

Summary
Crohn's disease (CD) is a chronic inflammatory condition of the human gastrointestinal tract whose aetiology remains largely unknown. Dysregulated adaptive immune responses and defective innate immunity both contribute to this process. In this study, we demonstrated that the interleukin (IL)‐17A+interferon (IFN)‐γ+ and IL‐22+IFN‐γ+ T cell subsets accumulated specifically in the inflamed terminal ileum of CD patients. These cells had higher expression of Ki‐67 and were active cytokine producers. In addition, their proportions within both the IL‐17A‐producer and IL‐22‐producer populations were increased significantly. These data suggest that IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets might represent the pathogenic T helper type 17 (Th17) population in the context of intestinal inflammation for CD patients. In the innate immunity compartment we detected a dramatic alteration of both phenotype and function of the intestinal innate lymphoid cells (ILCs), that play an important role in the maintenance of mucosal homeostasis. In the inflamed gut the frequency of the NKp44–CD117–ILC1s subset was increased significantly, while the frequency of NKp44+ILC3s was reduced. Furthermore, the frequency of human leucocyte antigen D‐related (HLA‐DR)‐expressing‐NKp44+ILC3s was also reduced significantly. Interestingly, the decrease in the NKp44+ILC3s population was associated with an increase of pathogenic IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the adaptive compartment. This might suggest a potential link between NKp44+ILC3s and the IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the terminal ileum of CD patients.
Keywords: Crohn's disease, innate lymphoid cells, T cells
Introduction
Crohn's disease is a chronic inflammatory disease of the human intestine, the pathogenesis of which is not completely understood 1. Accumulating evidence indicates that dysregulated mucosal immune responses towards the commensal microbiota contribute to this process and lead to tissue damage 2. In CD patients, various components of the gut immune system are dysregulated, especially the abnormal T cell activity along with excess cytokine production. This may be the result of disruption of a negative regulatory mechanism.
Traditionally, interferon (IFN)‐γ‐producing T helper type 1 (Th1) cells are believed to be the major effector cells in the pathogenesis of CD 3. Several lines of evidence also suggest that interleukin (IL)‐17A‐producing Th17 cells contribute to intestinal inflammation 4, 5, 6. However, reports from clinical trials were surprising, as the anti‐IFN‐γ antibody fontolizumab resulted in only minor improvement of inflammation in CD patients 7, and the anti‐IL‐17A antibody secukinumab was less effective than placebo in patients with moderate to severe CD 8. However, the trial with the anti‐IL‐12/23‐p40 antibody ustekinumab, that targeted cytokines that drive both Th1 and Th17 differentiation, reported significantly increased response rates in patients with moderate to severe CD 9. These differential results from clinical trials of CD patients demonstrate that a strategy that targets both the Th1 and Th17 pathways might be more promising.
Innate lymphoid cells (ILCs) are newly identified innate effector cells that are prevalent in mucosal tissues, such as the gastrointestinal tract. They are divided into three groups (ILC1s, ILC2s and ILC3s), based on their cytokine production and transcription factor expression. In the normal gut, ILC3s are the most abundant ILC subset at steady state (approximately 60–75% of total ILCs) and play essential roles in host defence, regulation of inflammation, tissue repair and maintenance of epithelial barrier function 10. Recent research has illustrated that ILC3s can regulate adaptive immune responses, particularly those of CD4+ T cells. Qiu et al. reported that ILC3s could inhibit T cell‐mediated intestinal inflammation through aryl hydrocarbon receptor signalling and regulation of microflora 11. Hepworth et al. showed that, in their mouse model, major histocompatibility complex class II (MHC‐II)+ILC3s could present antigens to CD4+ T cells in the absence of co‐stimulatory signals, and this interaction limited pathological CD4+ T cell response(s) to intestinal commensal bacteria 12. Additional work showed that human intestinal ILC3s also expressed MHC‐II 13; thus, it seems likely that human MHC‐II+ILC3s have direct cellular contact with effector T cells and regulate their response. However, certain fundamental questions remain elusive, such as which subset of T cells are and where the interaction occurs. Interestingly, several studies have also demonstrated that in the context of chronic intestinal inflammation of CD patients, there is a phenotype change of ILCs 14, 15, 16. This phenotype change possibly results in immunopathology.
Thus, in this study, we evaluated the alteration of both phenotype and function of mucosal ILC3s and the composition and features of effector T cells that produced IFN‐γ, IL‐17A and/or IL‐22 in the inflamed terminal ileum of CD patients. This effort will help us to explore a potential missing link between innate immunity and adaptive immunity and have a clearer understanding for the pathogenesis of CD.
Materials and methods
Subjects
Fresh terminal ileum surgical tissues were obtained from UF Health Shands Hospital (Gainesville, FL, USA), with the assistance of the UF Clinical and Translational Science Institute. All experiments were performed according to University of Florida Institutional Review Board‐approved protocol, with written informed consent from all subjects. Twenty CD patients who responded poorly to various medications, developed intestinal complications and needed surgery therapy were included into this study. Their clinical characteristics are listed in Table 1.
Table 1.
