitrogen, Carlsbad, CA, USA) for 35 cycles (95°C for 1 min, 55°C for 1 min and 72°C for 1 min) and a holding temperature of 4°C. Purity and size of amplicons were confirmed by 2% agarose gel electrophoresis.
Quantitative real‐time PCR
Quantitative real‐time PCR (qPCR) reactions were performed using TaqMan Universal Master Mix II (Thermo Fisher Scientific), according to the manufacturer's instructions, with primers for FAM‐labelled murine glyceraldehyde 3‐phosphate dehydrogenase (Mm99999915_g1) and mIL‐1β (Mm00434228_m1), and VIC‐labelled mP2X7 (Mm01199503_m1), and primers for FAM‐labelled human hypoxanthine phosphoribosyl transferase 1 (Hs99999909_m1) and hIL‐1β (Hs01555410_m1) and VIC‐labelled hIFN‐γ (Hs00989291_m1), hIL‐17 (Hs00936345_m1) and hP2X7(B) (AIOIXC2) (Thermo Fisher Scientific), as indicated. qPCR cycles consisted of two initial steps of 50°C for 2 min and 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. qPCR reactions were conducted in triplicate, and were performed on a Roche Diagnostics (Indianapolis, IN, USA) LightCycler 480, and analysis was conducted using LightCycler480 software version 1.5.1.
Immunoblotting
Cell lysates were prepared and immunoblotting was performed as described 18. Briefly, cell lysates (15 μg protein per lane) were loaded into a Mini‐PROTEAN TGXTM Precast Gel (Bio‐Rad, Hercules, CA, USA) and electrophoresed. Proteins were transferred to a nitrocellulose membrane (Bio‐Rad) using a Bio‐Rad Trans‐Blot Turbo Blotting System. Membranes were washed with Tris‐buffered saline (250 mM NaCl and 50 mM Tris, pH 7·5) containing 0·2% Tween‐20 (TBST) and blocked overnight at 4°C with blocking solution (TBST containing 5% milk powder). Membranes were incubated with rabbit anti‐mP2X7 (extracellular epitope) antibody (1 : 500) (Alomone Laboratories, Jerusalem, Israel) for 2 h at room temperature (RT), washed with TBST and incubated with peroxidase‐conjugated goat anti‐rabbit immunoglobulin (Ig)G (1 : 1000) (Rockland Immunochemicals, Limerick, PA, USA) for 1 h at RT. Membranes were washed with TBST and incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and images collected using an Amersham Imager600 (GE Healthcare).
ATP‐induced YO‐PRO‐12+ uptake assay
P2X7 pore formation was quantified by measuring ATP‐induced YO‐PRO‐12+ uptake, as described previously 19. Briefly, cells in NaCl medium (145 mM NaCl, 5 mM KCl, 5 mM glucose and 10 mM HEPES, pH 7·4) at 37°C were incubated in the absence or presence of BBG for 15 min, and then incubated with 1 μM YO‐PRO‐1 iodide (Molecular Probes, Eugene, OR, USA) in the absence or presence of 1 mM ATP (Sigma‐Aldrich) for 10 min. Incubations were stopped by addition of ice‐cold NaCl medium containing 20 mM MgCl2 (MgCl2 medium) and centrifugation (300 g for 3 min). Cells were incubated with PE‐conjugated anti‐mCD11c or APC‐conjugated anti‐hCD3 mAb, respectively, where indicated, for 15 min and washed with NaCl medium. Data were collected using an LSRII Flow Cytometer (band‐pass filter 515/20 for YO‐PRO‐12+, 575/25 for PE and 660/20 for APC) and FACSDiva software version 8.0. Geometric mean fluorescence intensity of YO‐PRO‐12+ uptake was analysed using FlowJo software (Supporting information, Fig. S3).
Immunohistochemistry
Paraffin‐embedded formalin‐fixed tissues were sectioned (5 μm) and mounted on Snowcoat slides (Leica) coated with porcine gelatin (Sigma‐Aldrich). To identify T cells, tissue sections were deparaffinized and heat‐induced epitope retrieval was performed by incubating in sodium citrate buffer [10 mM trisodium citrate dihydrate (Sigma‐Aldrich) and 0.05% Tween‐20, pH 6] for 20 min at 95°C. Before all subsequent steps, tissue sections were washed with Tris‐buffered saline (138 mM NaCl, 2·7 mM KCl and 50 mM Tris, pH 7·5) containing 0·05% Tween‐20 (TBST2). Tissue sections were blocked with PBS containing 20% goat serum (Thermo Fisher Scientific) and 3% bovine serum albumin (BSA) (Amresco, Solon, OH, USA) for 30 min at RT, and incubated subsequently with rabbit anti‐hCD3 mAb (clone: EP449E) (1 : 50) (Abcam, Cambridge, UK) in TBST2 containing 1% BSA overnight at 4°C. Endogenous peroxidase was quenched by incubating with 3% hydrogen peroxide in PBS containing 1% BSA for 20 min at RT. Tissue sections were then incubated with horseradish peroxidase‐conjugated goat anti‐rabbit IgG (1 : 250) (Invitrogen) in TBST2 containing 1% BSA for 15 min at RT. Finally, tissue sections were incubated with 3,3‐diaminobenzidine tetrachloride (Sigma‐Aldrich) for 5 min at RT, counterstained with haematoxylin and dehydrated. To identify apoptotic cells, an in‐situ Apoptosis Detection Kit (Abcam) was used as per the manufacturer's instructions. Immunohistochemistry images were captured using a Leica DMRB microscope and Leica Application Software version 4.3.
Enzyme‐linked immunosorbent assay (ELISA)
Blood, collected via cardiac puncture from euthanized mice, was incubated for 1 h at RT and centrifuged (1700 g for 10 min). Supernatants were recentrifuged (1700 g for 10 min) and sera stored at −80°C. Serum cytokine concentrations were measured using hIFN‐γ, hIL‐17, hIL‐4 and mIL‐1β Ready‐Set‐Go! ELISA Kits (eBioscience, San Diego, CA, USA), as per the manufacturer's instructions. Absorbance (450 and 570 nm) was measured using a SpectraMax Plus 384 (Molecular Devices, Sunnyvale, CA, USA).
Intracellular staining
Splenocytes, following ammonium chloride potassium buffer lysis, were incubated for 4 h in RPMI‐1640 medium containing 2 mM L‐glutamine (Thermo Fisher Scientific), 10% FBS, 1% non‐essential amino acids (Thermo Fisher Scientific), 55 μM mercaptoethanol (Thermo Fisher Scientific), 100 U/ml penicillin/100 μg/ml streptomycin (Thermo Fisher Scientific), 50 ng/ml phorbol 12‐myristate 13‐acetate (Sigma‐Aldrich), 1 μg/ml ionomycin (Sigma‐Aldrich) and 1 μg/ml GolgiStopTM (BD Biosciences). Cells were centrifuged (300 g for 5 min) and washed once with PBS (300 g for 5 min). Staining was performed using the BD Biosciences Intracellular Stain Kit, as per the manufacturer's instructions, mAb added and incubated for 30 min at RT. Cells were washed with PBS and data were collected using an LSRII Flow Cytometer (band‐pass filter 515/20 for FITC, 575/25 for PE, 675/40 for PerCP‐Cy5.5 and 660/20 for APC) and FACSDiva software version 8.0. The percentage of hIFN‐γ+ and hIL‐17+ cells was analysed using FlowJo software (Supporting information, Fig. S4).
Statistical analysis
Data are given as mean ± standard error of the mean (s.e.m.). Statistical differences were calculated using Student's t‐test for single comparisons or one‐way analysis of variance (anova) with Tukey's post‐hoc test for multiple comparisons. Weight and clinical score were analysed using a repeated‐measures two‐way anova. Survival was compared using the log‐rank (Mantel–Cox) test. Correlations between cytokines were assessed using Spearman's correlation. All statistical analyses and graphs were generated using GraphPad Prism version 5 for PC (GraphPad Software, La Jolla, CA, USA). P < 0·05 was considered significant for all tests.
Results
NSG mice injected with hPBMCs engraft predominantly human CD3+ T cells and exhibit clinical signs of GVHD
NSG mice injected with hPBMCs develop GVHD from 4 weeks 20. In the current study, NSG mice were injected i.p. with 10 × 106 freshly isolated hPBMCs (hPBMC group) or saline (control group), and observed for signs of GVHD development for up to 8 weeks. At 3 weeks post‐hPBMC injection, mice were checked for engraftment of human cells. hPBMC‐injected mice demonstrated engraftment of human leucocytes (14·4 ± 2·7% hCD45+mCD45– cells, n = 9) while, as expected, no human leucocytes were observed in three randomly selected control mice (Fig. 1a). In hPBMC‐injected mice the majority of hCD45+ cells were T cells (97·5 ± 0·3% hCD3+hCD19– cells, n = 9) (Fig. 1b). NSG mice injected with hPBMCs demonstrated no human B cell (hCD3–hCD19+) engraftment (data not shown), and the remaining human leucocytes were negative for both hCD3 and hCD19 (2·5 ± 0·3%, n = 9) (Fig. 1c).
