

Summary
The mammalian target of rapamycin (mTOR) is a serine‐threonine kinase that has been shown to be essential for the differentiation and function of various immune cells. Earlier in vitro studies showed that mTOR signalling regulates B‐cell biology by supporting their activation and proliferation. However, how mTOR signalling temporally regulates in vivo germinal centre B (GCB) cell development and differentiation into short‐lived plasma cells, long‐lived plasma cells and memory cells is still not well understood. In this study, we used a combined conditional/inducible knock‐out system to investigate the temporal regulation of mTOR complex 1 (mTORC1) in the GCB cell response to acute lymphocytic choriomeningitis virus infection by deleting Raptor, a main component of mTORC1, specifically in B cells in pre‐ and late GC phase. Early Raptor deficiency strongly inhibited GCB cell proliferation and differentiation and plasma cell differentiation. Nevertheless, late GC Raptor deficiency caused only decreases in the size of memory B cells and long‐lived plasma cells through poor maintenance of GCB cells, but it did not change their differentiation. Collectively, our data revealed that mTORC1 signalling supports GCB cell responses at both early and late GC phases during viral infection but does not regulate GCB cell differentiation into memory B cells and plasma cells at the late GC stage.
Keywords: B cell, cell differentiation, gene regulation
Abbreviations
- BCR
B‐cell receptor
- CGG
chicken gamma globulin
- GCB
germinal centre B cell
- GC
germinal centre
- LCMV
lymphocytic choriomeningitis virus
- mTORC1
mTOR complex 1
- mTORC2
mTOR complex 2
- mTOR
mammalian target of rapamycin
- NP
4‐hydroxy‐3‐nitrophenylacetyl
- RT‐qPCR
real‐time quantitative polymerase chain reaction
- Tfh
follicular helper T cell
- YFP
yellow fluorescent protein
Introduction
Humoral immunity is the central component of vaccination through which vaccines elicit long‐lived plasma cells and memory B cells. As both of them are differentiated from germinal centre B (GCB) cells, GCB cell development is key to vaccine efficiency and effectiveness.1, 2
GCB cell development involves complex extracellular signalling and intercellular interaction. During early B‐cell responses, antigen‐specific B cells are activated and interact with follicular helper T (Tfh) cells to form the GC.3, 4 GCB cells become highly proliferative in the dark zone and undergo somatic hyper‐mutation before moving to the light zone bearing the B‐cell receptor (BCR) affinity selection via follicular dendritic cells and immunoglobulin heavy‐chain class‐switch.5, 6 The B cells with high‐affinity BCRs are selected and differentiated into antibody‐producing plasma cells or memory B cells.4, 7 However, how B cells integrate the signals from the microenvironment and are prompted to GCB cell differentiation is still unknown.
The mammalian target of rapamycin (mTOR) is a serine‐threonine kinase that plays key roles in responding to extracellular signalling and controlling intracellular signalling and metabolism processes.8 The signalling downstream of mTOR consists of the mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) pathways.9 mTORC1 mainly regulates autophagy, protein synthesis and glucose–lipid metabolism by S6K1, 4E‐BP1, and PPARγ. mTORC2 plays important roles in cytoskeleton organization, cell survival and migration.8, 10
Recently, mTOR signalling has been shown to be deeply implicated in immune cell responses and differentiation, including memory establishment of CD8 cells,11 differentiation of various CD4 sub‐lineages,12 responses of macrophages,13 dendritic cells,14 natural killer cells and granular cells.8 However, the number of relevant reports on mTOR function in B cells is still limited, and the results remain controversial.15
Rapamycin treatment has been a classic strategy used to study mTOR signalling in immune cell responses. Early studies showed that in vitro rapamycin treatment impeded B‐cell proliferation and plasma cell differentiation.16, 17, 18, 19 However, the strong BCR or Toll‐like receptor signalling induced by agonists hardly reflected the B‐cell responses in complex physiological conditions; moreover, rapamycin only partially inhibits mTOR signalling, mTORC1.20
Using conditional knockout mice provides the possibility to investigate the role of mTOR in vivo. Constitutively inactivated mTOR knock‐in mice showed decreased capacity of differentiation from B cells to plasma cells.1 CD19‐Cre‐induced mTOR‐deleted mice showed impaired GC development and attenuated plasma cell differentiation.3 Meanwhile, other reports have shown controversial results. Increasing mTOR activity by deleting its upstream inhibitor tuberous sclerosis 1 (TSC‐1) resulted in unbiased plasma cell differentiation from B cells,21 whereas another report showed that TSC‐1 promotes plasma cell differentiation but is dispensable for GC formation.22 One explanation for this discordance is that conventional or CD19‐Cre‐induced mTOR variation causes a B‐cell development handicap before normal GC development.15, 21, 23 A more precise knockout strategy should be used to elucidate the role of mTOR in GC establishment and plasma cell and memory B‐cell differentiation.
Meanwhile, mTOR signalling converts different extracellular stimuli and intracellular signals into appropriate cell responses. B‐cell responses are initiated and regulated precisely by successive signals from extracellular space or Tfh cells.24 At each precise time‐point, mTOR signalling should be adapted to ensure consistency in the temporal regulation with B‐cell responses. However, very few studies addressing the temporal control of B‐cell responses have been reported.
In this study, we investigated the temporal regulation of mTORC1 in B‐cell responses to lymphocytic choriomeningitis virus (LCMV) infection using conditional and inducible knockout systems. We found that both pre‐ and late GC deletion of mTORC1 signalling strongly stunted the GCB cell response, but the differentiation of memory B cells and long‐lived plasma cells from GCB cells was not impaired by late GC mTORC1 deficiency.
