

Summary
Inflammatory responses mediated by macrophages are part of the innate immune system, whose role is to protect against invading pathogens. Lipopolysaccharide (LPS) found in the outer membrane of Gram‐negative bacteria stimulates an inflammatory response by macrophages. During the inflammatory response, extracellular LPS is recognized by Toll‐like receptor 4, one of the pattern recognition receptors that activates inflammatory signalling pathways and leads to the production of inflammatory mediators. The innate immune response is also triggered by intracellular inflammasomes, and inflammasome activation induces pyroptosis and the secretion of pro‐inflammatory cytokines such as interleukin‐1β (IL‐1β) and IL‐18 by macrophages. Cysteine‐aspartic protease (caspase)‐11 and the human orthologues caspase‐4/caspase‐5 were recently identified as components of the ‘non‐canonical inflammasome’ that senses intracellular LPS derived from Gram‐negative bacteria during macrophage‐mediated inflammatory responses. Direct recognition of intracellular LPS facilitates the rapid oligomerization of caspase‐11/4/5, which results in pyroptosis and the secretion of IL‐1β and IL‐18. LPS is released into the cytoplasm from Gram‐negative bacterium‐containing vacuoles by small interferon‐inducible guanylate‐binding proteins encoded on chromosome 3 (GBP chr3)‐mediated lysis of the vacuoles. In vivo studies have clearly shown that caspase‐11 −/− mice are more resistant to endotoxic septic shock by excessive LPS challenge. Given the evidence, activation of caspase‐11 non‐canonical inflammasomes by intracellular LPS is distinct from canonical inflammasome activation and provides a new paradigm in macrophage‐mediated inflammatory responses.
Keywords: Gram‐negative bacteria, inflammation, intracellular lipopolysaccharide, macrophage, non‐canonical inflammasome
Abbreviations
- AIM2
absent in melanoma 2
- CARD
caspase recruit domain
- CTB
cholera toxin B
- GBP
guanylate‐binding protein
- GSDMD
gasdermin‐D
- IFN
interferon
- IL
interleukin
- LPS
lipopolysaccharide
- LRR
leucine‐rich repeats
- NLR
NOD‐like receptor
- NOD
nucleotide‐binding oligomerization domain
- OMV
outer membrane vesicle
- PAMP
pathogen‐associated molecular pattern
- PRR
pattern recognition receptor
- PYD
pyrin domain
- TLR
Toll‐like receptor
Introduction
Inflammation is a series of biological processes that evolved to protect the body from invading pathogens and is characterized by heat, redness, swelling, pain and loss of function.1, 2, 3, 4 Inflammation as part of the innate immune response is mediated primarily by macrophages and initiated by a highly conserved group of receptors known as pattern recognition receptors (PRRs) that recognize pathogen‐associated molecular patterns (PAMPs) derived from pathogens.3, 4, 5 Several types of PRR families have been identified including Toll‐like receptors (TLRs), c‐type lectin receptors and scavenging receptors, which are localized on the outer cell surface or endosomal membrane, in addition to retinoic acid inducible gene‐I‐like receptors, absent in melanoma 2 (AIM2)‐like receptors (ALRs), and nucleotide‐binding oligomerization domain (NOD)‐like receptors (NLRs) that are located in the cytoplasm.6, 7 One of the most extensively studied PRRs is TLR4, which forms an extracellular receptor complex with MD2 to recognize lipopolysaccharide (LPS), the most inflammatory component of Gram‐negative bacteria cell walls.7 TLR4‐mediated recognition of extracellular LPS immediately activates inflammatory pathways in macrophages, such as nuclear factor‐κB), activator protein (AP)‐1, and interferon regulatory factor (IRF) pathways, by initiating signal transduction cascades of intracellular inflammatory molecules,8, 9, 10, 11, 12 resulting in the production of pro‐inflammatory cytokines, interferons, inflammatory proteins, and microbicidal factors.5 Interestingly, a new paradigm of LPS‐induced inflammatory responses in macrophage‐mediated innate immunity against pathogen infection has emerged.13
In response to PAMPs, a subset of NLRs and AIM2‐like receptors induces a unique innate immune response. These intracellular receptors assemble cytoplasmic complexes, known as inflammasomes, to activate the inflammatory caspase, caspase‐1.14, 15, 16 The activation of caspase‐1 induces pyroptosis, which is an inflammatory form of programmed cell death that frequently occurs upon infection with intracellular pathogens. In addition, caspase‐1 in macrophages induces the maturation and secretion of the pro‐inflammatory cytokines interleukin‐1β (IL‐1β) and IL‐18.15, 17, 18 Interestingly, recent studies have shown that caspase‐11 can also induce caspase‐1‐dependent maturation and secretion of IL‐1β and IL‐18 as well as caspase‐1‐independent pyroptosis in response to intracellular LPS derived from cytoplasmic Gram‐negative bacteria.19 This caspase‐11‐mediated innate immune response is analogous to caspase‐1 activation by canonical inflammasome scaffolds, but has different and unique characteristics compared with canonical inflammasome activation during macrophage‐mediated inflammatory responses. This caspase‐11 scaffold is therefore referred to as a ‘non‐canonical inflammasome’.17, 19, 20, 21, 22, 23, 24, 25 From this unexpected observation, the role of caspase‐11 non‐canonical inflammasome has been actively investigated in macrophage‐mediated inflammatory responses.
This review aims to introduce briefly the structures and activation of inflammasomes and to discuss and summarize recent research progress in our understanding of caspase‐11 non‐canonical inflammasome activation during macrophage‐mediated inflammatory responses, including discovery of caspase‐11 non‐canonical inflammasome, the molecular mechanism of caspase‐11 non‐canonical inflammasome activation, and subsequent inflammatory events mediated by caspase‐11 non‐canonical inflammasome activation. Moreover, this review highlights how this could lead to the development of therapeutics for the treatment of inflammatory and infectious diseases.
Structures and activation of canonical and non‐canonical inflammasomes
Canonical inflammasomes
Canonical inflammasomes induce inflammatory responses by processing inactive pro‐caspase‐1 into cleaved active caspase‐1.15, 18 These canonical inflammasomes are protein complexes of either NLRs or pyrin domain (PYD)‐containing non‐NLRs assembling apoptosis‐associated speck‐like protein containing a C‐terminal caspase recruit domain (CARD) (ASC), and pro‐caspase‐1.15, 18 Different types of NLR family members, such as NLRC4, NLRP1, NLRP3 and AIM2 have been identified, and in‐depth studies have demonstrated that each NLR member is differentially stimulated by different ligands.15, 18
NLRC4 inflammasome
NLRC4 was initially identified as a structurally similar molecule to apoptotic‐protease activating factor 1 and was demonstrated to form an inflammasome in response to bacterial flagellin, a building block of the bacterial locomotion machinery, as well as needle subunits, which are used to inject virulent factors into host cells.26, 27, 28, 29, 30, 31, 32 NLRC4 consists of three major motifs: an N‐terminal CARD motif, a nucleotide‐binding and oligomerization domain (NACHT) motif, and C‐terminal leucine‐rich repeats (LRRs), and interacts with pro‐caspase‐1 through its CARD motif to assemble an NLRC4 inflammasome (Fig. 1a). Interestingly, because NLRC4 has a CARD motif itself, it generally does not need the adaptor molecule, ASC, for direct interaction with pro‐caspase‐1, which also contains a CARD motif. Two research groups, however, also reported that ASC is required for the activation of an NLRC4 inflammasome. They showed endogenous NLRC4‐ASC‐Casp‐1 specks in macrophages and that ASC has been shown to be essential for robust IL‐1β secretion when NLRC4 is activated.33, 34
Figure 1.