The clinical characteristics of Crohn's disease (CD) patients
| Subjects | Gender | Age (years) | Complication(s) in the gut | Medication |
|---|---|---|---|---|
| 1 | M | 25 | Extensive ulceration | Anti‐TNF‐α adalimumab |
| 2 | M | 58 | Ulceration | Anti‐TNF‐α infliximab |
| 3 | M | 28 | Fistulae | Anti‐TNF‐α infliximab |
| 4 | M | 37 | Fissure | Anti‐TNF‐α infliximab and adalimumab |
| 5 | M | 25 | No | Anti‐IL‐12/23 p40 ustekinumab |
| 6 | F | 31 | Adenocarcinoma | Anti‐α4β7 vedolizumab |
| 7 | F | 46 | Obstruction and narrowing | Adalimumab and azathioprine |
| 8 | F | 60 | Mural fibrosis and adhesions | Anti‐α4β7 vedolizumab |
| 9 | M | 23 | Fissure | Anti‐IL‐12/23 p40 ustekinumab |
| 10 | F | 26 | Fistulae | Anti‐IL‐12/23 p40 ustekinumab |
| 11 | F | 57 | Submucosal oedema | Anti‐α4β7 vedolizumab |
| 12 | M | 42 | Fistulae | Anti‐TNF‐α infliximab |
| 13 | F | 35 | Severe stricture | Anti‐IL‐12/23 p40 ustekinumab |
| 14 | M | 37 | Stricture and fistulae | Anti‐IL‐12/23 p40 ustekinumab |
| 15 | F | 36 | Fistulae, pelvic abscess and strictures | Anti‐IL‐12/23 p40 ustekinumab |
| 16 | F | 20 | Non‐specific bowel wall thickening and oedema | Natalizumab anti‐α4 |
| 17 | F | 40 | Perforation and stricture | Anti‐TNF‐α infliximab |
| 18 | F | 35 | Stricture | Mercaptopurine and vedolizumab |
| 19 | M | 65 | Perforation, fistulae with intramuscular abscess | Infliximab and azathioprine |
| 20 | F | 28 | Extensive ulceration and mural fibrosis | Anti‐IL‐12/23 p40 ustekinumab |
TNF = tumour necrosis factor; IL = interleukin.
Tissue collection and processing
After the surgery, pathologists from the UF Health Shands Hospital performed gross examination of the surgical samples from CD patients, cut one piece of macroscopically unaffected terminal ileum (if available) or affected terminal ileum and passed the tissue to us. The range of collected tissue amounts was between 2·0 and 8·0 g. The time of tissue harvest after surgery is 1 h, on average. Harvested surgical ileum tissue was placed into stripping buffer [Mg2+ and Ca2+‐free Hanks's balanced salt solution (HBSS) media containing 100 U/ml penicillin, 100 µg/ml streptomycin, 25 µg/ml gentamycin and 0·5 mM dithiothreitol (DTT)], delivered to the laboratory on ice. The outer fat tissue was removed immediately, 0·3 g of tissue was put into a 1·5‐ml collection tube with 1 ml of RNAlater buffer (Thermo Fisher Scientific, Grand Island, NY, USA) for RNA extraction and 0·3 g of tissue was fixed in 10% neutral‐buffered formalin (Thermo Fisher Scientific, Grand Island, NY, USA) for histology evaluation. The residual tissue was cut into small strips, placed into 25‐ml stripping buffer (as described above) and washed twice at 37°C in the shaker for 10 min at a constant speed of 8 g. The supernatant was discarded. The remaining tissue was then minced further with a sterile surgical scalpel and digested in 25 ml digestion buffer [Dulbecco's modified Eagle's medium (DMEM) without sodium pyruvate supplemented with 75 U/ml collagenase type XI, 20 µg/ml Dispase neutral protease II, 500 U/ml DNase, 0·5mM DTT and 1% fetal bovine serum (FBS)] at 37°C in the shaker for 15 min at a constant speed of 8 g. The tissue was filtered through a 70‐µm cell strainer, again dissociated mechanically on top of the strainer, and digested in 25‐ml digestion buffer for another 15 min. The supernatant was collected and centrifuged at 287 g for 8 min. All cells were cryopreserved slowly in cell freezing medium (Gibco, Grand Island, NY, USA) and transferred to liquid nitrogen for future side‐by‐side analysis.
Flow cytometry
After thawing, single‐cell suspensions were incubated with LIVE/DEAD™ Fixable Yellow dye (Thermo Fisher Scientific) on ice for 30 min, then stained with antibodies to the following markers [all antibodies were from BioLegend (San Diego, CA, USA) unless stated otherwise]: anti‐human Lineage Cocktail 3 (CD3, clone SK7; CD19, clone SJ25C1; CD20, clone L27 and CD14, clone MφP9; BD Bioscience), anti‐CD45 (clone HI30; Thermo Fisher Scientific), anti‐CD56 (clone HCD56), anti‐CD117 (clone 104D2; eBioscience, San Diego, CA, USA), anti‐CD127 (clone eBioRDR5; eBioscience), anti‐NKp44 (clone P44–8), anti‐CRTH2 (clone BM16), anti‐CCR6 (clone G034E3), anti‐human leucocyte antigen D‐related (HLA‐DR) (clone LN3), anti‐CD80 (clone L307.4) and anti‐CD86 (clone 2331). For intracellular staining, cells were fixed and permeabilized using the FIX & PERM® Cell Permeabilization Kit (Thermo Fisher Scientific) and stained with anti‐Ki‐67(clone Ki‐67). For cytokine production analysis, cells were stimulated with 200 ng/ml phorbol 12‐myristate 13‐acetate (Sigma‐Aldrich, St Louis, MO, USA), 1 µg/ml ionomycin (Sigma‐Aldrich) and 1 µl/ml monensin (Biolegend, San Diego, CA, USA) for 4·5 h at 37°C in a 5% CO2 incubator, stained with surface markers, fixed and permeabilized as described above, and stained with anti‐IFN‐γ (clone 4S.B3), anti‐IL‐17A (clone BL168) or anti‐IL‐22 (clone 22URTI). Data were acquired on a LSRII (BD Biosciences) in the Cytometry Core at the University of Florida, and analysed using FlowJo software version 9.8.1 (TreeStar Inc., Ashland, OR, USA).