Materials and methods
Mice and treatment
C57BL/6J (B6), Aicda‐Cre (stock number 018422), B1‐8hi (stock number 007775) and Rptor flox (stock number 013188) strains were purchased from Jackson Laboratory (Bar Harbor, ME). Homozygous Aicda‐Cre‐Rptor flox strains were obtained by back‐crossing Aicda‐Cre to Rptor flox strain, and genotypes were checked by standard PCR procedures as supplied by Jackson Laboratory. Aicda‐ERT2‐Cre mice were a gift from Dr Claude‐Agnès Reynaud from Institut National de la Santé et de la Recherche Médicale (INSERM).25 To induce deletion of Raptor, 1 mg sunflower‐oil‐diluted tamoxifen was injected intraperitoneally into Aicda‐ERT2‐Cre‐Rptor flox mice at the indicated time‐points. An acute viral infection model was established by infecting mice with 2 × 105 plaque‐forming units Armstrong LCMV via intraperitoneal injection. In rapamycin treatment experiments, mice were treated with 75 mg/kg rapamycin via intraperitoneal injection during the indicated post‐infection periods. For vaccination with 4‐hydroxy‐3‐nitrophenylacetyl conjugated with chicken gamma globulin (NP‐CGG), mice were subcutaneously injected in both hind legs with 50 μg NP‐CGG (Biosearch Tech., Novato, CA) emulsified 1 : 1 with complete Freund's adjuvant (Sigma, St Louis, MO); on day 8 post‐vaccination the inguinal lymph nodes were extracted for cytometry analysis.
All handling of animals was performed following the guidelines of the Institutional Animal Care and Use Committees of the Third Military Medical University.
Flow cytometry and antibodies
Flow cytometry analysis was carried out using a FACSCanto II (BD Biosciences, Franklin Lakes, NJ) and the data were analysed using flowjo software (Tree Star, Ashland, OR). Surface staining was performed in PBS containing 2% fetal bovine serum (weight/volume). Staining for intracellular IgG and IgM were performed using a Cytofix/Cytoperm Fixation/Permeabilization Kit (554714; BD Biosciences). Intra‐nuclear staining of Bcl‐6, Foxp3 and Ki67 was performed using a Foxp3/Transcription Factor Staining Buffer Set (00‐5523; eBioscience, San Diego, CA).
The antibodies and reagents used for flow cytometry staining are listed in the Supplementary material (Table S1).
Enzyme‐linked immunosorbent assay
To determine LCMV‐specific IgG titres, Nunc MaxiSorp® flat‐bottom 96‐well plates (44‐2404‐21) were coated with 50 μg LCMV nucleoprotein. After blocking, diluted mouse serum and secondary antibody horseradish peroxidase‐conjugated goat anti‐mouse IgG (1036‐05; SouthernBiotech, Birmingham, AL) were successively incubated for 90 min at room temperature. The signal was revealed using o‐phenylenediamine dihydrochloride (P8787; Sigma).
Virus titration
LCMV viral loads in tissue samples were quantified using a quantitative PCR (qPCR) assay as described previously.26 In brief, the viral RNA in serum, spleen or diluted virus solution with known titre was extracted and retro‐transcribed to cDNA. The primers NP‐R 5′‐cagaccttggcttgctttacacag‐3′ and NP‐F 5′‐cagaaatgttgatgctggactgc‐3′ were used to detect and quantify the viral RNA copies by standard RT‐qPCR.
Bone‐marrow chimeric mice
The bone marrow of C57BL/6J (CD45.1) and Aicda‐Cre‐Rptor flox (CD45.2) mice was collected and transferred at a ratio of 4 : 6 into lethally irradiated C57BL/6J (CD45.1) mice via intravenous injection with a total bone marrow cell count of 5 million per mouse. The bone marrow was reconstructed for 10 weeks before the chimeric mice were infected with LCMV.
RT‐qPCR
To compare gene expression changes in GCB cells from wild‐type and Aicda‐Cre‐Rptor flox mice, B220+ PNA+ CD95+ cells were sorted directly into TRIzol LS reagent (10296; Life Technologies, Carlsbad, CA) and extracted with isopropyl ethanol. The cDNA was obtained by reverse‐transcribing total RNA with a RevertAid H Minus First Strand cDNA Synthesis Kit (K1632; Thermo Scientific, Waltham, MA). To quantify the relative gene copy number of Rptor, wild‐type and Aicda‐Cre‐Rptor flox cells were lysed and genomic DNA was extracted with a genomic DNA extraction kit (HF223; Yuanpinghao Biotech, Tianjin, China) following the manufacturer's instructions. Quantitative PCR was carried out with AceQ qPCR SYBR Green Master Mix (Q111; Vazyme, Jiangsu Sheng, China) on a CFX96 Touch Real‐Time System (Bio‐Rad, Hercules, CA), and the sequence of primers for qPCR was: Aicda‐Forward, 5′‐gccaccttcgcaacaagtct‐3′, Aicda‐Reverse, 5′‐ccgggcacagtcatagcac‐3′; Bcl6‐Forward, 5′‐agacgcacagtgacaaacca‐3′, Bcl6‐Reverse, 5′‐agtgtgggtcttcaggttgg‐3′; Prdm1‐Forward, 5′‐agtgcaatgtctgtgccaag‐3′, Prdm1‐Reverse, 5′‐ttgagattgcttgtgctgct‐3′; Rptor‐Forward, 5′‐ctcggtacaagcagagcct‐3′, Rptor‐Reverse, 5′‐tcttgatgggagtggtgagg‐3′.