Figure 1.
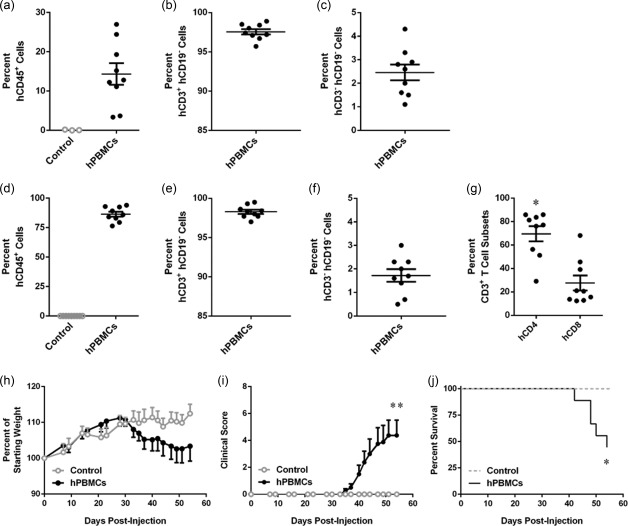
Engraftment of human leucocytes and development of GVHD in non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice. (a‐g) NSG mice were injected intraperitoneally (i.p.) with either 10 × 106 human (h) peripheral blood mononuclear cells (PBMCs) (n = 9) or an equal volume of saline (control) (n = 9), and monitored for clinical signs of graft‐versus‐host disease (GVHD) over 8 weeks. (a–g) The percentage of human leucocytes in (a–c) blood at 3 weeks post‐hPBMC injection and (d–f) spleens at end‐point were determined by flow cytometry. (a,d) hCD45+ leucocytes are expressed as a percentage of total mCD45+ and hCD45+ leucocytes. (b,e) hCD3+ hCD19– cells and (c,f) hCD3– hCD19– cells are expressed as a percentage of total hCD45+ leucocytes. (g) hCD4+ and hCD8+ T cell subsets are expressed as a percentage of total hCD3+ leucocytes. Data represent group mean ± standard error of the mean (s.e.m.); symbols represent individual mice; *P < 0·05 compared to hCD8+ T cells. (h–j) NSG mice were monitored for (h) weight loss, (i) clinical score and (j) survival. Data represent (h,i) group means ± s.e.m. or (j) percentage survival (control, n = 9, hPBMC n = 9); *P < 0·05 and **P < 0·005 compared to control mice.
Analysis of spleens from mice euthanized at end‐point confirmed the absence of human leucocytes in control mice (n = 9) (Fig. 1d). hPBMC‐injected mice engrafted large amounts of human leucocytes (86·4 ± 2·0% hCD45+mCD45– cells, n = 9) (Fig. 1d). The majority of human leucocytes in the spleen were T cells (98·3 ± 0·2% hCD3+hCD19– cells, n = 9) (Fig. 1e). No human B cells (hCD3‐ hCD19+) were found in the spleen (data not shown), and the remaining cells were negative for both hCD3 and hCD19 (1·7 ± 0·3%, n = 9) (Fig. 1f). Analysis of human T cells (hCD3+) revealed that hPBMC‐injected mice demonstrated a significantly higher percentage of hCD4+ T cells (69·6 ± 6·6%, n = 9) compared to hCD8+ T cells (27·6 ± 6·4%, n = 9) (P = 0·0121) (Fig. 1g).
As stated above, mice were observed for signs of GVHD for up to 8 weeks. Weight changes were similar between hPBMC‐injected and control mice from weeks 1 to 4, with both groups demonstrating weight gain. However, hPBMC‐injected mice steadily began to lose weight from day 30, while saline‐injected control mice maintained a weight of more than 110% of their starting weight throughout the 8‐week observation period (P = 0·4154) (Fig. 1h). Saline‐injected control mice did not show any signs of GVHD, and maintained a clinical score of 0 during the observation period (n = 9). In contrast, hPBMC‐injected mice displayed clinical signs of GVHD from day 30, first evident as fur ruffling, followed by hunching, reduced activity, weight loss and/or areas of scaly skin patches. hPBMC‐injected mice had a mean clinical score of 4·3 ± 1·1 at end‐point. The clinical scores between the two groups were significantly different throughout 8 weeks (n = 9) (P = 0·0038) (Fig. 1i). hPBMC‐injected mice succumbed to disease from day 40, with a median survival time (MST) of 54 days, and exhibited a 55% mortality rate by end‐point, compared to 100% survival of control mice (P = 0·0101) (Fig. 1j).
NSG mice injected with hPBMCs show histological evidence of GVHD and increased murine P2X7 expression
Histological analysis of the target organs, liver, small intestine and skin, revealed that mice injected with hPBMCs displayed greater leucocyte infiltration and tissue damage compared to tissues from control mice (Fig. 2a). Compared to control mice, hPBMC‐injected mice demonstrated mildly increased leucocyte infiltration, loss of structural integrity and apoptosis of cells in the liver (Fig. 2a), mild damage in the small intestine with increased apoptotic cells and rounded villi (Fig. 2a), and lastly mild leucocyte infiltration and epidermal thickening in the skin compared to control mice (Fig. 2a).
Figure 2.
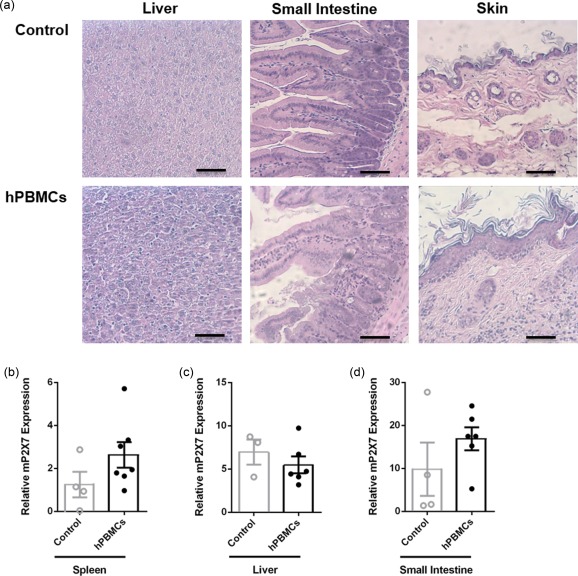
Non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice injected with human leucocytes develop graft‐versus‐host disease (GVHD). (a–d) NSG mice injected intraperitoneally (i.p.) with either human (h) peripheral blood mononuclear cells (hPBMCs) or saline (control) (day 0) (from Fig. 1) were monitored for clinical signs of graft‐versus‐host disease (GVHD) over 8 weeks. (a) Tissue sections (liver, small intestine and skin) from control (top panel) or hPBMC‐injected mice (bottom panel) at end‐point were stained with haematoxylin and eosin and images captured by microscopy. Each image is representative of two mice per group; bar represents 100 μm. (b–d) The relative expression of murine (m) P2X7 in (b) spleen, (c) liver and (d) small intestine from mice at end‐point was examined by quantitative polymerase chain reaction (qPCR). Data represent group mean ± standard error of the mean (s.e.m.) (n = 3–7); symbols represent individual mice.
P2X7 expression is up‐regulated in host tissues in allogeneic mouse models of GVHD 12, 14. Therefore, mP2X7 expression in the spleen, liver and small intestine was analysed by qPCR. mP2X7 expression was increased twofold in the spleens from hPBMC‐injected mice (2·6 ± 0·6, n = 7) compared to spleens from control mice (1·2 ± 0·6, n = 4), but this difference did not reach statistical significance (P = 0·1656) (Fig. 2b). Hepatic mP2X7 expression was similar in hPBMC‐injected (5·5 ± 0·6, n = 6) and control mice (6·9 ± 1·4, n = 3) (P = 0·4160) (Fig. 2c). mP2X7 expression was approximately twofold greater in the small intestines from hPBMC‐injected mice (16·9 ± 2·7, n = 6) compared to control mice (9·9 ± 6·2, n = 4), but this did not reach statistical significance (P = 0·2668) (Fig. 2d).
NSG mice express full‐length functional P2X7 receptors
The data above (Fig. 2b–d) demonstrate that NSG mice express P2X7, but it remained unknown if NSG mice express functional P2X7. Immunoblotting using an antibody against the extracellular domain of P2X7 demonstrated a major band at 81 kDa, corresponding to full‐length P2X7 21 in the positive control RAW264.7 macrophages and in NSG splenocytes (n = 2) (Fig. 3a).
Figure 3.