Reverse transcription–polymerase chain reaction (RT–PCR)
A 0·3‐g sample of terminal ileum tissue was minced into small pieces and disrupted with the tissue homogenizer. Total RNA was isolated using QIAzol Lysis reagent (Qiagen, Valencia, CA, USA), following the manufacturer's instructions. The SuperScript® VILO cDNA Synthesis Kit (Thermo Fisher Scientific) was used for cDNA synthesis. PCR was performed using an ABI PRISM StepOnePlus Real time PCR System (Thermo Fisher Scientific) in a 20‐ml volume containing the cDNA, 2 × TaqMan® Gene Expression Master Mix, nuclease‐free water and 1× Taqman Gene Expression Assay for target genes including T‐box transcription factor (TBX21), RAR‐related orphan receptor C (RORC), IFN‐γ, IL‐17A, IL‐22, IL‐12, IL‐23 and tumour necrosis factor (TNF)‐α (Thermo Fisher Scientific). Reactions were run in triplicate in three independent experiments. The mean of housekeeping gene glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as an internal control to normalize target gene expression levels.
Histology
Fresh surgical intestinal tissue samples were fixed in formalin for 20 h at 4°C, embedded in paraffin, sectioned and stained with haematoxylin and eosin for inflammation evaluation.
Statistical analyses
Statistical significance was determined using the Mann–Whitney U‐test and Student's t‐test, which were performed using GraphPad Prism version 6 software (GraphPad Software, La Jolla, CA, USA). As shown in the figures, the asterisk (*) levels indicate significant differences (*P‐value < 0·05, **P < 0·01 and ***P < 0·001).
Results
Dysregulated inflammatory gene expression in the inflamed terminal ileum of CD patients
We collected samples of inflamed and unaffected (when available) terminal ileum resections from 20 adult CD patients who failed medical treatment and developed intestinal complications (Table 1). When compared to the unaffected ileums, the inflamed ileums presented with histological signs of intestinal inflammation, including epithelial monolayer damage, crypt architectural distortion and increased lymphocyte infiltration in the lamina propria (Fig. 1a). As a further assessment of the inflammation, we compared the expression patterns of inflammatory genes within these tissues. The mRNA gene expression of proinflammatory cytokines IFN‐γ, IL‐17A, IL‐22, IL‐1β, IL‐23A, IL‐12p35 and TNF‐α, as well as transcription factor TBX21, were increased significantly in the inflamed terminal ileum of CD patients (Fig. 1b). There was no difference regarding the mRNA gene expression of transcription factor RORC (Fig. 1b). Moreover, the mRNA expression ratios of TBX21/RORC and IFN‐γ/IL‐17A were increased significantly in the inflamed tissues, while there was a trend towards elevations in the ratio of IFN‐γ/IL‐22 (Fig. 1c).
Figure 1.
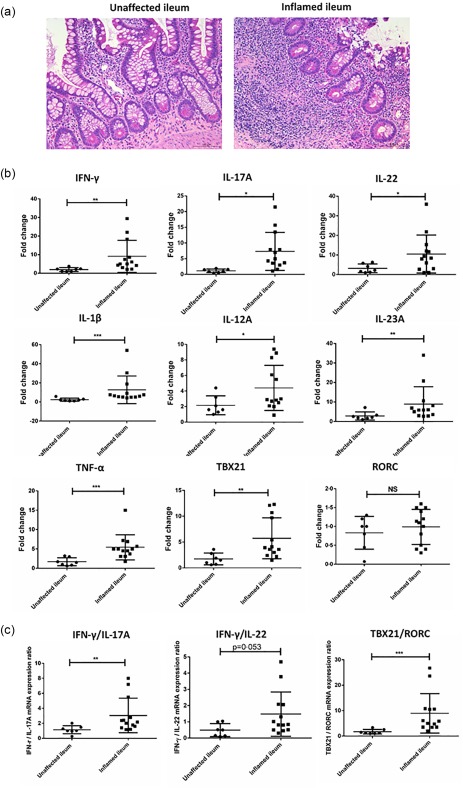
Dysregulated inflammatory gene expression in the inflamed terminal ileum of Crohn's disease (CD) patients. (a) Representative haematoxylin and eosin (H&E) staining of the unaffected and inflamed terminal ileum of our CD patients. Scale bar: 100 µm. (b) Comparison of interferon (IFN)‐γ, interleukin (IL)‐17A, IL‐22, IL‐1β, tumour necrosis factor (TNF)‐α, IL‐23A and IL‐12p35 as well as T‐box transcription factor (TBX21) and RAR‐related orphan receptor C (RORC) mRNA genes expression in the unaffected (n = 7) and inflamed (n = 13) terminal ileum of the CD patients by reverse transcription–polymerase chain reaction (RT–PCR). (c) Comparison of TBX21/RORC, IFN‐γ/IL‐17A, IFN‐γ/IL‐22 mRNA expression ratio in the unaffected (n = 7) and inflamed (n = 13) terminal ileum of the CD patients by RT–PCR. Statistical analyses were performed using a two‐tailed Mann–Whitney U‐test. *P < 0·05; **P < 0·01.