Results
mTORC1 signalling sustained the GCB cell response to acute LCMV infection
To keep B‐cell development intact with mTOR activity but temporally inactivate mTORC1 signalling during GC response, we crossed the Aicda‐Cre mouse strain with the Rptor flox strain to create the Aicda‐Cre/Rptor flox strain (Rptor‐KO). RT‐qPCR results showed that Rptor expression was strongly decreased in GCB cells but not in naive B cells or CD4 cells (Fig. 1a). Cytometry results showed that CD98, a downstream marker of mTORC1 signalling,27 had much lower expression in GCB cells from Rptor‐KO mice (Fig. 1a).
Figure 1.
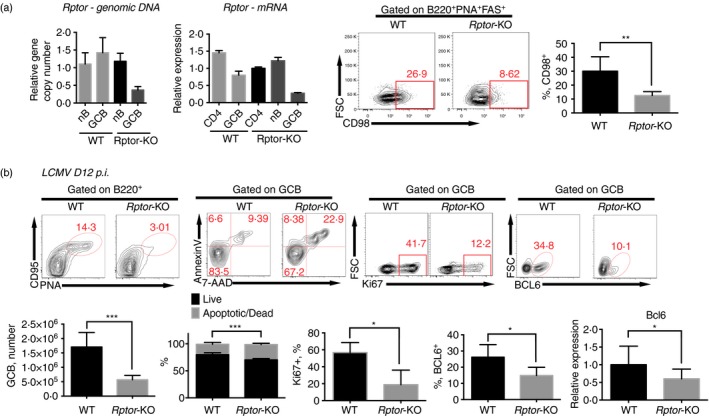
Mammalian target of rapamycin complex 1 (mTORC1) signalling sustains B‐cell responses to lymphocytic choriomeningitis virus (LCMV) infection. (a) Quantification of Rptor genomic DNA (left) and mRNA (middle left) copy number by quantitative PCR in sorted lymphocytes as indicated; staining of CD98 in germinal centre B (GCB) (PNA + CD95+ B220+) cells (middle right) and frequency of CD98+ population in GCB cells (right) from wild‐type C57BL/6J (WT) and Aicda‐Cre‐Rptor flox (Rptor‐KO) mice 8 days after infection with the Armstrong strain of LCMV. (b) Flow cytometry of B220+ B cells (top, left) and PNA + CD95+ B220+ GCB cells (top, middle and right); and quantification of PNA + CD95+ B220+ GCB cell number, apoptotic/dead cell frequency, Ki67+ frequency and BCL6+ frequency in PNA + CD95+ B220+ GCB cells (bottom, left, middle left and middle) in spleens from WT and Rptor‐KO mice on day 12 after LCMV infection. Quantification of frequency and total number per spleen of indicated subsets and quantification of Bcl6 mRNA in GCB cells in the indicated mice (bottom middle right and right). *P < 0·05 **P < 0·005 ***P < 0·002 (unpaired two‐tailed t‐test). Data are representative of three independent experiments with three to six mice per group (error bars, SEM). [Colour figure can be viewed at wileyonlinelibrary.com]
After LCMV infection, we observed a significant decrease in GCB cell frequency and total number in spleens of Rptor‐KO mice on day 8. Annexin‐V assays revealed increased apoptosis in Rptor‐deficient GCB cells (Fig. 1b). Furthermore, Rptor‐KO mice showed lower Ki67+ GCB cell frequency. This lower GCB cell frequency was accompanied by lower BCL6 expression in Rptor‐deficient GCB cells (Fig. 1b). RT‐qPCR results confirmed the down‐regulation of Bcl6 expression at the transcriptional level (Fig. 1b), which explains the impaired GCB cell differentiation.
These results showed that mTORC1 sustained the GCB cell response during acute LCMV infection.
mTORC1 supported plasma cell differentiation and humoral response against acute LCMV infection
To confirm whether mTORC1 deficiency impairs humoral immunity, splenocytes from LCMV‐infected Rptor‐KO and wild‐type mice were analysed by flow cytometry. Lower frequency and numbers of plasma cells were observed (Fig. 2a), consistent with lower Prdm1 expression in Rptor‐deficient plasma cells, implying impaired plasma cell differentiation from GCB cells (Fig. 2a). LCMV‐specific antibody in serum was much lower in Rptor‐KO mice from day 8 to day 35 post‐infection (Fig. 2b). The impaired humoral immune response also led to decreased anti‐viral protection, with higher LCMV titres in serum and spleens of Rptor‐KO mice (Fig. 2b).
Figure 2.