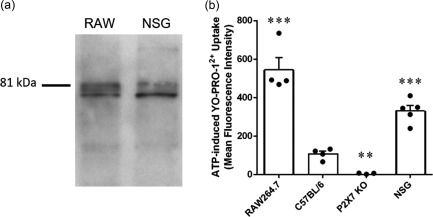
Non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice express full‐length functional P2X7. (a) Lysates of RAW264.7 (RAW) macrophages and splenocytes from NSG mice were examined by immunoblotting using an anti‐P2X7 antibody. Image is representative of two independent experiments. (b) RAW264.7 cells or splenocytes from C57BL/6, P2X7 knock‐out (KO) or NSG mice in NaCl medium containing 1 μM YO‐PRO‐12+ were incubated in the absence or presence of 1 mM adenosine triphosphate (ATP) for 10 min at 37°C. The mean fluorescence intensity of YO‐PRO‐12+ uptake into RAW264.7 cells or CD11c+ splenic dendritic cells (DCs) was then assessed by flow cytometry. ATP‐induced YO‐PRO‐12+ uptake was determined as the difference between YO‐PRO‐12+ uptake in the presence and absence of ATP. Data represent group mean ± standard error of the mean (s.e.m.) (n = 3–5); symbols represent individual replicates (RAW264.7 cells) or mice (splenic CD11c+ DCs); ** P < 0·005 and *** P < 0·0001 compared to C57BL/6.
To determine if NSG mice express functional P2X7, RAW264.7 cells or splenic leucocytes from mice were incubated in the absence or presence of ATP and YO‐PRO‐12+ uptake assessed by flow cytometry. ATP induced YO‐PRO‐12+ uptake into positive control RAW264.7 cells (n = 4) and CD11c+ splenic DCs from C57BL/6 mice (n = 4), but not negative control CD11c+ splenic DCs from P2X7 KO mice (n = 3) (Fig. 3b). Notably, ATP also induced YO‐PRO‐12+ uptake into CD11c+ splenic DCs from NSG mice (n = 5) (Fig. 3b). This uptake was 50% lower than that of RAW264.7 macrophages (P < 0·005), but threefold greater than that of C57BL/6 CD11c+ splenic DCs (P < 0·005) (Fig. 3b).
BBG prevents ATP‐induced cation uptake into human and murine leucocytes
Since its initial use in rodent models of multiple sclerosis 22, spinal cord injury 23 and Huntington's disease 24, BBG has been used in more than 40 studies to establish the role of P2X7 in various disorders 25, including our group in a murine model of amyotrophic lateral sclerosis 26. To confirm that BBG can block hP2X7 and mP2X7, human RPMI8226 and murine RAW264.7 cells, both of which express endogenous P2X7 27, 28, were preincubated with increasing concentrations of BBG and then ATP‐induced YO‐PRO‐12+ uptake was assessed. BBG exhibited a concentration‐dependent inhibition of ATP‐induced YO‐PRO‐12+ uptake, with maximal inhibition at 1 μM in both cell types, and half maximal inhibitory concentrations (IC50) of 99·0 ± 1·2 nM and 59·7 ± 1·2 nM in RPMI8226 cells and RAW264.7 cells, respectively (Fig. 4a). These IC50 values are similar to those known for recombinant hP2X7 and mP2X7 25.
Figure 4.
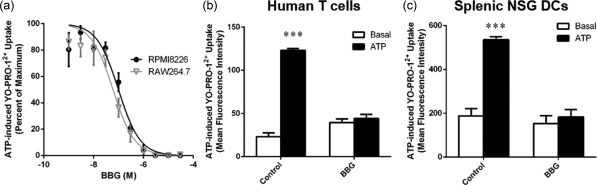
Brilliant Blue G (BBG) prevents adenosine triphosphate (ATP)‐induced cation uptake into human and murine leucocytes. (a) Human RPMI8226 and murine RAW264.7 cells, (b) human peripheral blood mononuclear cells (PBMCs) or (c) non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) splenocytes in NaCl medium were preincubated for 15 min at 37°C in the absence or presence of (a) BBG as indicated or (b,c) 1 μM BBG. Cells were then incubated with 1 μM YO‐PRO‐12+ in the absence or presence of 1 mM adenosine triphosphate (ATP) for 10 min at 37°C. The mean fluorescence intensity of YO‐PRO‐12+ uptake into (a) RPMI8226 or RAW264.7 cells, (b) hCD3+ T cells or (b) NSG splenic CD11c+ dendritic cells (DCs) was then assessed by flow cytometry. (a) ATP‐induced YO‐PRO‐12+ uptake is represented as a percentage of maximal ATP response in the absence of BBG. (a–c) Data represent group mean ± standard error of the mean (s.e.m.) (n = 3); *** P < 0·0001 compared to respective basal.
To confirm that BBG can block P2X7 in primary leucocytes, hPBMCs and NSG splenocytes were preincubated in the absence or presence of BBG, and then ATP‐induced YO‐PRO‐12+ uptake was assessed. ATP induced significant YO‐PRO‐12+ uptake into hCD3+ T cells, which was reduced by 95% by preincubation with BBG (P < 0·0001, n = 3) (Fig. 4b). ATP induced significant YO‐PRO‐12+ uptake into NSG CD11c+ splenic DCs, which was reduced 92% by preincubation with BBG (Fig. 4c) (P < 0·0001, n = 3). Basal uptake of YO‐PRO‐12+ in the absence or presence of BBG was similar for each respective cell subtype (Fig. 4b,c).
BBG does not affect engraftment of human cells or prevent GVHD in NSG mice
To investigate whether P2X7 blockade can prevent disease in a humanized mouse model of GVHD, NSG mice were injected with hPBMCs and the P2X7 antagonist BBG (50 mg/kg) was injected subsequently every 2 days from days 0 to 8. Mice were monitored for weight loss and signs of GVHD for up to 10 weeks. To determine if BBG affected engraftment of human cells, blood was collected at 3 weeks post‐hPBMC injection and cells analysed by flow cytometry. BBG‐injected mice demonstrated a similar level of human leucocytes (10·4 ± 1·3% hCD45+mCD45– cells, n = 14) compared to saline‐injected (vehicle control) mice (10·6 ± 1·4% hCD45+mCD45– cells, n = 16) (P = 0·9179) (Fig. 5a). In both groups of mice the majority of hCD45+ cells were T cells, which did not differ between BBG‐injected mice (97·2 ± 0·4% hCD3+hCD19– cells, n = 14) and saline‐injected mice (97·4 ± 0·4% hCD3+hCD19– cells, n = 16) (P = 0·7014) (Fig. 5b). Neither group of mice demonstrated engraftment of B cells (hCD3–hCD19+) (data not shown). The remaining hCD45+ cells were negative for both hCD3 and hCD19, and the percentage of these cells did not differ between BBG‐injected mice (2·7 ± 0·4%, n = 14) and saline‐injected mice (2·5 ± 0·4%, n = 16) (P = 0·8291) (Fig. 5c).
Figure 5.
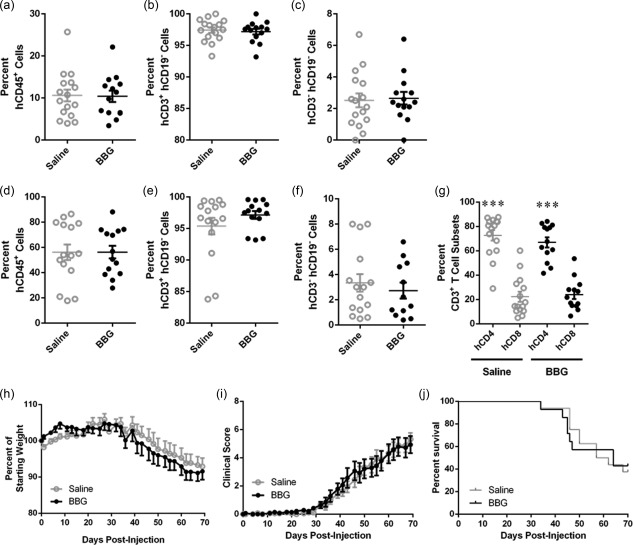
Brilliant Blue G (BBG) does not affect engraftment of human cells. (a–j) Non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice were injected intraperitoneally (i.p.) with 10 × 106 human (h) peripheral blood mononuclear cells (PBMCs), and subsequently with saline (control) (n = 14) or saline containing Brilliant Blue G (BBG) (50 mg/kg) (n = 16) every 2 days (from days 0 to 8), and monitored for clinical signs of graft‐versus‐host disease (GVHD) over 10 weeks. (a–g) The percentage of human leucocytes in (a–c) blood at 3 weeks post‐hPBMC injection and (d–f) spleens at end‐point were determined by flow cytometry. (a,d) hCD45+ leucocytes are expressed as a percentage of total mCD45+ and hCD45+ leucocytes. (b,e) hCD3+hCD19– cells and (c,f) hCD3–hCD19– cells are expressed as a percentage of total hCD45+ leucocytes. (g) hCD4+ and hCD8+ T cell subsets are expressed as a percentage of total hCD3+ leucocytes. Data represent mean ± standard error of the mean (s.e.m.); symbols represent individual mice; ***P < 0·0001 compared to hCD8+ T cells. (h–j) NSG mice were monitored for (a) weight loss, (b) clinical score, and (c) survival. Data represent (h, i) group means ± s.e.m. or (j) percentage survival (saline n = 14, BBG n = 16).