Enrichment of IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the inflamed gut
Lamina propria mononuclear cells (LPMC) were also isolated from the surgical tissues and the profile of each T cell subset was investigated. It is important to note that our anti‐IL‐17A and anti‐IL‐22 antibodies were in the same channel. Therefore, when we performed intracellular cytokine staining, only the combinations of anti‐IFN‐γ and IL‐17A antibodies or anti‐IFN‐γ and IL‐22 antibodies were used. In the CD45+lineage+ compartment, the total number of isolated living lymphocytes per gram of tissue was significantly higher in the inflamed terminal ileum (Fig. 2a). Consistent with previous studies, the frequencies of IL‐17A– IFN‐γ+ and IL‐22–IFN‐γ+ T cell subsets within the CD45+lineage+ population were increased remarkably, which implicated them as active mediators of intestinal inflammation (Fig. 2b,c) 5, 17. Surprisingly, the frequency of the IL‐17A+IFN‐γ– T cell subset within CD45+lineage+LPMC was comparable between inflamed tissues and unaffected tissues (Fig. 2b,c). A trend in reduced frequency of the IL‐22+IFN‐γ– T cell subset was observed in the inflamed terminal ileum of CD patients (Fig. 2b,c). Interestingly, the frequencies of IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets were up‐regulated significantly in the inflamed tissues (Fig. 2b,c). Furthermore, the fraction of the IL‐17A+IFN‐γ+ T cell subset within IL‐17A‐producers, as well as the IL‐22+IFN‐γ+ T cell subset within IL‐22‐producers, were both up‐regulated significantly in inflamed tissues. In a small number of patients, the frequency of the IL‐22+IFN‐γ+ T cell subset comprised almost 80% of the IL‐22 producers (Fig. 2b–d). This might be a result of the possible preferential proliferation of IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets, as we noticed that in the inflamed tissues both had relatively higher Ki‐67 expression compared to IL‐17A+IFN‐γ– and IL‐22+IFN‐γ– T cell subsets (Fig. 3a,b). In addition, we evaluated the expression of homing receptor CCR6 on these populations. CCR6 is important for Th17 cell migration in the gut and is part of the Th17 cell signature 18. IL‐17A+IFN‐γ– and IL‐22+IFN‐γ– T cell subsets had the highest CCR6 expression, while IL‐17A–IFN‐γ+ and IL‐22–IFN‐γ+ T cell subsets were negative for this marker. The IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets had intermediate CCR6 expression levels (Fig. 3c,d).
Figure 2.
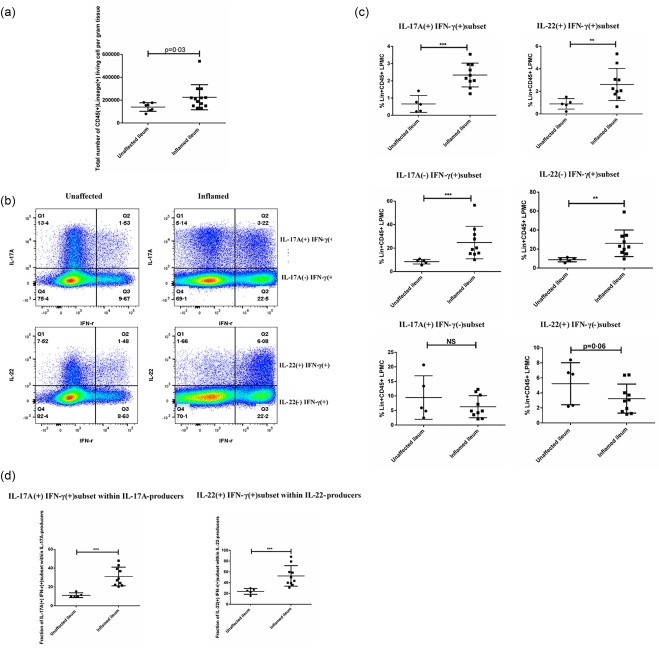
Enrichment of interleukin (IL)‐17A+interferon (IFN)‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the inflamed terminal ileum of Crohn's disease (CD) patients. (a) Total number of isolated living lymphocytes in the CD45+lineage+ lamina propria mononuclear cell (LPMC) compartment per gram tissue from both unaffected (n = 7) and inflamed groups (n = 13). (b) Representative flow‐plot of the frequency of IFN‐γ+IL‐17A+, IFN‐γ+IL‐17A–, IFN‐γ+IL‐22+, IFN‐γ+IL‐22–, IFN‐γ–IL‐17A+ and IFN‐γ– IL‐22+ T cell subsets within CD45+lineage+LPMC compartment from both unaffected and inflamed terminal ileum. (c) Quantification of the frequency of various T cell subsets within CD45+lineage+LPMC compartment between unaffected tissue (n = 5) and inflamed tissue (n = 10). (d) Comparison of the fraction of IL‐17A+IFN‐γ+ T cell subset within IL‐17A‐producers and IL‐22+IFN‐γ+ T cell subset within IL‐22–producers between unaffected (n = 5) and inflamed terminal ileum (n = 10). Statistical analyses were performed using a two‐tailed Mann–Whitney U‐test. *P < 0·05; **P < 0·01.
Figure 3.
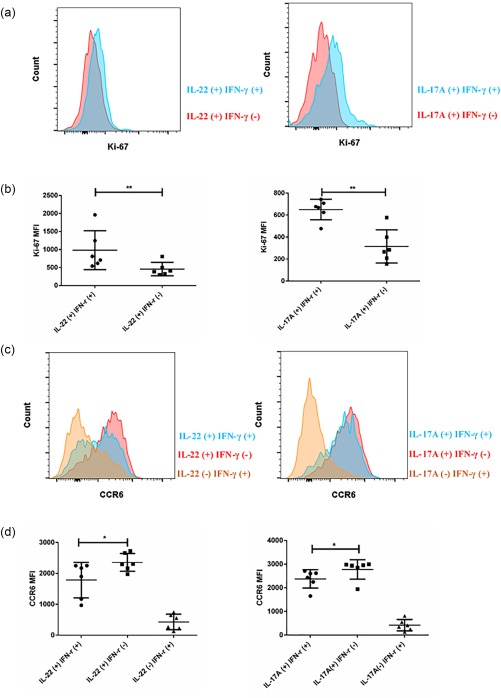
Interleukin (IL)‐17A+interferon (IFN)‐γ + and IL‐22+IFN‐γ+ T cell subsets had relatively higher Ki‐67 expression. (a) Representative histogram of Ki‐67 expression between IL‐17A+IFN‐γ+ (blue) and IL‐17A+IFN‐γ– T cell subset (red), IL‐22+IFN‐γ+ T cell subsets (blue) and IL‐22+IFN‐γ– T cell subsets (red) in the inflamed tissues. (b) Quantification of Ki‐67 expression between IL‐17A+IFN‐γ+ (blue) and IL‐17A+IFN‐γ– T cell subset (red), IL‐22+IFN‐γ+ T cell subsets (blue) and IL‐22+IFN‐γ– T cell subsets (red) (n = 6) in the inflamed tissues. (c) Representative histogram of chemokine receptor type 6 (CCR6) expression among various T cell subsets in the inflamed tissues. (d) Quantification of CCR6 expression among various T cell subsets in the inflamed tissues (n = 6). Statistical analyses were performed using Student's t‐test. *P < 0·05; **P < 0·01.