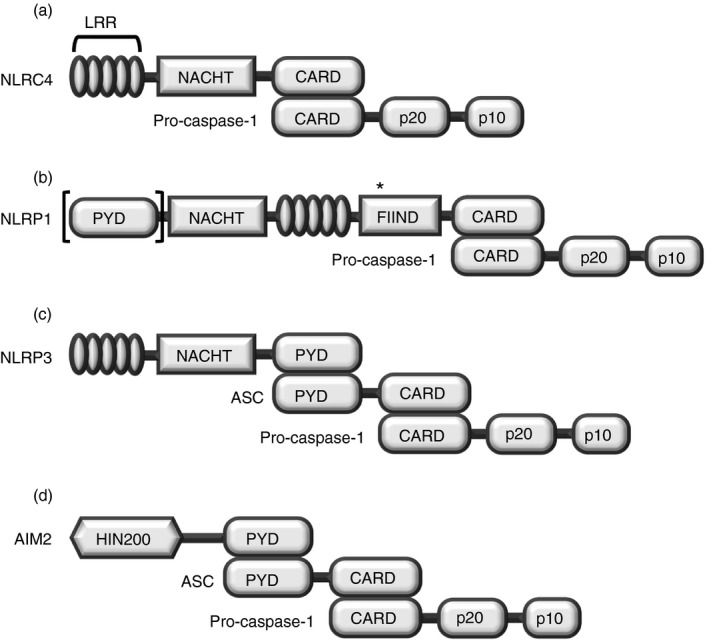
Structures of canonical inflammasomes. (a–c) Three different NLRs, and (d) AIM2 assembly of canonical inflammasomes through binding with pro‐caspase‐1. (a) NLRC4 and (b) NLRP1 (lack of [PYD] in mouse NLRP1 isoforms) assemble canonical inflammasomes by interacting directly with inactive pro‐caspase‐1 through the CARD motif, whereas (c) NLRP3 and (d) AIM2 assemble canonical inflammasomes by interacting indirectly with pro‐caspase‐1 via the bipartite PYD–CARD adaptor protein ASC. After assembly of canonical inflammasomes, pro‐caspase‐1 is cleaved and matures to an active form. Abbreviations used: LRR, leucine‐rich repeat; NRL, nucleotide‐binding oligomerization domain (NOD)‐like receptor; caspase, cysteine‐aspartic protease; CARD, caspase recruit domain; NACHT, nucleotide‐binding and oligomerization domain; FIIND, function to find domain; AIM2, absent in melanoma 2; PYD, pyrin domain; HIN, haematopoietic interferon‐inducible nuclear proteins. *Autocatalytic cleavage.
NLRP1 inflammasome
NLRP1 is the first NLR family member identified to form an inflammasome complex.35 Human NLRP1 has only one family member characterized by an N‐terminal PYD, NACHT, LRRs, a function‐to‐find domain, and a C‐terminal CARD (Fig. 1b). By contrast, multiple isoforms of NLRP1, such as NLRP1A, NLRP1B and NLRP1C, have been identified in mice, and their structures are evolutionarily conserved, but they lack a PYD motif.18 NLRP1 interacts directly with pro‐caspase‐1 through its CARD motif (Fig. 1b) and does not need an ASC to assemble the NLRP1 inflammasome with pro‐caspase‐1 (Fig. 1b). Although the activation of NLRP1 inflammasome is generally ASC‐independent for pyroptosis and IL‐1β secretion, two research groups observed that ASC is necessary for the formation of NLRP1 inflammasome and autoproteolysis of pro‐caspase‐1.36, 37 Van Opdenbosch et al.37 further demonstrated that ASC enhanced cytokine secretion by the activation of NLRP1b inflammasome in response to low‐level LeTx stimulation. In mouse macrophages, NLRP1B was activated and forms an inflammasome in response to lethal toxins of Bacillus anthracis; therefore, the macrophages isolated from Nlrp1b knock‐out mice were not capable of generating the active form of caspase‐1, secreting IL‐1β, or inducing pyroptosis.38, 39
NLRP3 inflammasome
NLRP3, also known as cryopyrin, has three major motifs: an N‐terminal PYD motif, a NACHT motif and C‐terminal LRRs (Fig. 1c). NLRP3 responds to a variety of stimuli, including crystals/particles such as uric acid, silica, alum, β‐amyloid and asbestos, nucleic acid hybrids such as RNA–DNA hybrids, pore‐generating toxins, extracellular ATP, hyaluronan and various pathogens including viruses, bacteria, fungi and protozoans.40, 41 Binding of these stimuli to extracellular receptors expressed on macrophages, which is the first signal called priming, and subsequent triggering, which is an additional signal, initiate the activation of the NLRP3 inflammasome by assembly of an NLRP3–ASC–pro‐caspase‐1 complex (Fig. 1c). NLRP3 homotypically interacts with a bipartite adaptor molecule, ASC, through the PYD motif, and pro‐caspase‐1 is in turn recruited to the NLRP3–ASC complex to bind with ASC through its CARD motif.15
AIM2 inflammasome
AIM2 was identified as an intracellular sensor protein to be activated by directly recognizing double‐stranded DNAs based on the observation that the double‐stranded DNAs derived from intracellular microbial and host cells induced the activation and autoproteolysis of pro‐caspase‐1 with the involvement of ASC but did not activate previously demonstrated effector molecules, such as NLRP3 and TLRs, or interferon (IFN) signalling.42 AIM2 is a member of the p200 protein family of proteins. These proteins have a PYD at the N‐terminus and a haematopoietic IFN‐inducible nuclear protein (HIN) domain at the C‐terminus (Fig. 1d). Sensing of intracellular double‐stranded DNAs derived from invading pathogens, such as Francisella tularensis, cytomegalovirus and vaccinia virus, results in the activation of AIM2 and assembly of an AIM2 canonical inflammasome by homotypic interaction with the PYD motif of the adaptor molecule, ASC and another homotypic interaction between ASC and pro‐caspase‐1 through their CARD motifs (Fig. 1d).43, 44, 45, 46 The activation of AIM2 inflammasomes occurs with the formation of a speck, which is a single large perinuclear aggregate.47 Speck is formed by polymerization of AIM2 and ASC through the PYD motif and nucleation of ASC fibres by AIM2 into star‐shaped branched filaments that serve as a platform for the clustering of pro‐caspase‐1.47, 48, 49 Speck formation for the activation of canonical inflammasomes has also been observed in NLRC4, NLRP1 and NLRP3,33, 37, 50 suggesting that speck formation might be an essential process for the activation of canonical inflammasomes, even though the stimulating agents for the various canonical inflammasomes are quite different.