Analysis of spleens from all mice euthanized at end‐point demonstrated that human leucocytes comprised the majority of total murine and human leucocytes. BBG‐injected mice engrafted a similar level of human leucocytes (56·3 ± 5·0% hCD45+mCD45– cells, n = 13) compared to saline‐injected mice (56·2 ± 6·2% hCD45+mCD45– cells, n = 15) (P = 0·9899) (Fig. 5d). The majority of human leucocytes in the spleen were T cells, and both BBG‐injected mice (97·1 ± 0·6% hCD3+hCD19– cells, n = 13) and saline‐injected mice (95.4 ± 0.6% hCD3+hCD19– cells, n = 15) demonstrated similar percentages of T cells (P = 0·2481) (Fig. 5e). No human B cells (hCD3‐ hCD19+) were found in the spleens (data not shown). The remaining hCD45+ cells were negative for both hCD3 and hCD19, and percentages of these cells did not differ between BBG‐injected mice (2·7 ± 0·6%, n = 15) and saline‐injected mice (2·7 ± 0·6%, n = 15) (P = 0·5325) (Fig. 5f). Analysis of the human T cells (hCD3+) revealed that BBG‐ and saline‐injected mice contained similar percentages of hCD4+ T cells (67·0 ± 4·1%, n = 13 versus 72·7 ± 4·3%, n = 15, respectively, P = 0·3515). BBG‐ and saline‐injected mice also contained similar percentages of hCD8+ T cells (24·0 ± 3·6%, n = 13 versus 22·3 ± 4·1%, n = 15, respectively, P = 0·7583). In both BBG‐ and saline‐injected mice, the percentage of hCD4+ T cells was significantly higher than hCD8+ T cells (P < 0·0001) (Fig. 5g).
As stated above, mice were observed for signs of GVHD for up to 10 weeks. Both BBG‐ and saline‐injected humanized mice began to lose weight from day 30 (P = 0·8450) (Fig. 5h). Both BBG‐ and saline‐injected mice began to exhibit signs of mild GVHD at day 35, and both BBG‐ and saline‐injected mice exhibited similar clinical scores at end point (5·3 ± 0·5, n = 16 versus 4·9 ± 0·6, n = 14, respectively). The clinical scores between the two groups were not significantly different throughout 10 weeks (P = 0·8356) (Fig. 5i). Survival was also similar in both BBG‐injected mice (MST of 64 days, and mortality rate of 57%, n = 16) and saline‐injected mice (MST of 60 days and mortality rate of 62%, n = 14) (P = 0·9874 for mortality rate) (Fig. 5j).
BBG reduces histological evidence of GVHD in humanized mice
BBG‐ and saline‐injected humanized mice demonstrated similar damage to the liver with some leucocyte infiltration, apoptosis of cells and fibrosis (Fig. 6a). Both BBG‐ and saline‐injected humanized mice demonstrated structural damage to the small intestine; however, BBG‐injected humanized mice exhibited decreased leucocyte infiltration compared to saline‐injected humanized mice (Fig. 6a). BBG‐injected humanized mice also exhibited mildly decreased leucocyte infiltrates in the dermis, decreased apoptotic cells and reduced basal vacuolar change of the skin compared to that of saline‐injected humanized mice (Fig. 6a).
Figure 6.
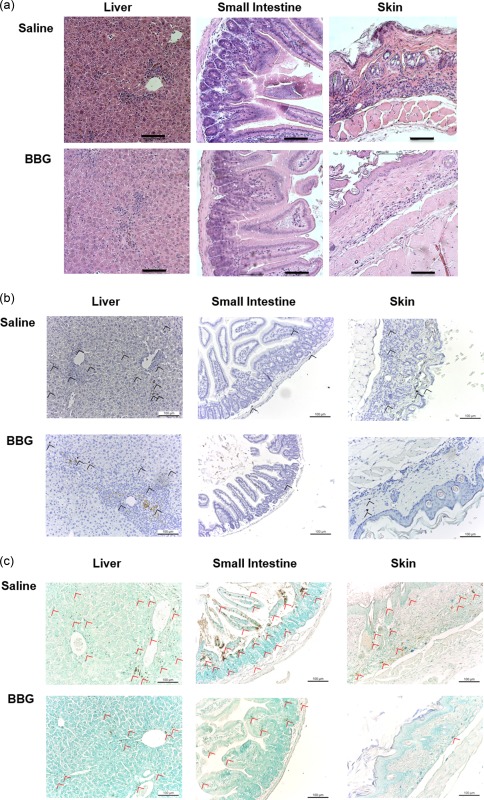
Brilliant Blue G (BBG) does not prevent graft‐versus‐host disease (GVHD) in humanized mice. (a–c) Non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice injected intraperitoneally (i.p.) with 10 × 106 human (h) peripheral blood mononuclear cells (PBMCs) (day 0), and with saline (control) or 50 mg/kg Brilliant Blue G (BBG) (from Fig. 5) were monitored for clinical signs of GVHD over 10 weeks. Tissue sections (liver, small intestine and skin) from hPBMC‐injected mice injected with saline (control) or BBG at end‐point were stained with (a) haematoxylin and eosin, (b) anti‐hCD3 monoclonal antibody with 3,3′‐diaminobenzidine tetrachloride detection system and haematoxylin or (c) terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick‐end labelling with 3,3′‐diaminobenzidine tetrachloride detection system and methyl green. Images were captured by microscopy with each image representative of (a) four or (b,c) two mice per group; bars represent 100 μm; (b) black arrow heads indicate T cells and (c) red arrow heads indicate apoptotic cells.
Immunohistochemistry of the target organs, liver, small intestine and skin, was used to determine if the leucocyte infiltrates observed above included human T cells. T cells (indicated by black arrows) were identified in the liver, and to a lesser extent in the small intestine and skin (Fig. 6b). T cell infiltration in the liver, small intestine and skin appeared to be reduced in BBG‐injected mice compared to saline‐injected mice (Fig. 6b). To confirm tissue damage in the liver, small intestine and skin, these target organs were analysed for evidence of apoptosis (fragmented DNA) by immunohistochemistry. Apoptotic cells (indicated by red arrows) were evident in the liver and small intestine and, to a lesser extent, in the skin (Fig. 6c). Apoptosis in these organs appeared to be reduced in BBG‐injected mice compared to saline‐injected mice (Fig. 6c).
BBG‐injected mice demonstrate similar mP2X7 and hP2X7 expression in GVHD target organs
To determine whether injection of BBG altered expression of murine (host) or human (donor) P2X7 expression in the spleens and target organs of GVHD, tissues from humanized mice were analysed by qPCR. mP2X7 expression was similar in spleens of BBG‐injected mice (4·6 ± 1·0, n = 6) and saline‐injected mice (5·7 ± 0·5, n = 6) (P = 0·5446) (Fig. 7a). mP2X7 expression in the liver was reduced by 58% in BBG‐injected mice (1·6 ± 0·3, n = 4) compared to saline‐injected mice (3·9 ± 1·2, n = 5); however, this difference did not reach statistical significance (P = 0·1619) (Fig. 7b) Conversely, mP2X7 expression was increased almost two‐fold in the small intestines of BBG‐injected mice (2·8 ± 0·7, n = 6) compared to saline‐injected mice (1·6 ± 0·3, n = 5), but this did not reach statistical significance (P = 0·1763) (Fig. 7c). hP2X7 expression was decreased by 74% in the spleens of BBG‐injected mice (3·0 ± 0·9, n = 5) compared to saline‐injected mice (11·4 ± 3·5, n = 6) and this difference approached statistical significance (P = 0·0595) (Fig. 7d). In contrast, hP2X7 expression was similar in the livers and small intestines of BBG‐injected mice (6·3 ± 2·6, n = 5, and 7·6 ± 2·9, n = 5, respectively) compared to saline‐injected mice (3·7 ± 1·1, n = 4, and 5·7 ± 1·8, n = 6, respectively) (P = 0·4296 and 0·5642, respectively) (Fig. 7e,f).
Figure 7.