The alteration of ILC3s phenotype and function in the inflamed terminal ileum of CD patients
Emerging evidence demonstrates that ILCs are important innate effector and regulatory cells in tissue defence, along with the repair and maintenance of homeostasis, especially along the mucosal surfaces 19. In the context of intestinal inflammation in CD patients, Bernink et al. showed previously that IFN‐γ‐producing ILC1s accumulated in inflamed mucosal tissues 14. However, their study included only seven patients. A relatively large cohort of CD patients would be helpful to evaluate and confirm further the reported changes of ILC phenotype and function in the gut of CD patients.
In the present study, we recruited 20 patients. The gating strategy used for our analysis is outlined in Fig. 4a. The numbers of total ILCs isolated per gram of tissue were comparable between groups (Fig. 4c). In the unaffected terminal ileum, NKp44+ILC3s were the dominant ILC subset, and comprised approximately 60% of the total ILCs (Fig. 4b,c). These cells were potent IL‐22 producers, but did not produce IL‐17A and IFN‐γ (Fig. 4d). However, in the inflamed tissue, the frequency of NKp44+ILC3s was reduced and the remaining ILC3s produced less IL‐22 (Fig. 4d,e). At the same time, we noticed that NKp44+ILC3s from the inflamed terminal ileum were also able to produce low amounts of IFN‐γ, but not IL‐17A (Fig. 4d–f). Additionally, the frequency of NKp44–CD117–ILC1s increased in the inflamed tissue (Fig. 4b,c), along with the elevated frequency of IFN‐γ‐producing cells within ILC1s. We also noticed that ILC1s from both unaffected tissues and inflamed tissues did not produce IL‐22 (Fig. 4g–i).
Figure 4.
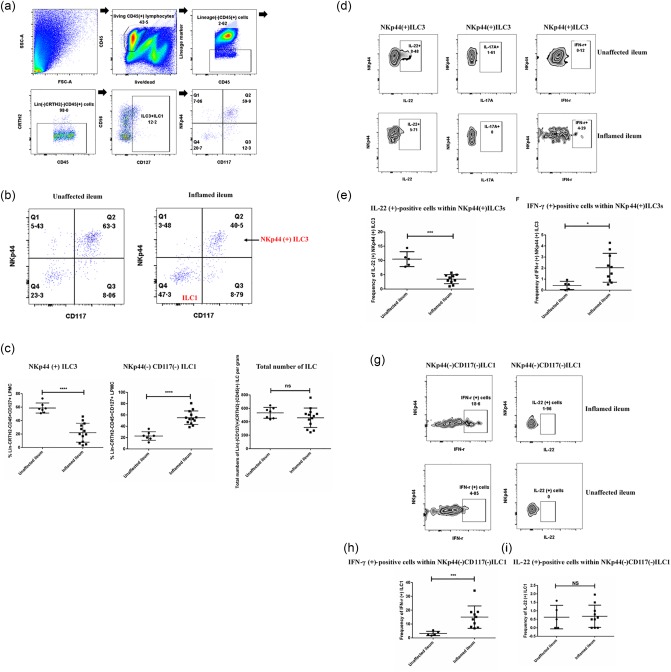
The alteration of ILC3s' phenotype and function in the inflamed terminal ileum of Crohn's disease (CD) patients. (a) Gating strategy for the identification of human intestinal innate lymphoid cell (ILC) subsets in the terminal ileum of CD patients. (b) Representative flow plot of the frequency of NKp44+ILC3s and NKp44–CD117– ILC1s within CD45+CD127+CRTH2– lineage– lamina propria mononuclear cells from the unaffected ileum and inflamed ileum. (c) Quantification of the frequency of NKp44+ILC3s and NKp44–CD117–ILC1s from the unaffected ileum (n = 7) and inflamed ileum (n = 13) as well as the total number of isolated ILCs per gram tissue between unaffected group and inflamed group. (d) Representative flow plot of IL‐22, IL‐17A and IFN‐γ expression from NKp44+ILC3s isolated from both inflamed and unaffected human terminal ileum. (e,f) Quantification of the frequency of IFN‐γ‐expressing‐NKp44+ILC3s and IL‐22‐expressing NKp44+ILC3s between the unaffected tissues (n = 5) and inflamed tissues (n = 10). (g) Representative flow‐plot of IL‐22 and IFN‐γ expression from NKp44–CD117– ILC1s isolated from both inflamed and unaffected human terminal ileum. (h,i) Quantification of the frequency of IFN‐γ‐expressing‐ILC1s and IL‐22‐expressing ILC1s between the unaffected tissues (n = 5) and inflamed tissues (n = 10). Statistical analyses were performed using a two‐tailed Mann–Whitney U‐test. *P < 0·05; **P < 0·01.