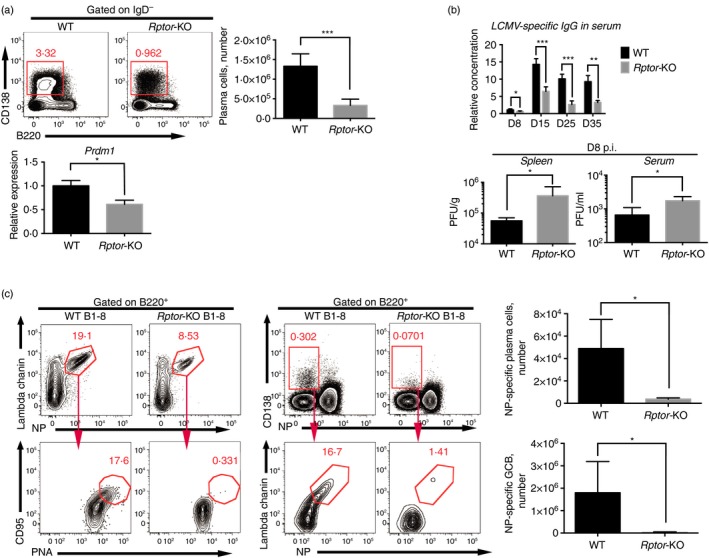
Mammalian target of rapamycin complex 1 (mTORC1) supported plasma cell differentiation and humoral response against acute lymphocytic choriomeningitis virus (LCMV) infection. (a) Flow cytometry of IgD− lymphocytes (top left), CD138+ B220mi/lo plasma cell total number (top right), and quantification of Prdm1 in plasma cells from wild‐type (WT) and Rptor‐KO mice on day 12 after LCMV infection. (b) Quantification of LCMV‐specific IgG in serum of Rptor‐KO and WT mice after the indicated post‐infection time (top). The concentrations were normalized to that of WT mice on post‐infection day 8. Quantification of LCMV titres in spleens and serum of Rptor‐KO and WT mice on post‐infection day 8 (bottom). (c) Flow cytometry of B220+ B cells and quantification of 4‐hydroxy‐3‐nitrophenylacetyl (NP)‐specific GCB (NP + Ig‐λ + PNA + CD95+ B220+) cells and NP‐specific plasma cells (NP + Ig‐λ + CD138+ B220mi/lo) among total cell number in spleens from wild‐type B1‐8hi (WT B1‐8) and Aicda‐Cre‐Rptor flox B1‐8hi (Rptor‐KO B1‐8) mice on day 8 after LCMV infection. *P < 0·05 **P < 0·005 ***P < 0·002 (unpaired two‐tailed t‐test). Data are representative of three independent experiments with three to six mice per group (error bars, SEM). [Colour figure can be viewed at wileyonlinelibrary.com]
Then, we assessed whether mTORC1 deficiency impaired antigen‐specific B‐cell responses in a protein immunization model. The Aicda‐Cre/Rptor flox strain was further crossed with B1‐8hi (wild‐type B1‐8) mice, which possess NP‐specific BCR, to obtain Rptor‐KO B1‐8. On day 8 following vaccination with NP‐CGG via subcutaneous injection, the drained lymph nodes of Rptor‐KO B1‐8 and wild‐type B1‐8 were analysed by cytometry. The results showed that NP‐specific GCB and plasma cells almost disappeared in Rptor‐KO B1‐8 mice (Fig. 2c), suggesting that antigen‐specific B‐cell responses were highly dependent on mTORC1 signalling. These results showed that mTORC1 signalling supports B‐cell responses to LCMV infection by sustaining GCB cell proliferation, differentiation and early plasma cell differentiation.
The attenuated humoral response in mTORC1‐deficient mice showed that the humoral immune response was dependent on mTORC1 signalling.
GCB cell‐intrinsic mTORC1 signalling regulated the GCB cell response
To more precisely assess the intrinsic role of mTORC1 in regulating B‐cell responses, we generated chimeras by reconstituting irradiated wild‐type (CD45.1+) recipient mice with a mixture of bone marrow cells from Aicda‐Cre‐Rptor flox (CD45.2+) and wild‐type donor mice (CD45.1+) at a ratio of 4 : 6, in which the mTORC1‐deficient B cells were in the same condition as wild‐type B cells (Fig. 3a). The chimeric mice infected with the Armstrong strain of LCMV and the splenocytes were analysed by cytometry on day 8 post‐infection. Similar to Rptor‐KO mice, CD45.2+ (Rptor‐KO) cells had much lower frequency of GCB (Fig. 3b) and plasma cells (Fig. 3c) compared with CD45.1+ (wild‐type) cells. These data showed that the mTORC1 signal regulated B‐cell responses to acute LCMV infection in a B‐cell intrinsic manner.
Figure 3.
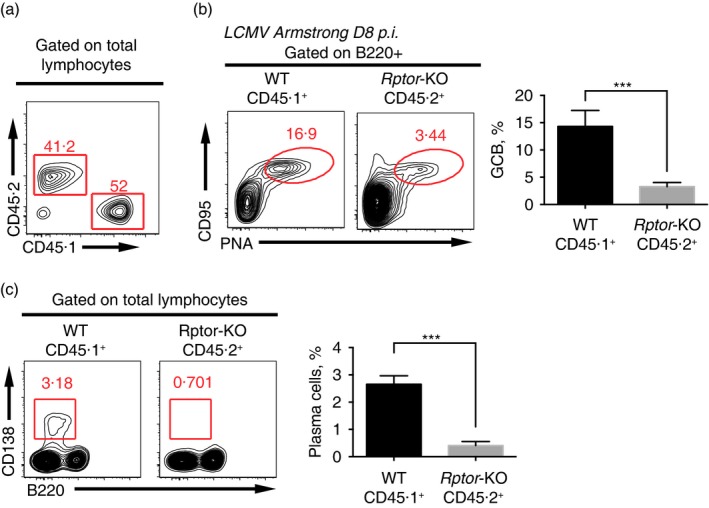