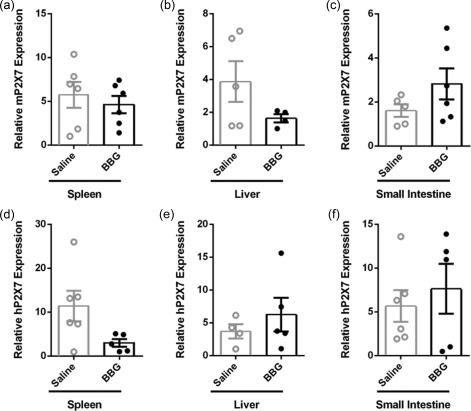
Brilliant Blue G (BBG) does not impact expression of mP2X7 or hP2X7 in tissues in humanized mice. (a–f) Non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice injected intraperitoneally (i.p.) with 10 × 106 human peripheral blood mononuclear cells (day 0), and with saline (control) or 50 mg/kg Brilliant Blue G (BBG) (from Fig. 5) were monitored for clinical signs of graft‐versus‐host disease (GVHD) over 10 weeks. The relative expression of (a–c) murine (m) P2X7 and (d‐f) hP2X7 in (a,d) spleen, (b,e) liver and (c,f) small intestine from mice at end‐point were examined by quantitative polymerase chain reaction (qPCR). Data represent group mean ± standard error of the mean (s.e.m.) (n = 4–6); symbols represent individual mice.
BBG reduces serum human IFN‐γ in humanized mice
IFN‐γ and IL‐17 are important proinflammatory cytokines implicated in the pathogenesis of GVHD 4, 5. To determine if BBG treatment altered the amount of these cytokines, serum hIFN‐γ and hIL‐17 concentrations in humanized mice were assessed by ELISA. hIFN‐γ was present in the serum of all mice for which samples were available (Fig. 8a). Notably, treatment with BBG reduced significantly the amount of serum hIFN‐γ in BBG‐injected mice (10·0 ± 2·4 ng/ml, n = 12), which was 54% lower than that of saline‐injected mice (21·8 ± 2·4 ng/ml, n = 13) (P = 0·0023) (Fig. 8a). In contrast, hIL‐17 was detected only in the serum from four BBG‐injected mice (33%) and two saline‐injected mice (15%) for which samples were available (Fig. 8b). The amount of hIL‐17 in the serum of BBG‐injected mice (40·8 ± 21·1 pg/ml, n = 4) was almost 10‐fold greater than that of saline‐injected mice (4·4 ± 4·0 pg/ml, n = 2); however, this difference did not reach statistical significance (P = 0·1031) (Fig. 8b).
Figure 8.
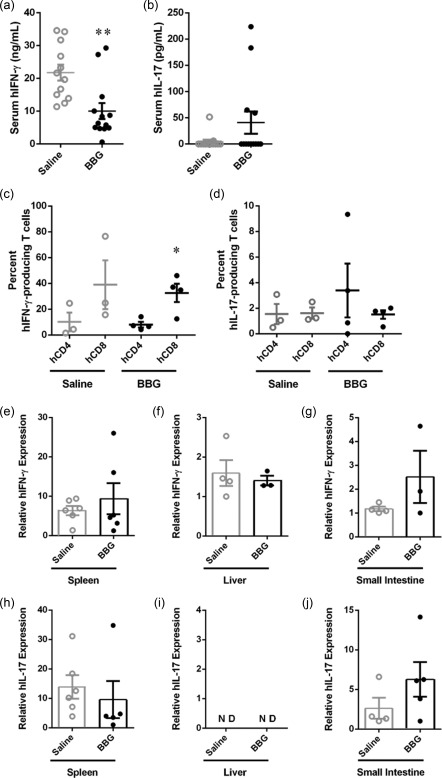
Brilliant Blue G (BBG) reduces serum human interferon (IFN)‐γ significantly in humanized mice. (a–j) Non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice were injected intraperitoneally (i.p.) with 10 × 106 human (h) peripheral blood mononuclear cells (PBMCs) (day 0) and with saline (control) or 50 mg/kg Brilliant Blue G (BBG) (from Fig. 5). Concentrations of serum (a) hIFN‐γ, and (b) hIL‐17 were analysed by enzyme‐linked immunosorbent assay (ELISA). Data represents group mean ± standard error of the mean (s.e.m.) (saline n = 12, BBG n = 13); **P < 0·005 compared to saline‐injected mice. (c,d) Splenocytes were incubated in complete RPMI‐1640 medium containing phorbol 12‐myristate 13‐acetate and ionomycin for 4 h, and the percentage of hCD4+ and hCD8+ T cells producing intracellular (c) hIFN‐γ or (d) hIL‐17 was determined by flow cytometry. Data represents group mean ± s.e.m. (saline n = 3, BBG = 4); symbols represent individual mice; *P < 0·05 compared to corresponding hCD4+ T cells. (e–j) The relative expression of (e–g) hIFN‐γ and (h–j) hIL‐17 in (e,h) spleen, (f,i) liver and (g,j) small intestine from mice at end‐point were examined by quantitative polymerase chain reaction (qPCR). Data represents group means ± s.e.m. (n = 2–6); symbols represent individual mice; n.d. = not detected.
To determine the potential source of hIFN‐γ and hIL‐17, splenocytes were isolated from humanized mice and the intracellular expression of hIFN‐γ and hIL‐17 was analysed by flow cytometry. Both hCD4+ and hCD8+ T cells produced hIFN‐γ and, to a lesser extent, hIL‐17 (Fig. 8c,d). The percentage of hIFN‐γ‐producing hCD8+ T cells was fourfold greater than hIFN‐γ‐producing hCD4+ T cells from BBG‐injected mice (32·6 ± 7·1% versus 8·1 ± 2·1%, respectively, n = 4) and saline‐injected mice (39·0 ± 18·9% versus 10·2 ± 7·3%, respectively, n = 3), but this only reached statistical significance in the BBG‐injected group (P = 0·0165) (Fig. 8c). The percentage of hIL‐17‐producing hCD4+ and hCD8+ T cells in BBG‐injected mice (3 ± 4% versus 2 ± 1%, n = 4, respectively) was similar to saline‐injected mice (2 ± 1% versus 2 ± 1%, n = 3, respectively) (P = 0·5046 and 0·8437, respectively) (Fig. 8d).
To determine if expression of IFN‐γ and IL‐17 were altered in the spleens and target organs of GVHD, tissues from humanized mice were analysed by qPCR. hIFN‐γ expression was 1·5‐fold greater in the spleens of BBG‐injected mice (9·4 ± 3·9, n = 6) compared to saline‐injected mice (6·4 ± 1·2, n = 6), but this difference did not reach statistical significance (P = 0·4825) (Fig. 8e). hIFN‐γ expression was similar in livers of BBG‐injected mice (1·4 ± 0·1, n = 6) and saline‐injected mice (1·6 ± 0·3, n = 6) (P = 0·6549) (Fig. 8f). hIFN‐γ expression was twofold greater in the small intestines of BBG‐injected mice (2·5 ± 1·1, n = 3) compared to saline‐injected mice (1·2 ± 0·1, n = 4) but again this difference did not reach statistical significance (P = 0·2050) (Fig. 8g). hIL‐17 expression in spleens of BBG‐injected mice (9·6 ± 6·3, n = 5) was similar to saline‐injected mice (13·9 ± 4·0, n = 6) (P = 0·5441) (Fig. 8h). hIL‐17 was not detected in the livers of either BBG‐injected mice (n = 2) or saline‐injected mice (n = 4) (Fig. 8i). hIL‐17 expression was twofold greater in the small intestines of BBG‐injected mice (6·3 ± 2·2, n = 5) compared to saline‐injected mice (2·6 ± 1·3, n = 4), but this difference did not reach statistical significance (P = 0·2263) (Fig. 8j).
BBG does not alter murine or human IL‐1β expression in humanized mice
Activation of P2X7 causes IL‐1β maturation and release from antigen‐presenting cells 29, a proinflammatory cytokine implicated in the pathogenesis of GVHD 30. Moreover, P2X7‐mediated IL‐1β release is involved in the production of IFN‐γ 31 and IL‐17 32. Given that the identity of the antigen‐presenting cells involved in human T cell activation in our humanized model of GVHD remains unknown, and as mIL‐1β can stimulate human cells 33, 34, the expression of both mIL‐1β and hIL‐1β in the spleen, liver and small intestine from these mice was analysed by qPCR. mIL‐1β expression was threefold greater in the spleens of BBG‐injected mice (6·3 ± 2·8, n = 3) compared to saline‐injected mice (2·0 ± 0·9, n = 5), but this difference did not reach statistical significance (P = 0·1183) (Fig. 9a). mIL‐1β expression was similar in the livers and small intestines of BBG‐injected (2·7 ± 1·2, n = 5 and 2·5 ± 0·5, n = 6, respectively) and saline‐injected mice (2·7 ± 0·7, n = 4 and 1·9 ± 0·6, n = 6, respectively) (P = 0·9552 and 0·4829, respectively) (Fig. 9b,c). In contrast to mIL‐1β, hIL‐1β expression was approximately 50% lower in the spleens of BBG‐injected mice (2·2 ± 1·1, n = 5) compared to saline‐injected mice (5·0 ± 2·1, n = 5), but this difference did not reach statistical significance (P = 0·2986) (Fig. 9d). hIL‐1β expression was not detected in livers or small intestines from either BBG‐ or saline‐injected mice (both n = 5) (data not shown). Therefore, given the broad expression of mIL‐1β within the humanized mice, mIL‐1β in the serum of these mice was assessed by ELISA. However contrary to mIL‐1β expression in tissues, mIL‐1β was not detected in the serum from BBG‐injected (0·0 ± 0·0, n = 12) or saline‐injected mice (0·0 ± 0·0, n = 11) using an ELISA with a reported sensitivity of 8 pg/ml (eBioscience).