NKp44+ILC3s were correlated negatively with the enrichment of IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets
In addition to their role in the first line of defence against invading pathogens through rapid cytokine production, it has been suggested that ILCs can also influence adaptive immune responses, particularly those of effector T cells 20. In mouse models, it was shown that ILC3‐intrinsic MHC‐II expression restrained pathogenic CD4+ T cell responses specific for commensal gastrointestinal bacteria 12. In our cohort, the frequencies of NKp44+ILC3s were correlated negatively with the enrichment of IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the inflamed tissue (Fig. 5a). Furthermore, a fraction of NKp44+ILC3s expressed HLA‐DR, which is required for antigen‐specific interactions with effector T cells. Consistent with other studies, the NKp44+ILC3s did not express the two classical co‐stimulatory molecules CD80 and CD86 (Fig. 5b,c) 12, 13. Interestingly, the frequency of HLA‐DR‐expressing ILC3s was decreased significantly in the inflamed tissue compared to the unaffected area (Fig. 5c,d). Together these data may suggest that NKp44+ILC3s were less capable of mediating T cell responses under the inflamed conditions.
Figure 5.
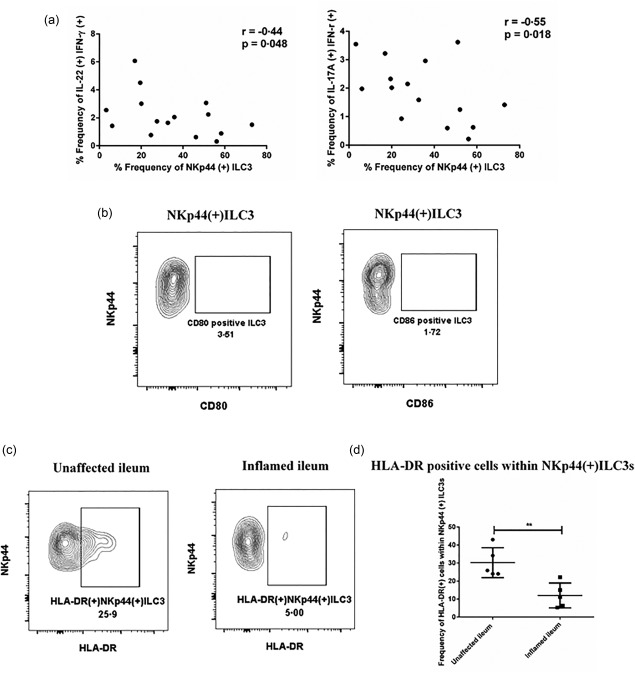
NKp44+ innate lymphoid cells (ILCs) were correlated negatively with the enrichment of interleukin (IL)‐17A+interferon (IFN)‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the terminal ileum of Crohn's disease (CD) patients. (a) Correlation was tested by Spearman's rank test between the frequency of NKp44+ILC3s and the frequency of IL‐17A+IFN‐γ+ or IL‐22+IFN‐γ+ T cell subsets in the terminal ileum of CD patients (n = 15). (b) Representative flow‐plot of CD80 and CD86 expression from NKp44+ILC3s. (c) Representative flow‐plot of the frequency of human leucocyte antigen D‐related (HLA‐DR)‐expressing cells within NKp44+ILC3s from the unaffected and inflamed terminal ileum tissue. (d) Quantification of the frequency change of HLA‐DR‐expressing cells within NKp44+ILC3s from the unaffected group (n = 5) and inflamed group (n = 5). Statistical analyses were performed using Student's t‐test. *P < 0·.05. **P < 0·01.
Discussion
In this study, we observed the accumulation of IL‐17A+IFN‐γ+ and IL‐22+FN‐γ+ T cell subsets in the inflamed terminal ileum of CD patients in the presence of high concentrations of cytokines IL‐12, IL‐1β and IL‐23. Surrounded by the same inflammatory microenvironment, NKp44+ILC3s not only showed reduced numbers, but also demonstrated less IL‐22 production and acquired IFN‐γ‐producing ability. Interestingly, we noticed a negative correlation between the frequencies of NKp44+ILC3s and the enrichment of IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the inflamed tissue. Moreover, we showed that a fraction of NKp44+ILC3s expressed HLA‐DR, but had low or undetectable expression levels of the two classical co‐stimulatory molecules CD80 and CD86. The frequency of HLA‐DR‐expressing NKp44+ILC3s was reduced significantly in the inflamed area, which might indicate a possible link between NKp44+ILC3s and the IL‐17A+IFN‐γ+, IL‐22+IFN‐γ+ T cell subsets in the gut of CD patients.
In the adaptive immunity compartment, it is well accepted that the IFN‐γ‐producing T cells contribute to the intestinal inflammation in CD patients. However, it has been somewhat controversial regarding the role of IL‐17A‐producing cells in this process 21, 22. Here, our data provide further evidence for a preferential skewing of T cells towards a dual IL‐17A+IFN‐γ+ or IL‐22+IFN‐γ+ phenotype during active intestinal inflammation in CD patients. Our study also confirmed that the proinflammatory cytokines IL‐12, IL‐23 and IL‐1β, which play key roles in the skewing of T cells, were expressed highly in the inflamed tissues 23, 24, 25. This could partially explain the positive results from the latest Phase 3 clinical trial of ustekinumab (the monoclonal antibody to the IL‐12/IL‐23 p40 subunit) among patients with moderately to severely active CD 26. In the Phase 2b trial mentioned earlier, ustekinumab treatment showed a benefit in terms of clinical response but not remission 9. In this 2016 trial, subcutaneous ustekinumab maintained remission in patients 26.