Non‐canonical inflammasome
Under certain conditions, induction of inflammatory responses bypasses the canonical inflammasome pathway; instead, the non‐canonical inflammasome, which refers to a complex of pro‐caspase‐11 and LPS, is activated in macrophages.17, 18, 24, 25 Recently, non‐canonical inflammasomes have been shown to have characteristics distinct from those of canonical inflammasomes. The non‐canonical inflammasome is activated by intracellular LPS released from Gram‐negative bacteria,21, 22 resulting in the formation of a molecular complex consisting of inactive pro‐caspase‐11 and LPS.17 In mouse macrophages, non‐canonical inflammasomes are assembled by direct interaction between pro‐caspase‐11 and LPS, and this direct interaction is accomplished by binding of the CARD motif of pro‐caspase‐11 and the lipid A tail of LPS (Fig. 2a). Caspase‐4 and caspase‐5 are the human counterparts of mouse caspase‐11. Human caspase‐4 and caspase‐5 were found to interact directly with intracellular LPS and activate the non‐canonical inflammasome in human myeloid cells.23, 51, 52, 53 Non‐canonical inflammasome assembly in human cells is accomplished in a similar manner to its assembly in mice. Pro‐caspase‐4 and/or pro‐caspase‐5 in human cells interact directly with the lipid A tail of LPS, and this interaction is mediated by binding of the CARD motifs of pro‐caspase‐4 and/or pro‐caspase‐5 and the lipid A tail of LPS (Fig. 2b). These pro‐caspase‐4/5/11 bindings with intracellular lipid A of LPS oligomerize (Fig. 2c) and in turn, induce not only pyroptosis but also maturation and secretion of pro‐inflammatory cytokines, IL‐1β and IL‐18. Interestingly, a recent study reported that caspase‐11 non‐canonical inflammasome activation can also induce the activation of the NLRP3–ASC–caspase‐1 pathway, newly identified as ‘non‐canonical NLRP3 inflammasome activation’, which is distinguished from the ‘canonical NLRP3 inflammasome activation’ induced by various ligands discussed in the section on ‘NLRP3 inflammasome’ through the activation of a nuclear factor‐κB) signalling pathway via TLR–MyD88 and/or tumour necrosis factor receptor.54 Activation of the non‐inflammasome pathway in macrophage‐mediated inflammatory responses will be discussed in the following sections in more detail; this discussion will focus mostly on mouse caspase‐11, because caspase‐11 was first identified as a critical modulator in the activation of the non‐canonical inflammasome in mouse macrophages, and most previous studies have focused on investigating the roles of caspase‐11 in regulating the activation of the non‐canonical inflammasome pathway during inflammatory responses in mouse macrophages.
Figure 2.
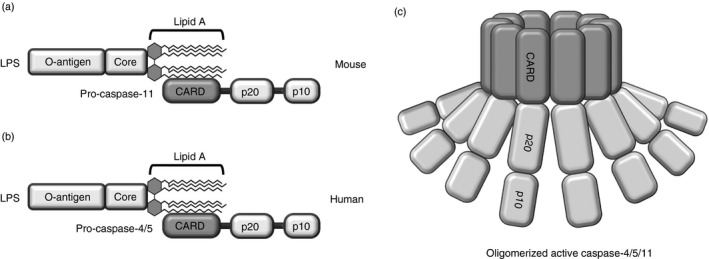
Structures of non‐canonical inflammasomes. Non‐canonical inflammasomes are assembled by direct interaction of the lipid A moiety of lipopolysaccharide (LPS) with the CARD motif of (a) pro‐caspase‐11 (mouse) or (b) pro‐caspase‐4/5 (human). (c) Activation of non‐canonical inflammasomes by oligomerization of caspase‐11 (mouse) or caspase‐4/5 (human) through their CARD motifs. Unlike canonical inflammasomes, non‐canonical inflammasomes form complexes in the absence of a bipartite adaptor protein, such as ASC; instead, the lipid A moiety of LPS directly interacts with the CARD motif of the caspase. Caspase, cysteine‐aspartic protease; CARD, caspase recruit domain.
Non‐canonical inflammasome activation during inflammatory responses in macrophages
Discovery of a caspase‐11 non‐canonical inflammasome
A number of previous studies have intensively investigated inflammasome activation in innate immune cells in response to various stimuli. Kayagaki et al.19 conducted a study of toxin‐stimulated activation of the inflammasome pathway and found that cholera toxin B (CTB) induced the secretion of one of the inflammasome‐stimulated pro‐inflammatory cytokines, IL‐1β, in an NLRP3/ASC inflammasome‐dependent manner in LPS‐primed mouse macrophages. Interestingly, this study reported the unexpected observation that CTB‐activated inflammasome responses and IL‐1β secretion were abolished in bone‐marrow‐derived macrophages isolated from the mouse strain 129S6, which has a polymorphism in the caspase‐11 gene locus resulting in a truncated and non‐functional caspase‐11 protein.19 Moreover, they also reported that the strain 129S6 mice encoding non‐functional caspase‐11 were much more resistant to a lethal dose of LPS that usually induces septic shock.19 These observations provided strong evidence that caspase‐11, which is not a component of the canonical inflammasome, plays a unique modulatory role in macrophage‐mediated innate immune responses, and that the molecular mechanism by which caspase‐11 induces inflammatory responses in macrophages is distinct from that of the canonical inflammasome activation pathway. A series of follow‐up studies has successfully established that this caspase‐11‐mediated inflammatory response is a ‘non‐canonical inflammasome pathway’ that is activated by a direct response to Gram‐negative bacteria, including Escherichia coli, Legionella pneumophila, Salmonella typhimurium, Citrobacter rodentium, Shigella flexneri, and Burkholderia spp.20, 55, 56, 57, 58, 59, 60
Caspase‐11: a direct sensor of intracellular LPS
As discussed above, the caspase‐11 non‐canonical inflammasome pathway is activated in response to invading Gram‐negative bacteria.20, 55, 56, 57, 58, 59 By contrast, it is not activated by Gram‐positive bacteria.56 This suggests that caspase‐11 recognizes intracellular LPS, inflammatory components derived from Gram‐negative bacteria. In support of this hypothesis, Kayagaki et al.22 demonstrated that treatment of macrophages with LPS (O111:B4 strain) along with CTB highly induced caspase‐11 activation. This was accomplished by CTB‐mediated transportation of extracellular LPS into the macrophages through interactions between CTB and the O‐antigen moiety of LPS. Therefore, CTB acted as a carrier for the intracellular delivery of LPS, which was, in turn, recognized by caspase‐11, resulting in caspase‐11‐mediated activation of the non‐canonical inflammasome pathway. Hagar et al.21 found that the caspase‐11 non‐canonical inflammasome pathway was activated in macrophages transfected with the lysates of Gram‐negative bacteria, but not with lysates of Gram‐positive bacteria, which could possibly support the notion that caspase‐11 recognizes intracellular LPS from Gram‐negative bacteria, resulting in activation of the caspase‐11 non‐canonical inflammasome pathway in macrophages.