Germinal centre B (GCB) cell‐intrinsic mammalian target of rapamycin complex 1 (mTORC1) signalling regulated the GCB cell response. (a) Flow cytometry of total lymphocytes in spleens of non‐infected chimeras generated with a mixture of wild‐type (CD45.1+) and Aicda‐Cre‐Rptor flox (CD45.2+) bone marrow cells. The numbers above the outlined areas indicate the percentages of CD45.1+ and CD45.2+ cells. (b) Flow cytometry of B220+ B cells in the LCMV‐infected chimeras in (a), assessed at day 8 after infection of the host with lymphocytic choriomeningitis virus (LCMV), and a summary of GCB cell frequency in B cells of CD45.1+ and CD45.2+ origin. (c) Flow cytometry of lymphocytes in the LCMV‐infected chimeras in (a), assessed at day 8 after infection of the host with LCMV, and a summary of plasma cell frequency in lymphocytes of CD45.1+ and CD45.2+ origin. ***P < 0·002 (unpaired two‐tailed t‐test). Data are representative of three independent experiments with three to six mice per group (error bars, SEM). [Colour figure can be viewed at wileyonlinelibrary.com]
Temporal deletion of mTORC1 signalling in early GC development resulted in an impaired early humoral response
To study the temporal regulation of mTORC1 signalling in B‐cell responses in different humoral response stages, an inducible Rptor‐KO system was established with an Aicda‐ERT2Cre‐YFP strain. The Aicda‐ERT2Cre‐YFP mice expressed a tamoxifen‐sensitive oestrogen receptor variant fused to Cre recombinase (ERT2‐Cre) under the control of the endogenous Aicda promoter, while the coding sequence of yellow fluorescent protein (YFP) with a floxed stop codon was knocked in at the Rosa26 locus.25 As Aicda is expressed after B‐cell activation and Cre‐mediated recombination occurs only in the presence of tamoxifen, deletion of the floxed gene in B cells can be induced by tamoxifen treatment after B‐cell activation or during GCB cell phase. Moreover, B cells with active Cre recombinase could be traced by YFP expression. To validate this inducible system, the Aicda‐ERT2Cre‐YFP mice were infected with LCMV and treated with tamoxifen from day 3 to day 7 (Fig. 4a). On day 12 post‐infection, YFP+ cells were found only among plasma cells and activated IgD− B cells (Fig. 4b), indicating bona fide Cre‐mediated deletion in early B‐cell activation. Then, we crossed the Aicda‐ERT2Cre‐YFP (control) strain with the Rptor flox strain to generate Aicda‐ERT2Cre‐YFP/Rptor flox (iRptor‐KO) mice, in which the deletion of Rptor could be induced by treatment with tamoxifen.
Figure 4.
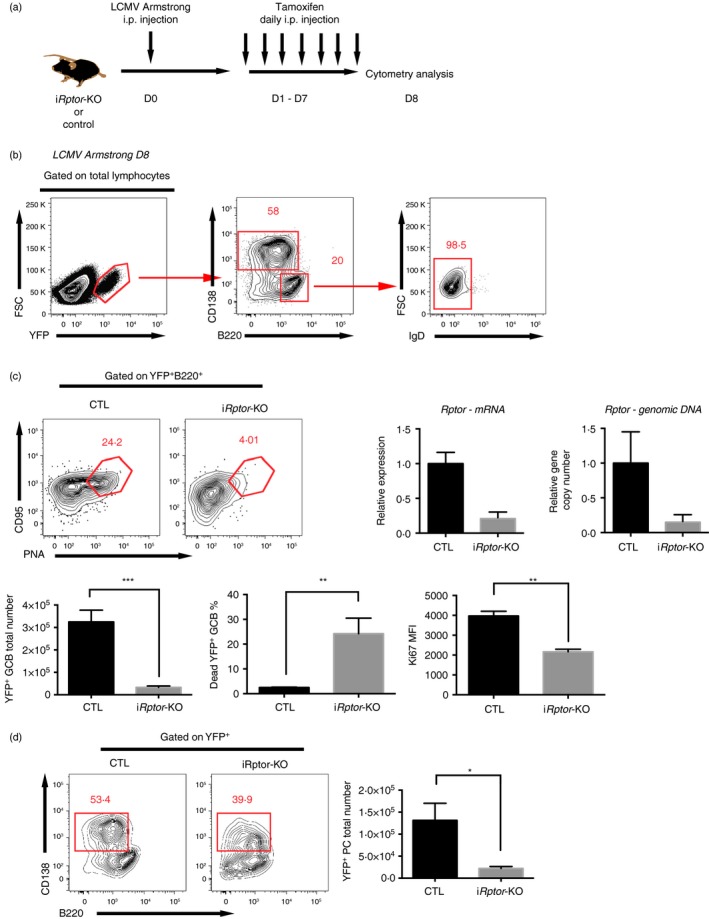
Temporal deletion of mammalian target of rapamycin complex 1 (mTORC1) signalling in early germinal centre (GC) development resulted in an impaired early humoral response. (a) Experimental set‐up for early GC induction of Rptor deletion. Aicda‐ERT2Cre‐YFP/Rptor flox (iR ptor‐KO) or control Aicda‐ERT2Cre‐YFP (CTL) mice were infected with the Armstrong strain of lymphocytic choriomeningitis virus (LCMV) and subsequently treated daily with tamoxifen via intraperitoneal injection during days 1–7 post‐infection. (b) Flow cytometry of lymphocytes in spleens of the CTL mice treated as described in (a) (left). The numbers above the outlined areas indicate the percentages of CD138+ plasma cells and B220+ B cells among YFP + cells (middle), and the percentage of IgD− B cells among YFP + B220+ B cells (right). (c) Flow cytometry of YFP + B220+ B cells (top left), quantification of Rptor genomic DNA and mRNA in sorted YFP + B220+ CD95+ PNA + GCB cells (top right), quantification of total YFP + B220+ CD95+ PNA + GCB cell number per spleen and quantification of viability dye‐labelled dead GCB cell frequency and Ki67 in YFP + B220+ CD95+ PNA + GCB cells (bottom) in the spleens of the CTL and iR ptor‐KO mice treated as described in (a). (d) Flow cytometry of YFP + cells and quantification of total YFP + B220mi/lo CD138+ plasma cell number in the spleen of the CTL and iR ptor‐KO mice treated as described in (a). *P < 0·05 **P < 0·005 ***P < 0·002 (unpaired two‐tailed t‐test). Data are representative of three independent experiments with three to six mice per group (error bars, SEM).