Figure 9.
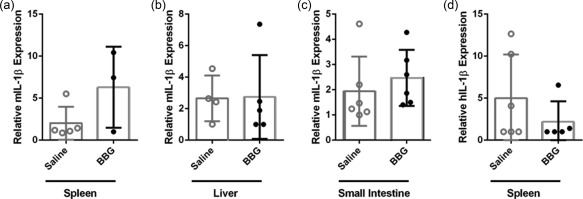
Brilliant Blue G (BBG) does not alter relative expression of human or murine interleukin (IL)‐1β in humanized mice. (a–d) Non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull (NSG) mice were injected intraperitoneally (i.p.) with 10 × 106 human (h) peripheral blood mononuclear cells (PBMCs) (day 0) and with saline (control) or 50 mg/kg Brilliant Blue G (BBG) (from Fig. 5). The relative expression of (a–c) murine (m) IL‐1β and (d) hIL‐1β in (a,d) spleen, (b) liver and (c) small intestine from mice at end‐point were examined by qPCR. Data represents group mean ± standard error of the mean (s.e.m.) (n = 3–6); symbols represent individual mice.
Discussion
This study aimed to investigate the effect of the P2X7 antagonist, BBG, in a preclinical humanized mouse model of GVHD. Injection of BBG into humanized mice did not alter human leucocyte engraftment or disease progression, as assessed by weight loss, clinical score and survival. However, treatment with BBG reduced significantly serum hIFN‐γ. Intracellular hIFN‐γ was detected in both splenic hCD4+ and hCD8+ T cells of humanized mice, with a larger proportion (fourfold) of hIFN‐γ+hCD8+ T cells than hIFN‐γ+hCD4+ T cells. However, splenic hCD4+ T cells were threefold more frequent than hCD8+ T cells in humanized mice, and therefore the relative contribution of each subset to circulating IFN‐γ production needs to be elucidated further. Pharmacological blockade or genetic deficiency of P2X7 has also been shown to reduce serum IFN‐γ in allogeneic mouse models of GVHD 12, 13. Additionally, P2X7 activation of the NLRP3 inflammasome and subsequent IL‐1β production promotes the generation of IFN‐γ+ CD8+ T cells in mice 31. Combined, these findings support a potential role for P2X7 in GVHD pathology in humans, possibly through the regulation of T cell activation and subsequent IFN‐γ production. To this end, IFN‐γ plays important roles in the up‐regulation of CXCR3 and subsequent trafficking of T cells to target tissues in allogeneic mouse models of GVHD 35. Conversely, others have suggested that IFN‐γ+CD8+ T cells and IFN‐γ+CD4+ T cells are important in the initiation and progression of disease, respectively, in an allogeneic mouse model of GVHD 36. Nevertheless, it remains to be determined if P2X7 functions through these mechanisms in allogeneic or humanized mouse models of GVHD.
The current study also identified the presence of serum hIL‐17 in a small proportion of humanized mice (15–30%), and intracellular hIL‐17 in an even smaller population of splenic hCD4+ and hCD8+ T cells (2–3%) from these mice. Due to the relatively small populations, it is not possible to determine positively the role of P2X7 on hIL‐17 production in this humanized mouse model of GVHD. However, the mean serum hIL‐17 concentration was 10‐fold greater in BBG‐injected mice compared to saline‐injected mice. These results may suggest that P2X7 blockade promotes hIL‐17 production in humanized mice, which contradicts previous invitro results in which P2X7 activation promoted hIL‐17 production in T cells 31. Others have shown that a shift from an IFN‐γ‐producing to an IL‐17‐producing T cell phenotype exacerbates GVHD in an allogeneic mouse model of GVHD 37. Similarly, increased numbers of IL‐17‐producing T cells were observed in patients with more severe GVHD 38. Investigating IL‐17‐producing T cells at time‐points prior to end‐point or through increased sample sizes may assist in addressing the role of P2X7 blockade in IL‐17 production in humanized mice with GVHD.
Due to the variation observed in hIL‐17 mRNA expression between humanized mice and the role of IL‐1β in the differentiation of IL‐17‐producing T cells 39, including models of P2X7 activation 32, mIL‐1β and hIL‐1β was also examined in the current study. mIL‐1β, but not hIL‐1β, mRNA expression was detected in the livers and small intestines of humanized mice, with no differences observed between BBG and saline treatments. In contrast, both mIL‐1β and hIL‐1β mRNA expression was detected in the spleens of humanized mice, with BBG treatment resulting in a threefold increase in mIL‐1β mRNA expression and a 50% decrease in hIL‐1β mRNA expression. However, there was no correlation between hIL‐17 and hIL‐1β or between hIL‐17 and mIL‐1β mRNA expression (results not shown). Due to the inability to detect serum mIL‐1β in these mice, serum mIL‐1β could not be compared with serum hIL‐17 (hIFN‐γ). The inability to detect serum mIL‐1β was not limited to this cytokine, as serum hIL‐4 was not detected in initial experiments to establish the humanized mouse model, despite detection of serum hIFN‐γ in these mice (results not shown).
As mentioned previously, injection of BBG into humanized mice did not alter disease progression, as assessed by weight loss, clinical score and survival. However, histological examination revealed that BBG reduced the infiltration of leucocytes into the small intestine and skin in this humanized mouse model of GVHD. Moreover, there was also histological evidence of reduced tissue damage in these tissues from BBG‐injected mice compared to saline‐injected mice, observations supported by immunohistochemical analysis of T cell infiltrates and apoptosis. In contrast, BBG did not alter the proportion of hCD4+ or hCD8+ T cells in blood and spleens of humanized mice. Others have reported that humanized NSG mice exhibit leucocyte infiltration and tissue damage in the liver, small intestine and skin, characteristic of GVHD 40. T cells are the main mediators of disease in this humanized mouse model of GVHD 41, 42. Consistent with these reports, immunohistochemical analysis with a human CD3 specific mAb detected T cells in the liver, small intestine and skin in humanized mice with GVHD. In support of this observation, hP2X7 mRNA was detected in the livers and small intestines of humanized mice using qPCR probes to the P2X7B splice variant, which is expressed predominantly in human T cells 43, with P2X7 present on both hCD4+ and hCD8+ T cells 44.
Collectively, the above data investigating the effect of the P2X7 antagonist, BBG, in a humanized mouse model of GVHD both confirm findings from previous studies using P2X7 antagonists in allogeneic mouse models of GVHD, and provide additional novel insights. Similar to allogeneic mouse models of GVHD 12, 13, 14, P2X7 antagonism reduced serum IFN‐γ, immune infiltrates and tissue damage in humanized mice with GVHD. By contrast, no therapeutic benefit (weight loss, clinical score or survival) was observed in humanized mice with GVHD, but therapeutic benefits were observed in allogeneic mouse models of GVHD 12, 13, 14. Besides obvious laboratory, species and mouse strain differences, a number of other notable differences remain between these models of GVHD. First, survival time in these allogeneic mouse models 12, 13 is at least half that compared to humanized mice. Thus, extended or delayed injection regimens may be required to show therapeutic benefits in humanized mice. Secondly, in the allogeneic mouse models, P2X7 antagonists were injected daily for the first 10 days 12, 13 or twice weekly for 28 days 14, in contrast to five injections during the first 8 days in our humanized mouse model. Thus, more frequent or extended injection regimens may be required to show therapeutic benefits with BBG in humanized mice with GVHD. Thirdly, the two studies that reported improved survival in the allogeneic mouse models 12, 13 used the P2X7 antagonists, pyridoxal‐phopshate‐6‐azophenyl‐2',4‐disulphonic acid, KN‐62 or stavudine, rather than BBG. Thus, testing of P2X7 antagonists other than BBG in humanized mice with GVHD may be warranted, although it should be noted that BBG at either 50 or 70 mg/kg (i.p. twice weekly for 28 days) prevented weight loss in one allogeneic mouse model of GVHD 14, indicating that BBG can display some therapeutic benefit in this disease. Finally, in each of the allogeneic mouse models, recipient mice were first irradiated 12, 13, 14, which causes ATP release 12. In contrast, the humanized mice in our study were not irradiated, potentially eliminating this initial release of ATP, which may subsequently activate P2X7 to promote disease. Thus, the therapeutic potential of P2X7 antagonists in irradiated humanized mice with GVHD could be investigated.