Caspase‐11 was demonstrated to not need the adaptor molecule ASC for activation,19 which suggested that caspase‐11 might interact directly with intracellular LPS. Studies to evaluate this supported this theory. Recombinant caspase‐11 expressed and purified from the Gram‐negative bacterium E. coli formed a huge caspase‐11 multimer with a molecular weight of ~600 000 due to LPS binding, whereas that from insect cells, which do not have LPS, was not multimeric, but monomeric.23 Moreover, direct interaction between caspase‐11 and LPS was observed in the macrophages electroporated with LPS, consequently resulting in the macrophage pyroptosis.23 The affinity between caspase‐11 and LPS was found to be comparable to that between TLR4 and MD2.61 Human caspase‐4 and caspase‐5 classified as inflammatory caspases are considered most homologous to mouse caspase‐11.23 Caspase‐4 was found to be highly expressed in human monocytes and induced pyroptosis in an LPS‐dependent manner.23 Similar to caspase‐11, recombinant caspase‐4/5 expressed and purified from E. coli formed a caspase‐4 multimer, whereas that from insect cells was monomeric.23 A biochemical study showed that mouse caspase‐11 (Fig. 2a) and human caspase‐4/5 (Fig. 2b) bind directly to lipid A of LPS through their CARDs, and that lipid A is sufficient for their bindings.23 Moreover, Hagar et al. reported that the structure of lipid A is a critical determinant of the activation of the caspase‐11 non‐canonical inflammasome pathway in macrophages. Penta‐ and hexa‐acylated lipid A successfully induced the activation of the caspase‐11 non‐canonical inflammasome pathway and pyroptosis of macrophages, whereas tetra‐acylated lipid A was not detected by caspase‐11.21, 62 These studies strongly indicate that mouse caspase‐11 and its human counterpart, caspase‐4/5, are oligomerized by direct interaction with intracellular LPS (Fig. 2c), especially the lipid A component of LPS, resulting in non‐canonical inflammasome activation in macrophages.
How is the intracellular LPS released into cytoplasm and detected by caspase‐11?
As discussed earlier, a number of studies have provided evidence that caspase‐11 directly senses intracellular LPS derived from a variety of Gram‐negative bacteria, resulting in the oligomerization of caspase‐11–LPS complexes. However, many steps involved in the activation of the caspase‐11 non‐canonical inflammasome still need to be elucidated. Recent studies have shown how caspase‐11 can detect intracellular LPS from infected Gram‐negative bacteria. Two groups showed that endogenous type I IFNs and their signalling are required for the activation of caspase‐11 and subsequent pyroptosis in macrophages infected with Gram‐negative bacteria.20, 56 When LPS was directly delivered into the macrophages, the delivered intracellular LPS significantly activated the caspase‐11 non‐canonical inflammasome pathway, leading to the induction of pyroptosis and IL‐1β secretion in a type I IFN signalling‐independent manner.59 This result suggests that gene expression induced by type I IFNs is required for cytoplasmic sensing of LPS by caspase‐11. These studies indicate that LPS from Gram‐negative bacteria are the key determinant of caspase‐11 activation in macrophages; however, it remains unclear how LPS of Gram‐negative bacteria enter the host cells, and how LPS is released from vacuoles containing internalized Gram‐negative bacteria into the cytoplasm. Recent studies have successfully answered this question. Recently, Vanaja et al. identified that Gram‐negative bacteria produced outer membrane vesicles (OMVs) and these OMVs delivered LPS of Gram‐negative bacteria into the cytoplasm of host cells through endocytosis, leading to induction of caspase‐11‐activated inflammatory responses.63 Pilla et al.64 demonstrated that guanylate‐binding proteins encoded on mouse chromosome 3 (GBPchr3) induced caspase‐11‐dependent pyroptosis in IFN‐stimulated macrophages infected with the Gram‐negative bacteria L. pneumophila, and that this caspase‐11‐dependent pyroptosis was dramatically reduced in GBPchr3‐deficient macrophages. Consistent with this study of Pilla et al., Meunier et al.65 reported that GBPchr3 lysed vacuoles containing the Gram‐negative bacteria S. typhimurium and released the bacteria into the cytoplasm, thus allowing the recognition of their LPS by caspase‐11, resulting in activation of the caspase‐11 non‐canonical inflammasome pathway and the secretion of IL‐1β and IL‐18 during infection. The activation of the caspase‐11 non‐canonical inflammasome pathway and the secretion of IL‐1β and IL‐18 were significantly reduced in GBPchr3‐deficient macrophages infected with S. typhimurium.65 These studies strongly suggest that GBPchr3 plays a critical role in the activation of the caspase‐11 non‐canonical inflammasome pathway during inflammatory responses by lysing Gram‐negative bacterium‐containing vacuoles and releasing bacterial LPS into the cytoplasm, where they encounter caspase‐11 (Figs 3a, b). Interestingly, the requirement of GBPchr3 in inflammasome activation is not unique to caspase‐11. Two research groups demonstrated that GBPchr3 was also involved in the activation of the AIM2 inflammasome.62, 66 A further study also demonstrated that GBPchr3 and the related member immunity‐related GTPase family member b10 (IRGB10) directly targeted and lysed the Gram‐negative bacterium Francisella novicida to further activate caspase‐11 and NLRP3 inflammasomes in the cytoplasm.67
Figure 3.