To induce the deletion in pre‐GC phase, the iRptor‐KO mice and control mice were infected with LCMV and treated with tamoxifen from day 3 to day 7. To confirm the Rptor deletion efficiency, RT‐qPCR assays were carried out with sorted YFP+ B220+ CD95+ PNA+ GCB cells on day 12 post‐infection; the results showed that both Rptor gene and mRNA copy number were strongly diminished in Rptor‐deficient GCB cells (Fig. 4c). When compared with the control mice, the iRptor‐KO mice had lower frequency and numbers of YFP+ GCB cells (Fig. 4c), similar to the Aicda‐Cre‐Rptor mice. Ki67 and viability dye staining showed that the lower number of GCB cells was due to a lower proliferation rate and higher cell death in the GCB cell population (Fig. 4c). Consequently, impaired plasma cell frequency and number occurred in iRptor‐KO mice (Fig. 4d).
The fact that induced deletion of Rptor from day 3 to day 7 post‐infection impaired GCB and plasma cell development confirmed that mTORC1 signalling was critical for an effective humoral response in the early phase.
Late mTORC1 signalling supported post‐GC humoral responses by maintaining the GCB cell population but was dispensable for splenic plasma cell, long‐lived plasma cell and memory B‐cell differentiation
Then, we sought to investigate whether mTORC1 signalling played equally supportive roles in the late phase of the humoral response, i.e. late GC response. To this end, iRptor‐KO and control mice were infected with LCMV and received tamoxifen treatment on days 10–15 post‐infection, during which GC remain active, but their activity starts declining. As Aicda expression is still present in GCB cells, the treatment with tamoxifen was expected to delete mTORC1 activity in GCB cells during the post‐GC stage, and at a later time‐point YFP+ cells included not only GCB cells that were still active but also the plasma cells, memory B cells and subsequent long‐lived plasma cells differentiated from the Rptor‐deleted GCB cells.
As described above, splenocytes from both iRptor‐KO and control mice were analysed on day 25 post‐infection (Fig. 5a). Rptor‐deleted B cells and Cre‐active control B cells were traced by YFP reporter gene expression. Total YFP+ number was decreased in iRptor‐KO mice (Fig. 5b). This decline might be a result of lower frequency of YFP+ GCB cells in Rptor‐deficient mice, which was decreased to one‐third that of control mice with the total number decreased to one‐tenth that of the level in control mice (Fig. 5c), indicating inefficient germinal centre support in the Rptor‐deficient mice. However, in contrast to the early GC Rptor deletion in which plasma cell frequency was strongly reduced in YFP+ cells, late GC Rptor deletion caused no change in frequency of CD138+ YFP+ plasma cells or B220+ IgD− CD38+ PNA− CD138− YFP+ memory B cells in Rptor‐deficient and in control mice, although a marked decrease in cell number was observed in respective subsets (Fig. 5c), suggesting that loss of mTORC1 signal later impairs the total number of plasma cells and memory B cells in spleen without biasing their differentiation.
Figure 5.
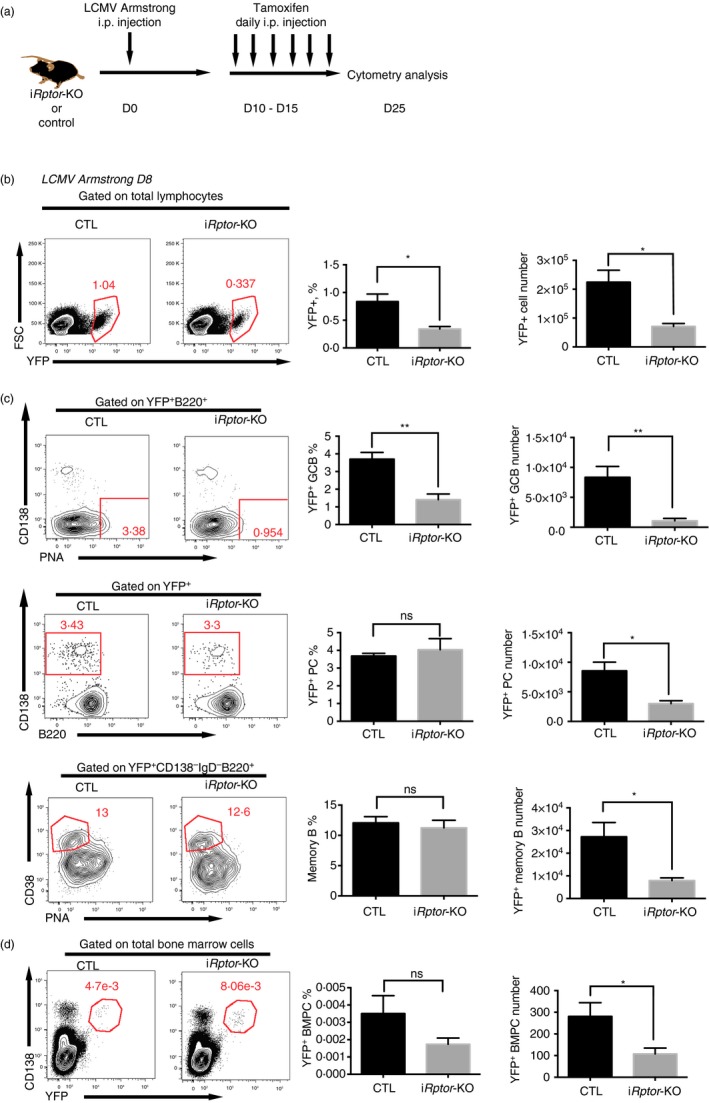
Late mammalian target of rapamycin complex 1 (mTORC1) signalling supported the post‐germinal centre (GC) humoral response by maintaining the GCB cell population. (a) Experimental set‐up for late GC induction of Rptor deletion. Aicda‐ERT2Cre‐YFP/Rptor flox (iR ptor‐KO) or control Aicda‐ERT2Cre‐YFP (CTL) mice were infected with the Armstrong strain lymphocytic choriomeningitis virus (LCMV) and subsequently treated daily with tamoxifen via intraperitoneal injection during days 10–15 post‐infection. (b) Flow cytometry of lymphocytes and quantification of YFP + lymphocyte frequency and number in the spleens of the CTL and iR ptor‐KO mice treated as described in (a). (c) Flow cytometry of YFP + B220+ B cells and quantification of YFP + B220+ CD95+ PNA + GCB cell frequency and number (top); flow cytometry of YFP + cells and quantification of YFP + B220mi/lo CD138+ plasma cell frequency and total number (middle), and flow cytometry of YFP + CD138− IgD− B220+ cells and quantification of YFP + CD138− IgD− B220+ CD38+ PNA − memory cell frequency and total number (middle) in the spleens of the CTL and iR ptor‐KO mice treated as described in (a). (d) Flow cytometry of YFP + CD138+ plasma cells and quantification of YFP + CD138+ plasma cell frequency and total number in the bone marrow of the CTL and iR ptor‐KO mice treated as described in (a). *P < 0·05 **P < 0·005 (unpaired two‐tailed t‐test). Data are representative of three independent experiments with three to six mice per group (error bars, SEM). [Colour figure can be viewed at wileyonlinelibrary.com]