Finally, this study showed that NSG mice demonstrate full‐length functional P2X7. First, qPCR revealed expression of P2RX7 mRNA in the spleens, livers and small intestines of NSG mice. Secondly, immunoblotting revealed a major band at 81 kDa, correlating with glycosylated, full‐length P2X7 45. Finally, ATP induced YO‐PRO‐12+ uptake into splenic DCs from NSG mice, a process impaired by BBG, and which was absent in splenic DCs from P2X7 KO mice. ATP‐induced YO‐PRO‐12+ uptake into splenic DCs from NSG mice was significantly higher than splenic DCs from C57BL/6 mice. This is due most probably to the absence and presence of a loss‐of‐function mutation (P451L) 46 in NSG and C57BL/6 mice, respectively. Although it is yet to be demonstrated formally that NSG mice are wild‐type at this allele, NSG mice are derived from NOD mice 47, a strain known to encode a proline residue at amino acid position 451 48.
In conclusion, the current study demonstrates that P2X7 blockade with BBG can reduce serum hIFN‐γ and inflammation and tissue damage significantly in a humanized mouse model of GVHD. The similar clinical course with skewing to Th17 suggests that clinically P2X7 blockade may be of additive benefit if combined with strategies that limit Th17, such as IL‐6 receptor blockade 49. This study suggests a potential benefit for P2X7 blockade, but highlights the need to also address alternative pathways of immune activation.
Disclosure
All authors declare that they have no disclosures.
Author contributions
N. J. G., L. B., D. L., S. R. A., M.S.S., S. I. A., R. S. and D. W. designed the experiments. N. J. G., L. B., D. L., S. R. A., V. S., R. S. and D. W. performed the experiments. N. J. G., L. B., D. L., S. R. A., R. S. and D. W. analysed the data. S. F. and W. V. provided histological analysis. N. J. G. prepared the figures and wrote the manuscript. R. S. and D. W. edited the manuscript. L. B., D. L., S. R. A, S. F., W. V., M. S. S. and S. I. A. co‐edited the manuscript.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Flow cytometric gating of leucocytes in blood from humanized mice. Blood cells from humanized mice were labelled with isotype control or CD‐specific monoclonal antibodies (mAb). Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify human leucocytes (hCD45+mCD45–), human T cells (hCD45+mCD45–hCD3+hCD19–) and human B cells (hCD45+mCD45–hCD3–hCD19+).
Fig. S2. Flow cytometric gating of leucocytes in spleens from humanized mice. Splenocytes from humanized mice were labelled with isotype control or CD‐specific monoclonal antibodies (mAb). Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify human leucocytes (hCD45+mCD45–), human T cells (hCD45+mCD45–hCD3+hCD19–) and human B cells (hCD45+mCD45–hCD3–hCD19+). Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify human CD4+ T cell (hCD3+hCD4+hCD8–) and human CD8+ T cell (hCD3+hCD4–hCD8+) subsets.
Fig. S3. Flow cytometric analysis of YO‐PRO‐12+ uptake into human hCD3+ T cells and murine CD11c+ splenic dendritic cells. Human peripheral blood mononuclear cells and splenocytes from non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull mice were incubated with YO‐PRO‐12+ in the absence (basal) (grey histograms) or presence of adenosine triphosphate (ATP) (black histograms) and labelled subsequently with anti‐human (h) CD3 or anti‐murine (m) CD11c monoclonal antibody. Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify hCD3+ T cells or mCD11c+ splenic DCs. YO‐PRO‐12+ uptake into these cells was then quantified using geometric mean fluorescence of histograms.
Fig. S4. Flow cytometric analysis of intracellular human interferon (IFN)‐γ and interleukin (IL)‐17 in human CD4+ and CD8+ T cells. Splenocytes from humanized mice were incubated with phorbol 12‐myristate 13‐acetate and ionomycin, and labelled with isotype control or specific monoclonal antibodies. Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify human CD4+ T cell (hCD3+hCD4+hCD8–), and human CD8+ T cell (hCD3+hCD4–hCD8+) subsets. The percentage of hIFN‐γ and hIL‐17 was quantified as the difference between cytokine specific (black histograms) and isotype control (grey histograms) monoclonal antibody (mAb) labelling (indicated by marker regions as shown).
Acknowledgements
This project was funded by the Illawarra Health and Medical Research Institute, the Centre for Medical and Molecular Biosciences (University of Wollongong) and AMP Foundation Australia. We would like to thank the technical staff of the Illawarra Health and Medical Research Institute, and the animal house staff at the Westmead Animal Facility for assistance. We would like to thank Professor David Steel (University of Wollongong) for statistical consulting.
Contributor Information
R. Sluyter, Email: rsluyter@uow.edu.au
D. Watson, Email: dwatson@uow.edu.au
References
- 1. Markey KA, MacDonald KPA, Hill GR. The biology of graft‐versus‐host disease: experimental systems instructing clinical practice. Blood 2014; 124:354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jagasia M, Arora M, Flowers MED et al Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood 2012; 119:296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ferrara JL, Levine JE, Reddy P, Holler E. Graft‐versus‐host disease. Lancet 2009; 373:1550–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yi T, Chen Y, Wang L et al Reciprocal differentiation and tissue‐specific pathogenesis of Th1, Th2, and Th17 cells in graft‐versus‐host disease. Blood 2009; 114:3101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gartlan KH, Markey KA, Varelias A et al Tc17 cells are a proinflammatory, plastic lineage of pathogenic CD8+ T cells that induce GVHD without antileukemic effects. Blood 2015; 126:1609–20. [DOI] [PubMed] [Google Scholar]
- 6. Holtan SG, Marcelo P, Weisdorf DJ. Acute graft‐versus‐host disease: a bench‐to‐bedside update. Blood 2014; 124:363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol 2016; 16:177–92. [DOI] [PubMed] [Google Scholar]
- 8. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 2011; 11:201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Marchi E, Orioli E, Dal Ben D, Adinolfi E. P2X7 receptor as a therapeutic target. Adv Protein Chem Struct Biol 2016; 104:39–79. [DOI] [PubMed] [Google Scholar]
- 10. Geraghty NJ, Watson D, Adhikary SR, Sluyter R. P2X7 receptor in skin biology and diseases. World J Dermatol 2016; 5:72–83. [Google Scholar]
- 11. Wiley J, Sluyter R, Gu B, Stokes L, Fuller S. The human P2X7 receptor and its role in innate immunity. Tissue Antigens 2011; 78:321–32. [DOI] [PubMed] [Google Scholar]
- 12. Wilhelm K, Ganesan J, Müller T et al Graft‐versus‐host disease is enhanced by extracellular ATP activating P2X7R. Nat Med 2010; 16:1434–8. [DOI] [PubMed] [Google Scholar]
- 13. Fowler BJ, Gelfand BD, Kim Y et al Nucleoside reverse transcriptase inhibitors possess intrinsic anti‐inflammatory activity. Science 2014; 346:1000–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhong X, Zhu F, Qiao J et al The impact of P2X7 receptor antagonist, brilliant blue G on graft‐versus‐host disease in mice after allogeneic hematopoietic stem cell transplantation. Cell Immunol 2016; 310:71–7. [DOI] [PubMed] [Google Scholar]
- 15. Lee K‐H, Park SS, Kim I et al P2X7 receptor polymorphism and clinical outcomes in HLA‐matched sibling allogeneic hematopoietic stem cell transplantation. Haematologica 2007; 92:651–7. [DOI] [PubMed] [Google Scholar]
- 16. Solle M, Labasi J, Perregaux DG et al Altered cytokine production in mice lacking P2X7 receptors. J Biol Chem 2001; 276:125–32. [DOI] [PubMed] [Google Scholar]
- 17. Tran JN, Pupovac A, Taylor RM, Wiley JS, Byrne SN, Sluyter R. Murine epidermal Langerhans cells and keratinocytes express functional P2X7 receptors. Exp Dermatol 2010; 19:e151–7. [DOI] [PubMed] [Google Scholar]
- 18. Bartlett R, Yerbury JJ, Sluyter R. P2X7 receptor activation induces reactive oxygen species formation and cell death in murine EOC13 microglia. Mediators Inflamm 2013; 2013:271813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spildrejorde M, Bartlett R, Stokes L et al R270C polymorphism leads to loss of function of the canine P2X7 receptor. Physiol Genomics 2014; 46:512–22. [DOI] [PubMed] [Google Scholar]
- 20. King M, Pearson T, Shultz LD et al A new Hu‐PBL model for the study of human islet alloreactivity based on NOD‐scid mice bearing a targeted mutation in the IL‐2 receptor gamma chain gene. Clin Immunol 2008; 126:303–14. [DOI] [PubMed] [Google Scholar]