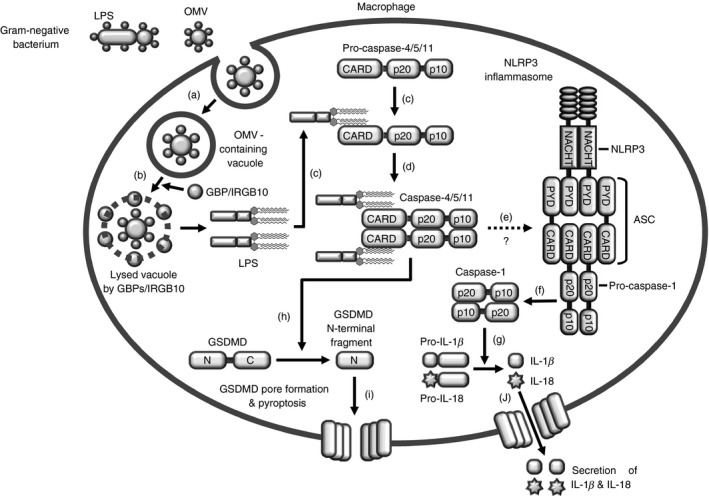
Intracellular LPS‐mediated caspase‐4/5/11 non‐canonical inflammasome activation. (a) LPS‐containing OMVs derived from Gram‐negative bacteria is internalized into a macrophage by endocytosis, and (b) LPS of the OMVs is released into the cytoplasm from the Gram‐negative bacterium‐containing vacuole by GBP chr3‐mediated lysis. (c) The released intracellular LPS binds directly to pro‐caspase‐11 and to pro‐caspase‐4/5/11 CARD motifs through the lipid A moiety, (d) leading to oligomerization of pro‐caspase‐4/5/11 to induce non‐canonical inflammasome activation. (e) Active caspase‐4/5/11 non‐canonical inflammasome activates the NLRP3 canonical inflammasome via an unknown mechanism, (f) inducing the proteolysis and maturation of pro‐caspase‐1. (g) Cleaved active caspase‐1, in turn, induces the proteolysis and maturation of the pro‐inflammatory cytokines IL‐1β and IL‐18. (h) Active caspase‐4/5/11 non‐canonical inflammasomes also cleave the linker loop of intact GSDMD to generate N‐ and C‐terminal fragments of GSDMD. (i) The cleaved N‐terminal fragments of GSDMD are localized to the cell membrane, bind to phosphoinositides of the cell membrane, and oligomerize to form membrane pores, resulting in cell swelling and lysis, known as pyroptosis. (j) Proteolysed mature IL‐1β and IL‐18 are secreted through GSDMD pores. Abbreviations used:OMV, outer membrane vesicle; Caspase, cysteine‐aspartic protease; GBP, guanylate‐binding protein; immunity‐related GTPase family member b10, IRGB10; LPS, lipopolysaccharide; GSDMD, gasdermin D; IL, interleukin; N, N‐terminal fragment of GSDMD; C, C‐terminal fragment of GSDMD; CARD, caspase recruit domain; NACHT, nucleotide‐binding and oligomerization domain; PYD, pyrin domain.
Caspase‐11 non‐canonical inflammasome activation
When pathogens invade the body, innate immune cells, especially macrophages, rapidly recognize the PAMPs of the invading pathogens through their extracellular PRRs, such as TLRs and scavenger receptors, leading to up‐regulation of the expression of inflammatory genes including those encoding pro‐inflammatory cytokines and anti‐microbial peptides. A subset of pathogen components, such as LPS, in turn stimulate much stronger inflammatory responses in a cooperative manner in response to signals transduced from outside the cells, resulting in activation of inflammatory caspases and inflammasome pathways. For the inflammatory response against invading pathogens, non‐canonical inflammasome activation requires two sequential signals in macrophages: (i) priming and (ii) triggering.19, 22, 68, 69 ‘Priming’ is the process by which PAMPs bind to extracellular PRRs on macrophage cell surfaces, resulting in the activation of signal transduction cascades to induce gene expression of the components required for non‐canonical inflammasome activation, including pro‐caspase‐11 and pro‐IL‐1β, and other proteins necessary for the caspase‐11 inflammasome responses, including pro‐caspase‐1 and pro‐IL‐18, which in most are constitutively expressed.19, 22, 68, 69 Because this priming signal neither activates a non‐canonical inflammasome pathway nor induces the processing and secretion of mature IL‐1β and IL‐18 or pyroptosis in macrophages, a second ‘triggering’ signal is required for full activation of inflammatory responses in macrophages. This triggering signal involves detection of intracellular LPS for subsequent activation of the non‐canonical inflammasome pathway in macrophages.21, 22 After LPS recognition, oligomerization of intracellular LPS–pro‐caspase‐11 complexes to form non‐canonical inflammasomes and consequent activation of the LPS–caspase‐11 inflammasome result in pyroptosis as well as the cleavage and secretion of the mature pro‐inflammatory cytokines IL‐1β and IL‐18 (Fig. 3). In more detail, intracellular LPS released from Gram‐negative bacteria‐containing vacuoles by GBPchr3‐mediated lysis are detected by pro‐caspase‐11, resulting in the formation of a pro‐caspase‐11–LPS complex (Fig. 3c). This complex, in turn, is oligomerized by homotypic interactions of the CARD motifs of pro‐caspase‐11 (Fig. 3d). Recent studies have reported that caspase‐11 requires the activation of the NLRP3 canonical inflammasome by an unknown mechanism (Fig. 3e) for proteolysis and maturation of pro‐caspase‐1 (Fig. 3f), and that the cleaved active caspase‐1 induces the subsequent processing of pro‐IL‐1β and pro‐IL‐18 to their active forms, IL‐1β and IL‐18 (Fig. 3g), whereas caspase‐11 is not required for caspase‐1‐mediated pyroptosis.19, 22 As discussed earlier, two hallmarks of caspase‐11 non‐canonical inflammasome activation are the secretion of IL‐1β and IL‐18 and pyroptosis; the mechanistic details of these events are described in the next section.