Regarding the polyfunctional T cells, there are some indications that they are superior in initiating active immune response and inflammation. Seder et al. argued that polyfunctional T cells were associated with enhanced protection and were optimized for effector function for vaccine design 27. Kannanqanat et al. showed that multiple‐cytokine‐producing anti‐viral CD4 T cells were functionally superior to single‐cytokine‐producing cells 28. Boyd et al. reported that pathogen‐specific T cell polyfunctionality correlated with T cell efficacy and immune protection 29. In the context of the intestinal inflammation of CD patients, their polyfunctionality could be detrimental. Globig et al. showed that the IL‐17A+IFN‐γ+ co‐producing CD4+ T cells with high expressions of T‐bet, CD26 and IL‐22 were enriched specifically in the inflamed mucosal tissues 30. Dunn et al. observed higher proportions of IL‐17A+IL‐22+ and IL‐22+IFN‐γ+CD4+ T cells in the inflamed colon of CD patients 31. Similarly, we showed the enrichment of IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the inflamed terminal ileum. One of the limitations in our study was that our anti‐IL‐17A and anti‐IL‐22 antibodies are in the same channel, we did not stain IL‐17A, IFN‐γ and IL‐22 together, and could not measure the frequencies of the IL‐17A+IL‐22+ T cell subset as well as the IL‐17A+IL‐22+IFN‐γ+ T cell subset in the inflamed tissues.
In the innate immunity compartment, our study supports results from previous investigations that reported the phenotype changes of ILCs in the inflamed gut of CD patients 14, 15, 32, 33, 34. Additionally, we observed an interesting functionality change of NKp44+ILC3s. In the unaffected tissues, NKp44+ILC3s produced only IL‐22 and did not produce IL‐17A or IFN‐γ. However, under inflamed conditions, they produced less IL‐22 and demonstrated IFN‐γ‐producing ability. Even though the level of IFN‐γ expression was still low, it was detectable. These IFN‐γ‐producing NKp44+ILC3s in the inflamed tissue might represent an intermediate population. This functional alteration of NKp44+ILC3s provides evidence of type 1 skewing of innate lymphocytes during CD, which was supported further by the increased frequency of IFN‐γ‐producing ILC1s in the inflamed tissues. It is important to note that the lineage marker antibody in our study for ILC phenotyping is limited in that it contains only CD3, CD14, CD19 and CD20, which makes it possible that there was potential contamination in our ILC1s by other cells.
Aside from their effector functions, multiple lines of evidence suggest a role for ILC3s in the regulation of adaptive immune responses, especially the CD4+ T cell response towards commensal antigens in the gut 12, 35, 36, 37. Our study demonstrated a direct correlation between the changes in NKp44+ILC3s and IL‐17A+IFN‐γ+ or IL‐22+IFN‐γ+ T cell subsets in the gut of CD patients. Additionally, the frequency of HLA‐DR‐expressing NKp44+ILC3s was decreased in the inflamed tissue. These data might indicate a potential regulatory role for HLA‐DR‐expressing NKp44+ILC3s for the induction of IL‐17A+IFN‐γ+ or IL‐22+IFN‐γ+ T cell subsets in CD. Additional studies are needed to investigate further the potential interaction as well as the mechanism between ILC3s and polyfunctional T cells in the human gut.
Moreover, until recently there was still limited information concerning the precise location of ILCs within tissues 38. Withers et al. showed that RORγt+CD3–ILC3s resided in the interfollicular region and the interface between B and T cell zones in the lymph node 36. Those are the specific regions that lymphocytes traverse during various stages of adaptive immune responses. Mackley et al. revealed further that constitutive trafficking of RORγt+ILC3s from the intestine to the draining mesenteric lymph nodes was CCR7‐dependent 37. Imaging studies of ILC3s' location and traffic in the human intestine are needed urgently and would be extremely helpful in the understanding of the potential cellular interactions between ILC3s and other mucosal immune cells.
In conclusion, our data highlight the relative pathogenic role of IL‐17A+IFN‐γ+ and IL‐22+IFN‐γ+ T cell subsets in the context of intestinal inflammation in CD patients. In the innate compartment, NKp44+ILC3s not only displayed frequency reduction, but also less IL‐22‐producing capacity as well as reduced HLA‐DR surface expression. The aberrant innate immunity possibly contributes to the dysregulated adaptive immune responses in the intestine of CD patients. Clinical strategies that target the NKp44+ILC3s subset in the human gut might deserve more attention.
Author contributions
J. L., A. L. D., S. M. W. and S. C. G. conceived and designed experiments. J. L., Y. T. and D. B. performed the experiments. A. I., M. J. C. and S. A. T. provided surgical samples from CD patients and their clinical feature analysis. J. L., S. M. W. and S. C. G. analysed the data and drafted the manuscript. A. L. D., A. I., M. J. C. and S. A. T. provided intellectual expertise in flow cytometry, innate, adaptive immunity and clinical immunology. All authors reviewed and edited the manuscript.
Disclosure
The authors declare no conflicts of interest in this study. Sarah Glover DO is a consultant for AbbVie, Janssen and Takeda. Sarah Glover DO has received grant support from AbbVie, Bristol Myers Squibb, Celgene, Gilead, Janssen, Genentech, Millennium, Pfizer, Receptos, Takeda and UCB.
Acknowledgements
This work was supported by the Gatorade Trust through funds distributed by the University of Florida, Department of Medicine. We also would thank the Cytometry Core of University of Florida for the help of flow cytometry and CTSI of University of Florida for the help of collecting surgical tissues.