- 21. Masin M, Young C, Lim K et al Expression, assembly and function of novel C‐terminal truncated variants of the mouse P2X7 receptor: re‐evaluation of P2X7 knockouts. Br J Pharmacol 2012; 165:978–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Matute C, Torre I, Pérez‐Cerdá F et al P2X7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci 2007; 27:9525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peng W, Cotrina ML, Han X et al Systemic administration of an antagonist of the ATP‐sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci USA 2009; 106:12489–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Díaz‐Hernández M, Díez‐Zaera M, Sánchez‐Nogueiro J et al Altered P2X7‐receptor level and function in mouse models of Huntington's disease and therapeutic efficacy of antagonist administration. FASEB J 2009; 23:1893–906. [DOI] [PubMed] [Google Scholar]
- 25. Bartlett R, Stokes L, Sluyter R. The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol Rev 2014; 66:638–75. [DOI] [PubMed] [Google Scholar]
- 26. Bartlett R, Sluyter V, Watson D, Sluyter R, Yerbury JJ. P2X7 antagonism using Brilliant Blue G reduces body weight loss and prolongs survival in female SOD1G93A amyotrophic lateral sclerosis mice. PeerJ 2017; 5:e3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Farrell AW, Gadeock S, Pupovac A et al P2X7 receptor activation induces cell death and CD23 shedding in human RPMI 8226 multiple myeloma cells. Biochim Biophys Acta 2010; 1800:1173–82. [DOI] [PubMed] [Google Scholar]
- 28. Constantinescu P, Wang B, Kovacevic K et al P2X7 receptor activation induces cell death and microparticle release in murine erythroleukemia cells. Biochim Biophys Acta 2010; 1798:1797–804. [DOI] [PubMed] [Google Scholar]
- 29. Englezou PC, Rothwell SW, Ainscough JS et al P2X7R activation drives distinct IL‐1 responses in dendritic cells compared to macrophages. Cytokine 2015; 74:293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCarthy PL Jr, Abhyankar S, Neben S et al Inhibition of interleukin‐1 by an interleukin‐1 receptor antagonist prevents graft‐versus‐host disease. Blood 1991; 78:1915–8. [PubMed] [Google Scholar]
- 31. Ghiringhelli F, Apetoh L, Tesniere A et al Activation of the NLRP3 inflammasome in dendritic cells induces IL‐1β–dependent adaptive immunity against tumors. Nat Med 2009; 15:1170–8. [DOI] [PubMed] [Google Scholar]
- 32. Zhao J, Wang H, Dai C et al P2X7 blockade attenuates murine lupus nephritis by inhibiting activation of the NLRP3/ASC/Caspase 1 pathway. Arthritis Rheum 2013; 65:3176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang JJ, Newton RC, Rutledge SJ et al Characterization of murine IL‐1β: isolation, expression, and purification. J Immunol 1988; 140:3838–43. [PubMed] [Google Scholar]
- 34. Vandenabeele P, Declercq W, Libert C, Fiers W. Development of a simple, sensitive and specific bioassay for interleukin‐1 based on the proliferation of RPMI 1788 cells comparison with other bioassays for IL‐1. J Immunol Methods 1990; 135:25–32. [DOI] [PubMed] [Google Scholar]
- 35. Choi J, Ziga ED, Ritchey J et al IFNγR signaling mediates alloreactive T‐cell trafficking and GVHD. Blood 2012; 120:4093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao K, Ruan S, Yin L et al Dynamic regulation of effector IFN‐γ‐producing and IL‐17‐producing T cell subsets in the development of acute graft‐versus‐host disease. Mol Med Rep 2016; 13:1395–403. [DOI] [PubMed] [Google Scholar]
- 37. Pan B, Zeng L, Cheng H et al Altered balance between Th1 and Th17 cells in circulation is an indicator for the severity of murine acute GVHD. Immunol Lett 2012; 142:48–54. [DOI] [PubMed] [Google Scholar]
- 38. Zhao XY, Xu LL, Lu SY, Huang XJ. IL‐17‐producing T cells contribute to acute graft‐versus‐host disease in patients undergoing unmanipulated blood and marrow transplantation. Eur J Immunol 2011; 41:514–26. [DOI] [PubMed] [Google Scholar]
- 39. Acosta‐Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1[beta] and 6 but not transforming growth factor‐[beta] are essential for the differentiation of interleukin 17‐producing human T helper cells. Nat Immunol 2007; 8:942–9. [DOI] [PubMed] [Google Scholar]
- 40. King M, Covassin L, Brehm M et al Human peripheral blood leucocyte non‐obese diabetic‐severe combined immunodeficiency interleukin‐2 receptor gamma chain gene mouse model of xenogeneic graft‐versus‐host‐like disease and the role of host major histocompatibility complex. Clin Exp Immunol 2009; 157:104–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Covassin L, Jangalwe S, Jouvet N et al Human immune system development and survival of non‐obese diabetic (NOD)‐scid IL2rγnull (NSG) mice engrafted with human thymus and autologous haematopoietic stem cells. Clin Exp Immunol 2013; 174:372–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abraham S, Choi JG, Ye C, Manjunath N, Shankar P. IL‐10 exacerbates xenogeneic GVHD by inducing massive human T cell expansion. Clin Immunol 2015; 156:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Adinolfi E, Cirillo M, Woltersdorf R et al Trophic activity of a naturally occurring truncated isoform of the P2X7 receptor. FASEB J 2010; 24:3393–404. [DOI] [PubMed] [Google Scholar]
- 44. Sluyter R, Wiley JS. P2X7 receptor activation induces CD62L shedding from human CD4+ and CD8+ T cells. Inflamm Cell Signal 2014; 1:e92. [Google Scholar]
- 45. Pupovac A, Geraghty NJ, Watson D, Sluyter R. Activation of the P2X7 receptor induces the rapid shedding of CD23 from human and murine B cells. Immunol Cell Biol 2015; 93:77–85. [DOI] [PubMed] [Google Scholar]
- 46. Adriouch S, Dox C, Welge V, Seman M, Koch‐Nolte F, Haag F. Cutting edge: a natural P451L mutation in the cytoplasmic domain impairs the function of the mouse P2X7 receptor. J Immunol 2002; 169:4108–12. [DOI] [PubMed] [Google Scholar]
- 47. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol 2007; 7:118–30. [DOI] [PubMed] [Google Scholar]
- 48. Syberg S, Schwarz P, Petersen S et al R. Association between P2X7 receptor polymorphisms and bone status in mice. J Osteoporos 2012; 2012:637986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen X, Das R, Komorowski R et al Blockade of interleukin‐6 signaling augments regulatory T‐cell reconstitution and attenuates the severity of graft‐versus‐host disease. Blood 2009; 114:891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Flow cytometric gating of leucocytes in blood from humanized mice. Blood cells from humanized mice were labelled with isotype control or CD‐specific monoclonal antibodies (mAb). Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify human leucocytes (hCD45+mCD45–), human T cells (hCD45+mCD45–hCD3+hCD19–) and human B cells (hCD45+mCD45–hCD3–hCD19+).
Fig. S2. Flow cytometric gating of leucocytes in spleens from humanized mice. Splenocytes from humanized mice were labelled with isotype control or CD‐specific monoclonal antibodies (mAb). Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify human leucocytes (hCD45+mCD45–), human T cells (hCD45+mCD45–hCD3+hCD19–) and human B cells (hCD45+mCD45–hCD3–hCD19+). Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify human CD4+ T cell (hCD3+hCD4+hCD8–) and human CD8+ T cell (hCD3+hCD4–hCD8+) subsets.
Fig. S3. Flow cytometric analysis of YO‐PRO‐12+ uptake into human hCD3+ T cells and murine CD11c+ splenic dendritic cells. Human peripheral blood mononuclear cells and splenocytes from non‐obese diabetic‐severe combined immunodeficiency‐interleukin (NOD‐SCID‐IL)‐2Rγnull mice were incubated with YO‐PRO‐12+ in the absence (basal) (grey histograms) or presence of adenosine triphosphate (ATP) (black histograms) and labelled subsequently with anti‐human (h) CD3 or anti‐murine (m) CD11c monoclonal antibody. Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify hCD3+ T cells or mCD11c+ splenic DCs. YO‐PRO‐12+ uptake into these cells was then quantified using geometric mean fluorescence of histograms.
Fig. S4. Flow cytometric analysis of intracellular human interferon (IFN)‐γ and interleukin (IL)‐17 in human CD4+ and CD8+ T cells. Splenocytes from humanized mice were incubated with phorbol 12‐myristate 13‐acetate and ionomycin, and labelled with isotype control or specific monoclonal antibodies. Forward‐ and side‐scatter was used to identify leucocytes, which were used subsequently to identify human CD4+ T cell (hCD3+hCD4+hCD8–), and human CD8+ T cell (hCD3+hCD4–hCD8+) subsets. The percentage of hIFN‐γ and hIL‐17 was quantified as the difference between cytokine specific (black histograms) and isotype control (grey histograms) monoclonal antibody (mAb) labelling (indicated by marker regions as shown).