Gasdermin D is a key molecule in caspase‐11 non‐canonical inflammasome‐induced pyroptosis and secretion of IL‐1β and IL‐18
Although priming is an essential process for induction of macrophage‐mediated inflammatory responses, the priming signal alone cannot induce pyroptosis and secretion of IL‐1β and IL‐18; a subsequent triggering signal is required.21, 22 A number of studies have elucidated the molecular mechanisms by which activation of the caspase‐11 non‐canonical inflammasome induces pyroptosis and secretion of mature IL‐1β and IL‐18 in macrophages infected with Gram‐negative bacteria. However, the question ‘how’ is still poorly understood and did not receive attention for a long period of time. Recently, however, a molecule, gasdermin‐D (GSDMD), has been identified as a critical player in these events. GSDMD is a highly conserved protein in mammals, although its function is unclear.70 GSDMD is approximately 480 amino acids long with two main domains: an N‐terminal GSDMD‐N domain and a C‐terminal GSDMD‐C domain connected by a linker loop71 and previously found to be cleaved by caspase‐1.72 Kayagaki et al.73 reported that GSDMD plays an essential role in caspase‐11 non‐canonical inflammasome‐dependent pyroptosis and secretion of IL‐1β. They generated Gsdmd −/− mice, and macrophages isolated from Gsdmd −/− mice had defects in pyroptosis and IL‐1β secretion in response to intracellular LPS or Gram‐negative bacterial infection and also exhibited resistance to LPS‐induced septic shock.73 In addition, they showed that caspase‐11 cleaved GSDMD, and that the N‐terminal fragment of the cleaved GSDMD facilitated pyroptosis and proteolytic maturation of caspase‐1 in an NLRP3‐dependent manner.73 Shi et al.74 made the same observation. They generated Gsdmd −/− mice and showed that intracellular LPS‐induced pyroptosis and IL‐1β secretion were significantly reduced in macrophages obtained from Gsdmd −/− mice. They further showed that both caspase‐1 and caspase‐4/5/11 cleaved the linker between the N‐terminal and C‐terminal GSDMD domain, and that the N‐terminal fragment of GSDMD was required and sufficient for pyroptosis, possibly by generating pores within the macrophage membrane.74 He et al.75 reported that the N‐terminal fragment of GSDMD processed by caspase‐1 was essential for pyroptosis and IL‐1β secretion, which were abolished in Gsdmd −/− Raw264.7 cells. Interestingly, GSDMD did not affect the maturation of either caspase‐11 or IL‐1β,75 suggesting that GSDMD is downstream of caspase‐1 and only aids in the induction of pyroptosis and the secretion of IL‐1β during the caspase‐11 non‐canonical inflammasome response.76 More direct evidence that GSDMD generates pores during the caspase‐11 non‐canonical inflammasome response have been reported. Ding et al.77 expressed and purified the N‐terminal fragments of GSDMD and demonstrated that the purified N‐terminal fragments of GSDMD lysed phosphoinositide/cardiolipin‐containing liposomes and formed structurally stable pores with a diameter of 10–14 nm and 17 symmetric protomers on the artificial and natural phospholipid membranes. Aglietti et al.78 observed by electron microscopy that N‐terminal fragments of GSDMD localized to the cell membrane with a higher order of oligomers and formed ring‐like structures within a few minutes of GSDMD cleavage. They further confirmed that localization of the cleaved N‐terminal fragments of GSDMD and formation of ring‐like structures within the cell membrane were only observed in macrophages after caspase‐11 activation by intracellular delivery of LPS.78 Liu et al.79 also demonstrated by electron microscopy that N‐terminal fragments of GSDMD induced pyroptosis by binding to phosphates and phosphatidylserine and cardiolipin and forming pores on the cell membrane. Taken together, these results strongly suggest that intact GSDMD, which is inactive, is cleaved at the internal linker loop by caspase‐11 (Fig. 3h), and that the cleaved N‐terminal fragments of GSDMD are essential for pyroptosis (Fig. 3g) and the secretion of IL‐1β and IL‐18 (Fig. 3j) by generating pores within the cell membrane during the caspase‐11 non‐canonical inflammasome response in macrophages.71, 80, 81
Non‐canonical inflammasome and LPS‐induced septic shock
During the inflammatory response, caspase‐1‐induced pyroptosis is the effector mechanism that clears invading pathogens by removing infected cells from the body.82 However, caspase‐1‐induced pyroptosis can be harmful under certain conditions because of the induction of systemic septic shock. Several studies have demonstrated that a caspase‐4/5/11 non‐canonical inflammasome cleaves pro‐caspase‐1 and induces caspase‐1‐mediated pyroptosis and IL‐1β secretion during macrophage‐mediated inflammatory responses,19, 20, 21, 22, 56, 60, 65, 83 suggesting the possibility that intracellular LPS‐induced activation of the caspase‐4/5/11 non‐canonical inflammasome could play a pivotal role in modulating endotoxic septic shock in vivo. This idea was recently explored by several research groups using mice challenged with excessive amounts of LPS to induce endotoxic septic shock, which is an established animal model for Gram‐negative bacteria‐induced sepsis. Earlier studies have demonstrated that mice with loss of either caspase‐1 or caspase‐11 were less susceptible to challenge with excessive LPS, and that these mice had higher survival rates in response to LPS‐induced lethality than wild‐type mice.84, 85 An additional study using several mouse strains with different polymorphisms clearly showed that caspase‐11 was more critical than caspase‐1 in the LPS‐induced death of mice.19 Aachoui et al.57 also reported that caspase‐11 −/− mice were more resistant to lethal challenge with the Gram‐negative bacteria Burkholderia thailandensis and Burkholderia pseudomallei than wild‐type mice. Two research groups showed that excessive LPS‐induced septic shock in mice was caspase‐11‐dependent but TLR4‐independent. Hagar et al.21 and Kayagaki et al.22 reported that caspase‐11 −/− mice primed with poly (I:C), a TLR3 ligand, were resistant to subsequent challenge with excessive LPS, whereas Tlr4 −/− mice primed with poly (I:C) were highly susceptible to subsequent LPS challenge, and their survival rates were very low. These results strongly indicate that priming the mice with TLR3 ligand up‐regulated the expression of the caspase‐11 gene by bypassing the requirement for TLR4 signalling; therefore, TLR4 is only required to induce the expression of the caspase‐11 gene during LPS‐induced endotoxic septic shock in mice, and LPS‐induced lethality is independent of TLR4. In support of this theory, Tlr4 −/− mice primed with poly (I:C) followed by excessive LPS challenge had very low survival rates, whereas mice primed with LPS, which are TLR4 ligands, followed by excessive LPS challenge exhibited resistance to LPS challenge and had high survival rates.21 Despite the clear evidence for the crucial role of caspase‐11 in endotoxic septic shock in vivo, how excessive LPS induces caspase‐11 activation and death of the mice by septic shock is still unclear, and detailed mechanistic studies in this regard are required. The role of GSDMD on the LPS‐induced LPS septic shock was also investigated in mice. As in caspase‐11 −/− mice, the mice lacking Gsdmd were highly resistant to LPS‐induced septic shock,73 suggesting that GSDMD is a critical pro‐pyroptotic substrate for caspase‐11 non‐canonical inflammasome and is essential for caspase‐11‐mediated lethal sepsis in vivo.
Conclusion and perspectives
An inflammatory response is a major host defence mechanism against invading pathogens, and host cells activate a variety of immune responses to prevent damage to the body. Numerous studies have investigated how inflammatory responses are initiated by recognition of extracellular PAMPs by PRRs, and some studies have also investigated how inflammatory responses are initiated in response to intracellular pathogens, especially intracellular Gram‐negative bacteria harbouring LPS, one of the most inflammatory components of bacteria. The inflammasome has been shown to play a pivotal role in the inflammatory response by activating the inflammatory caspase caspase‐1, which induces pyroptosis and the secretion of the pro‐inflammatory cytokine IL‐1β in macrophages. In these inflammasome studies, caspase‐11 was unexpectedly identified as a novel activator of caspase‐1, and the complex of caspase‐11 and intracellular LPS was referred to as the ‘non‐canonical inflammasome’,19 adding to the diversity of cytoplasmic innate immunity responses in macrophages.86, 87 Recent studies have not only identified intracellular LPS as agonists of the non‐canonical inflammasome,21, 22 but also demonstrated that inflammatory caspases, such as a mouse caspase‐11 and human caspase‐4/5, are direct molecular sensors of intracellular LPS and activate non‐canonical inflammasome responses.23 Intracellular LPS released from the Gram‐negative bacterium‐containing vacuoles by GBPchr3‐mediated proteolysis are directly detected by caspases‐4/5/11, and these caspases are oligomerized and activated, thereby inducing anti‐pathogenic inflammatory responses by triggering pyroptosis and secretion of IL‐1β and IL‐18 in macrophages. The activation of the caspase‐11 non‐canonical inflammasome by intracellular LPS during macrophage‐mediated inflammatory responses is illustrated in Fig. 3. The caspase‐11 non‐canonical inflammasome adds to the repertoire of host defence mechanisms that can defend against infection by intracellular Gram‐negative bacteria. Although a number of studies have successfully demonstrated the new roles of caspase‐11 non‐canonical inflammasome during macrophage‐mediated inflammatory responses, more studies to identify the crosstalk between caspase‐11 non‐canonical inflammasomes and other canonical inflammasomes will be required to explain how they cooperate and produce synergistic effects to protect our body from invading pathogens by inflammatory responses. Moreover, further studies on the useful strategies to effectively and selectively target and modulate the activation of the caspase‐11 non‐canonical inflammasome pathway are required to develop the promising diagnostics and therapeutics to prevent and treat infectious and inflammatory/autoimmune diseases.