As the long‐term plasma cells reside in bone marrow and serve as long‐term antibody‐secreting cells, we investigated the de novo generated plasma cells during LCMV infection by tracing YFP+ plasma cells in bone marrow. The total number of de novo generated plasma cells was decreased in Rptor‐deficient mice, whereas the IgG+ frequency remained intact when compared with the YFP+ plasma cells in control mice (Fig. 5d).
These data showed that the post‐GC deletion of mTORC1 in B cells impaired mainly GCB cell maintenance, leading to an attenuated number of plasma cells, memory B cells and bone marrow plasma cells without changing their differentiation.
Discussion
Because mTOR activity was shown to be necessary for early B‐cell development28 and B cells with constitutive or CD19‐Cre‐induced mTOR deficiency could not be activated correctly in in vitro or in vivo conditions,1, 3 the regulatory role of mTOR in further GCB cell, plasma cell and memory cell differentiation and function was difficult to study because the further differentiation and effector functions depend on appropriate B‐cell activation. In our current study, Aicda‐driven Cre‐recombinase induced Rptor deletion only during early GCB cell development, with early activation intact. This strategy kept the mTOR signalling intact during B‐cell development and early activation, ensuring correctly activated B cells before GCB cell development, which is an ideal mouse model for studying mTORC1 signalling in regulating B‐cell responses.
In addition, mTOR signalling integrates different intracellular and extracellular signals and modulates them into cell responses to these signals, and GCB cells interact with Tfh cells through a complex cytokine/membrane regulatory signalling manner. Hence, it is more reasonable to propose that mTOR signalling should play temporal roles instead of a constant monotonic function in B‐cell responses. How mTORC1 temporally regulates B‐cell responses in different phases becomes important.
Our data showed that early mTORC1 signalling deficiency caused a defect in GCB cell formation on day 8 post‐infection following acute LCMV infection, indicating that mTORC1 signalling played a critical role in early GCB cell development, which is critical to the entire humoral immune response because the long‐term antibody‐secreting plasma cells and memory B cells are derived from GCB cells.4 Importantly, early GC mTORC1 deficiency caused impaired plasma cell differentiation with lower frequency and lower Prdm1 expression. With the reduced GCB cell population and hampered differentiation, plasma cell size was consequently strongly diminished (Fig. 2a). With regard to those de novo generated plasma cells in the inducible model at early stage, the frequency of plasma cells was decreased among the YFP+ cells, which confirmed that early mTORC1 signalling is also critical for plasma cell differentiation in the early GC phase.
However, in the late GC phase, i.e. days 10–15 post‐infection, late GC mTORC1 signalling deficiency did not change the frequency of long‐lived plasma or memory B cells among total YFP+ cells (Fig. 5c), indicating intact differentiation of long‐lived plasma and memory B cells, even though their total numbers were decreased in tamoxifen‐treated iRptor‐KO mice. The total number of long‐lived plasma cells and memory B cells should be directly related to the poor maintenance of the GCB cell population, as the entire YFP+ population issued from the same Cre‐active GCB cells during day 10–15 post‐infection. These results are consistent with those of Jones et al.,29 suggesting that mTORC1 signalling is important for the differentiation of short‐lived plasma cells at the initial stage of GC reaction but dispensable for the differentiation of long‐lived plasma cells from GCB cells within a GC reaction.