References
- 1. Baumgart DC, Sandborn WJ. Crohn's disease. Lancet 2012; 380:1590–605. [DOI] [PubMed] [Google Scholar]
- 2. Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol 2006; 3:390–407. [DOI] [PubMed] [Google Scholar]
- 3. Shanahan F. Crohn's disease. Lancet 2002; 359:62–9. [DOI] [PubMed] [Google Scholar]
- 4. Fujino S, Andoh A, Bamba S et al Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003; 52:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kobayashi T, Okamoto S, Hisamatsu T et al IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut 2008; 57:1682–9. [DOI] [PubMed] [Google Scholar]
- 6. Rovedatti L, Kudo T, Biancheri P et al Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut 2009; 58:1629–36. [DOI] [PubMed] [Google Scholar]
- 7. Reinisch W, de Villiers W, Bene L et al Fontolizumab in moderate to severe Crohn's disease: a phase 2, randomized, double‐blind, placebo‐controlled, multiple‐dose study. Inflamm Bowel Dis 2010; 16:233–42. [DOI] [PubMed] [Google Scholar]
- 8. Hueber W, Sands BE, Lewitzky S et al Secukinumab, a human anti‐IL‐17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double‐blind placebo‐controlled trial. Gut 2012; 61:1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sandborn WJ, Gasink C, Gao LL et al Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med 2012; 367:1519–28. [DOI] [PubMed] [Google Scholar]
- 10. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol 2016; 17:765–74. [DOI] [PubMed] [Google Scholar]
- 11. Qiu J, Guo X, Chen ZM et al Group 3 innate lymphoid cells inhibit T‐cell‐mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 2013; 39:386–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hepworth MR, Fung TC, Masur SH et al Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria‐specific CD4(+) T cells. Science 2015; 348:1031–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hepworth MR, Monticelli LA, Fung TC et al Innate lymphoid cells regulate CD4+ T‐cell responses to intestinal commensal bacteria. Nature 2013; 498:113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bernink JH, Peters CP, Munneke M et al Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 2013; 14:221–9. [DOI] [PubMed] [Google Scholar]
- 15. Takayama T, Kamada N, Chinen H et al Imbalance of NKp44(+)NKp46(–) and NKp44(–)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn's disease. Gastroenterology 2010; 139:882–92, 892.e1–3. [DOI] [PubMed] [Google Scholar]
- 16. Li J, Doty AL, Iqbal A, Glover SC. The differential frequency of Lineage(–)CRTH2(–)CD45(+)NKp44(–)CD117(–)CD127(+)ILC subset in the inflamed terminal ileum of patients with Crohn's disease. Cell Immunol 2016; 304–305:63–8. [DOI] [PubMed] [Google Scholar]
- 17. Fuss IJ, Neurath M, Boirivant M et al Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN‐gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL‐5. J Immunol 1996; 157:1261–70. [PubMed] [Google Scholar]
- 18. Ito T, Carson WF 4th, Cavassani KA, Connett JM, Kunkel SL. CCR6 as a mediator of immunity in the lung and gut. Exp Cell Res 2011; 317:613–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 2015; 21:698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. von Burg N, Turchinovich G, Finke D. Maintenance of immune homeostasis through ILC/T cell interactions. Front Immunol 2015; 6:416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feng T, Qin H, Wang L, Benveniste EN, Elson CO, Cong Y. Th17 cells induce colitis and promote Th1 cell responses through IL‐17 induction of innate IL‐12 and IL‐23 production. J Immunol 2011; 186:6313–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol 2013; 8:477–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol 2014; 14:329–42. [DOI] [PubMed] [Google Scholar]
- 24. Bettelli E, Kuchroo VK. IL‐12‐ and IL‐23‐induced T helper cell subsets: birds of the same feather flock together. J Exp Med 2005; 201:169–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neurath MF. IL‐23: a master regulator in Crohn disease. Nat Med 2007; 13:26–8. [DOI] [PubMed] [Google Scholar]
- 26. Feagan BG, Sandborn WJ, Gasink C et al Ustekinumab as induction and maintenance therapy for Crohn's disease. N Engl J Med 2016; 375:1946–60. [DOI] [PubMed] [Google Scholar]
- 27. Seder RA, Darrah PA, Roederer M. T‐cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 2008; 8:247–58. [DOI] [PubMed] [Google Scholar]
- 28. Kannanganat S, Ibegbu C, Chennareddi L, Robinson HL, Amara RR. Multiple‐cytokine‐producing antiviral CD4 T cells are functionally superior to single‐cytokine‐producing cells. J Virol 2007; 81:8468–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boyd A, Almeida JR, Darrah PA et al Pathogen‐specific T cell polyfunctionality is a correlate of T cell efficacy and immune protection. PLOS ONE 2015; 10:e0128714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Globig AM, Hennecke N, Martin B et al Comprehensive intestinal T helper cell profiling reveals specific accumulation of IFN‐gamma+IL‐17+coproducing CD4+ T cells in active inflammatory bowel disease. Inflamm Bowel Dis 2014; 20:2321–9. [DOI] [PubMed] [Google Scholar]
- 31. Dunn ET, Taylor ES, Stebbings S, Schultz M, Butt AG, Kemp RA. Distinct immune signatures in the colon of Crohn's disease and ankylosing spondylitis patients in the absence of inflammation. Immunol Cell Biol 2016; 94:421–9. [DOI] [PubMed] [Google Scholar]
- 32. Glatzer T, Killig M, Meisig J et al RORgammat(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity 2013; 38:1223–35. [DOI] [PubMed] [Google Scholar]
- 33. Mizuno S, Mikami Y, Kamada N et al Cross‐talk between RORgammat+ innate lymphoid cells and intestinal macrophages induces mucosal IL‐22 production in Crohn's disease. Inflamm Bowel Dis 2014; 20:1426–34. [DOI] [PubMed] [Google Scholar]
- 34. Longman RS, Diehl GE, Victorio DA et al CX(3)CR1(+) mononuclear phagocytes support colitis‐associated innate lymphoid cell production of IL‐22. J Exp Med 2014; 211:1571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sonnenberg GF, Monticelli LA, Alenghat T et al Innate lymphoid cells promote anatomical containment of lymphoid‐resident commensal bacteria. Science 2012; 336:1321–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Withers DR, Gaspal FM, Mackley EC et al Cutting edge: lymphoid tissue inducer cells maintain memory CD4 T cells within secondary lymphoid tissue. J Immunol 2012; 189:2094–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mackley EC, Houston S, Marriott CL et al CCR7‐dependent trafficking of RORgamma(+) ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat Commun 2015; 6:5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Withers DR. Innate lymphoid cell regulation of adaptive immunity. Immunology 2016; 149:123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]