In conclusion, given the strong evidence of a critical role of caspase‐11 in the non‐canonical inflammasome response to intracellular LPS in macrophages, selective targeting of the caspase‐11 non‐canonical inflammasome pathway by various useful approaches, including caspase‐11‐targeting small interference RNAs, the agents interfering with the binding between caspase‐11 and intracellular LPS, and caspase‐11‐targeting antibody therapeutics using the recently developed intracellular molecule‐targeting antibody technology88 could be a novel and promising strategy for the prevention and treatment of a number of infectious and pathogen‐induced inflammatory/autoimmune diseases,89 such as sepsis, chronic respiratory/lung diseases, coronary artery disease, cystic fibrosis, rheumatoid arthritis, and some intestinal inflammatory diseases, including inflammatory bowel disease, Crohn's disease and ulcerative colitis.54
Disclosure
The author has no competing interests to declare.
Acknowledgements
The author thanks Professor Jae Youl Cho (Sungkyunkwan University, Korea) for his critical comments on the paper and support for an English edition.
References
- 1. Ferrero‐Miliani L, Nielsen OH, Andersen PS, Girardin SE. Chronic inflammation: importance of NOD2 and NALP3 in interleukin‐1β generation. Clin Exp Immunol 2007; 147:227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yi YS. Folate receptor‐targeted diagnostics and therapeutics for inflammatory diseases. Immune Netw 2016; 16:337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002; 20:197–216. [DOI] [PubMed] [Google Scholar]
- 4. Kayama H, Nishimura J, Takeda K. Regulation of intestinal homeostasis by innate immune cells. Immune Netw 2013; 13:227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140:805–20. [DOI] [PubMed] [Google Scholar]
- 6. Chen G, Shaw MH, Kim YG, Nunez G. NOD‐like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol 2009; 4:365–98. [DOI] [PubMed] [Google Scholar]
- 7. Song DH, Lee JO. Sensing of microbial molecular patterns by Toll‐like receptors. Immunol Rev 2012; 250:216–29. [DOI] [PubMed] [Google Scholar]
- 8. Yi YS, Son YJ, Ryou C, Sung GH, Kim JH, Cho JY. Functional roles of Syk in macrophage‐mediated inflammatory responses. Mediators Inflamm 2014; 2014:270302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu T, Yi YS, Yang Y, Oh J, Jeong D, Cho JY. The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediators Inflamm 2012; 2012:979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Byeon SE, Yi YS, Oh J, Yoo BC, Hong S, Cho JY. The role of Src kinase in macrophage‐mediated inflammatory responses. Mediators Inflamm 2012; 2012:512926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X et al Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998; 282:2085–8. [DOI] [PubMed] [Google Scholar]
- 12. Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y et al Cutting edge: toll‐like receptor 4 (TLR4)‐deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 1999; 162:3749–52. [PubMed] [Google Scholar]
- 13. Medzhitov R. Approaching the asymptote: 20 years later. Immunity 2009; 30:766–75. [DOI] [PubMed] [Google Scholar]
- 14. Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140:821–32. [DOI] [PubMed] [Google Scholar]
- 15. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014; 157:1013–22. [DOI] [PubMed] [Google Scholar]
- 16. Lee MS. Role of innate immunity in diabetes and metabolism: recent progress in the study of inflammasomes. Immune Netw 2011; 11:95–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang J, Zhao Y, Shao F. Non‐canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol 2015; 32:78–83. [DOI] [PubMed] [Google Scholar]
- 18. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016; 16:407–20. [DOI] [PubMed] [Google Scholar]
- 19. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J et al Non‐canonical inflammasome activation targets caspase‐11. Nature 2011; 479:117–21. [DOI] [PubMed] [Google Scholar]
- 20. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM et al Caspase‐11 increases susceptibility to Salmonella infection in the absence of caspase‐1. Nature 2012; 490:288–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 2013; 341:1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S et al Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013; 341:1246–9. [DOI] [PubMed] [Google Scholar]
- 23. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P et al Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514:187–92. [DOI] [PubMed] [Google Scholar]
- 24. Stowe I, Lee B, Kayagaki N. Caspase‐11: arming the guards against bacterial infection. Immunol Rev 2015; 265:75–84. [DOI] [PubMed] [Google Scholar]
- 25. Diamond CE, Khameneh HJ, Brough D, Mortellaro A. Novel perspectives on non‐canonical inflammasome activation. Immunotargets Ther 2015; 4:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP et al Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 2004; 430:213–8. [DOI] [PubMed] [Google Scholar]
- 27. Miao EA, Alpuche‐Aranda CM, Dors M, Clark AE, Bader MW, Miller SI et al Cytoplasmic flagellin activates caspase‐1 and secretion of interleukin 1β via Ipaf. Nat Immunol 2006; 7:569–75. [DOI] [PubMed] [Google Scholar]
- 28. Franchi L, Amer A, Body‐Malapel M, Kanneganti TD, Ozoren N, Jagirdar R et al Cytosolic flagellin requires Ipaf for activation of caspase‐1 and interleukin 1β in salmonella‐infected macrophages. Nat Immunol 2006; 7:576–82. [DOI] [PubMed] [Google Scholar]
- 29. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE et al Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 2010; 107:3076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H et al The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011; 477:596–600. [DOI] [PubMed] [Google Scholar]
- 31. Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011; 477:592–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rayamajhi M, Zak DE, Chavarria‐Smith J, Vance RE, Miao EA. Cutting edge: mouse NAIP1 detects the type III secretion system needle protein. J Immunol 2013; 191:3986–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella . J Exp Med 2010; 207:1745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P et al Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci USA 2014; 111:7403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐β . Mol Cell 2002; 10:417–26. [DOI] [PubMed] [Google Scholar]
- 36. Guey B, Bodnar M, Manie SN, Tardivel A, Petrilli V. Caspase‐1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc Natl Acad Sci USA 2014; 111:17254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti TD, Lamkanfi M. Activation of the NLRP1b inflammasome independently of ASC‐mediated caspase‐1 autoproteolysis and speck formation. Nat Commun 2014; 5:3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 2006; 38:240–4. [DOI] [PubMed] [Google Scholar]
- 39. Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z et al NLRP1‐dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol 2012; 189:2006–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 2012; 28:137–61. [DOI] [PubMed] [Google Scholar]
- 42. Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ et al The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008; 452:103–7. [DOI] [PubMed] [Google Scholar]
- 43. Hornung V, Ablasser A, Charrel‐Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al AIM2 recognizes cytosolic dsDNA and forms a caspase‐1‐activating inflammasome with ASC. Nature 2009; 458:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fernandes‐Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458:509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S et al HIN‐200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009; 323:1057–60. [DOI] [PubMed] [Google Scholar]
- 46. Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H et al An orthogonal proteomic‐genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 2009; 10:266–72. [DOI] [PubMed] [Google Scholar]
- 47. Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O'Rourke K et al Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis . Proc Natl Acad Sci USA 2010; 107:9771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R et al Prion‐like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014; 156:1207–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR et al Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell 2014; 156:1193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase‐1 autoproteolysis in pathogen‐induced cell death and cytokine processing. Cell Host Microbe 2010; 8:471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A. Human caspase‐4 and caspase‐5 regulate the one‐step non‐canonical inflammasome activation in monocytes. Nat Commun 2015; 6:8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM et al Human caspase‐4 mediates noncanonical inflammasome activation against gram‐negative bacterial pathogens. Proc Natl Acad Sci USA 2015; 112:6688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wacker MA, Teghanemt A, Weiss JP, Barker JH. High‐affinity caspase‐4 binding to LPS presented as high molecular mass aggregates or in outer membrane vesicles. Innate Immun 2017; 23:336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pellegrini C, Antonioli L, Lopez‐Castejon G, Blandizzi C, Fornai M. Canonical and non‐canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front Immunol 2017; 8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH et al Caspase‐11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 2012; 37:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM et al TRIF licenses caspase‐11‐dependent NLRP3 inflammasome activation by gram‐negative bacteria. Cell 2012; 150:606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE et al Caspase‐11 protects against bacteria that escape the vacuole. Science 2013; 339:975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA et al Caspase‐11 stimulates rapid flagellin‐independent pyroptosis in response to Legionella pneumophila . Proc Natl Acad Sci USA 2013; 110:1851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B et al Caspase‐11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog 2013; 9:e1003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z et al Toll or interleukin‐1 receptor (TIR) domain‐containing adaptor inducing interferon‐beta (TRIF)‐mediated caspase‐11 protease production integrates Toll‐like receptor 4 (TLR4) protein‐ and Nlrp3 inflammasome‐mediated host defense against enteropathogens. J Biol Chem 2012; 287:34474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shin HJ, Lee H, Park JD, Hyun HC, Sohn HO, Lee DW et al Kinetics of binding of LPS to recombinant CD14, TLR4, and MD‐2 proteins. Mol Cells 2007; 24:119–24. [PubMed] [Google Scholar]
- 62. Man SM, Karki R, Malireddi RK, Neale G, Vogel P, Yamamoto M et al The transcription factor IRF1 and guanylate‐binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 2015; 16:467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD et al Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase‐11 activation. Cell 2016; 165:1106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K et al Guanylate binding proteins promote caspase‐11‐dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci USA 2014; 111:6046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S et al Caspase‐11 activation requires lysis of pathogen‐containing vacuoles by IFN‐induced GTPases. Nature 2014; 509:366–70. [DOI] [PubMed] [Google Scholar]
- 66. Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Ruhl S et al Guanylate‐binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida . Nat Immunol 2015; 16:476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J et al IRGB10 liberates bacterial ligands for sensing by the AIM2 and caspase‐11‐NLRP3 inflammasomes. Cell 2016; 167():e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mariathasan S. ASC, Ipaf and Cryopyrin/Nalp3: bona fide intracellular adapters of the caspase‐1 inflammasome. Microbes Infect 2007; 9:664–71. [DOI] [PubMed] [Google Scholar]
- 69. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al Cutting edge: NF‐κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183:787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fujii T, Tamura M, Tanaka S, Kato Y, Yamamoto H, Mizushina Y et al Gasdermin D (Gsdmd) is dispensable for mouse intestinal epithelium development. Genesis 2008; 46:418–23. [DOI] [PubMed] [Google Scholar]
- 71. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin‐mediated programmed necrotic cell death. Trends Biochem Sci 2017; 42:245–54. [DOI] [PubMed] [Google Scholar]
- 72. Agard NJ, Maltby D, Wells JA. Inflammatory stimuli regulate caspase substrate profiles. Mol Cell Proteomics 2010; 9:880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S et al Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 2015; 526:666–71. [DOI] [PubMed] [Google Scholar]
- 74. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015; 526:660–5. [DOI] [PubMed] [Google Scholar]
- 75. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z et al Gasdermin D is an executor of pyroptosis and required for interleukin‐1β secretion. Cell Res 2015; 25:1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol 2017; 24:507–14 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J et al Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature 2016; 535:111–6. [DOI] [PubMed] [Google Scholar]
- 78. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N et al GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 2016; 113:7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H et al Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016; 535:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Aglietti RA, Dueber EC. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol 2017; 38:261–71. [DOI] [PubMed] [Google Scholar]
- 81. Shi J, Gao W, Shao F. Pyroptosis: gasdermin‐mediated programmed necrotic cell death. Trends Biochem Sci 2017; 42:245–54. [DOI] [PubMed] [Google Scholar]
- 82. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A et al Caspase‐1‐induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 2010; 11:1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C et al Noncanonical inflammasome activation of caspase‐4/caspase‐11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 2014; 16:249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J. Murine caspase‐11, an ICE‐interacting protease, is essential for the activation of ICE. Cell 1998; 92:501–9. [DOI] [PubMed] [Google Scholar]
- 85. Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C et al Mice deficient in IL‐1β‐converting enzyme are defective in production of mature IL‐1β and resistant to endotoxic shock. Cell 1995; 80:401–11. [DOI] [PubMed] [Google Scholar]
- 86. Crowley SM, Vallance BA, Knodler LA. Noncanonical inflammasomes: antimicrobial defense that does not play by the rules. Cell Microbiol 2017; 19: doi: 10.1111/cmi.12730. [DOI] [PubMed] [Google Scholar]
- 87. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 2017; 277:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shin SM, Choi DK, Jung K, Bae J, Kim JS, Park SW et al Antibody targeting intracellular oncogenic Ras mutants exerts anti‐tumour effects after systemic administration. Nat Commun 2017; 8:15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ercolini AM, Miller SD. The role of infections in autoimmune disease. Clin Exp Immunol 2009; 155:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]