The centre of B‐cell responses is the GC reaction, in which Tfh–GCB cell interactions are essential.4 Recently, the regulatory role of mTOR in Tfh–GCB cell interactions was clearly dissected.30 In fact, inactivation of mTORC1 signalling by rapamycin led to a halt of GCB cell responses and impaired helper CD4 T‐cell differentiation during viral infection.30 More recently, Tfh‐intrinsic mTOR signalling was shown to support Tfh cell differentiation and function by re‐modulating metabolism and promoting proliferation.31, 32 Indeed, all the evidence gained with our recent data suggest that mTOR signalling is critical for B‐cell responses by supporting both Tfh and GCB cell differentiation/proliferation and ensuring an appropriate interaction between Tfh and GCB cells.
Although several downstream targets of mTOR that regulate Tfh cell responses were found in recent studies,31, 32 the mechanism of regulation by mTOR in B‐cell responses is still unclear. Further studies are required to explore the downstream targets of mTOR signalling in the regulation of B‐cell responses.
Overall, this study disclosed the supportive role of mTORC1 signalling in B‐cell responses, demonstrated that late GC mTORC1 signalling is dispensable for long‐lived plasma cell and memory B‐cell differentiation, and revealed that mTORC1 signalling regulates B‐cell responses differentially in a time‐dependent manner.
Disclosures
The authors declare no competing financial interests.
Supporting information
Table S1. Antibodies and reagents used in flow cytometry.
Acknowledgements
We sincerely thank Dr Claude‐Agnès Reynaud from the Institut National de la Santé et de la Recherche Médicale (INSERM) for providing Aicda‐ERT2‐Cre mice and the core facility centre of the Third Military Medical University for performing the cell sorting experiments. This work was supported by the National Basic Research Programme of China (973 programme, 2013CB531500, to L.Y.), the National Natural Science Foundation of China (No. 31500733 to Q.B.) and Special Postdoctoral Science Fund of Chongqing (No. Xm2015007 to Q.B.).
References
- 1. Zhang S, Readinger JA, DuBois W, Janka‐Junttila M, Robinson R, Pruitt M et al Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood 2011; 117:1228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Plotkin SA, Orenstein AW, Offit PA. Vaccines, 6th edn. Amsterdam, Netherlands: Elsevier Inc., 2012. [Google Scholar]
- 3. Zhang S, Pruitt M, Tran D, Du Bois W, Zhang K, Patel R et al B cell‐specific deficiencies in mTOR limit humoral immune responses. J Immunol 2013; 191:1692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol 2012; 30:429–57. [DOI] [PubMed] [Google Scholar]
- 5. Neuberger MS, Lanoue A, Ehrenstein MR, Batista FD, Sale JE, Williams GT. Antibody diversification and selection in the mature B‐cell compartment. Cold Spring Harb Symp Quant Biol 1999; 64:211–6. [DOI] [PubMed] [Google Scholar]
- 6. Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Adv Immunol 2011; 110:1–26. [DOI] [PubMed] [Google Scholar]
- 7. Gourley TS, Wherry EJ, Masopust D, Ahmed R. Generation and maintenance of immunological memory. Semin Immunol 2004; 16:323–33. [DOI] [PubMed] [Google Scholar]
- 8. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol 2012; 30:39–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 2012; 12:325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF et al mTOR regulates memory CD8 T‐cell differentiation. Nature 2009; 460:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR et al The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 2011; 12:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang C‐S, Song C‐H, Lee J‐S, Jung S‐B, Oh J‐H, Park J et al Intracellular network of phosphatidylinositol 3‐kinase, mammalian target of the rapamycin/70 kDa ribosomal S6 kinase 1, and mitogen‐activated protein kinases pathways for regulating mycobacteria‐induced IL‐23 expression in human macrophages. Cell Microbiol 2006; 8:1158–71. [DOI] [PubMed] [Google Scholar]
- 14. Ohtani M, Nagai S, Kondo S, Mizuno S, Nakamura K, Tanabe M et al Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide‐induced interleukin‐12 production in dendritic cells. Blood 2008; 112:635–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu X, Ye L, Araki K, Ahmed R. mTOR, linking metabolism and immunity. Semin Immunol 2012; 24:429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakata A, Kuwahara K, Ohmura T, Inui S, Sakaguchi N. Involvement of a rapamycin‐sensitive pathway in CD40‐mediated activation of murine B cells in vitro . Immunol Lett 1999; 68:301–9. [DOI] [PubMed] [Google Scholar]
- 17. Aagaard‐Tillery KM, Jelinek DF. Inhibition of human B lymphocyte cell cycle progression and differentiation by rapamycin. Cell Immunol 1994; 156:493–507. [DOI] [PubMed] [Google Scholar]
- 18. Kay JE, Kromwel L, Doe SE, Denyer M. Inhibition of T and B lymphocyte proliferation by rapamycin. Immunology 1991; 72:544–9. [PMC free article] [PubMed] [Google Scholar]
- 19. Wicker LS, Boltz RC, Matt V, Nichols EA, Peterson LB, Sigal NH. Suppression of B cell activation by cyclosporin A, FK506 and rapamycin. Eur J Immunol 1990; 20:2277–83. [DOI] [PubMed] [Google Scholar]
- 20. Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol 1996; 14:483–510. [DOI] [PubMed] [Google Scholar]
- 21. Benhamron S, Tirosh B. Direct activation of mTOR in B lymphocytes confers impairment in B‐cell maturation and loss of marginal zone B cells. Eur J Immunol 2011; 41:2390–6. [DOI] [PubMed] [Google Scholar]
- 22. Ci X, Kuraoka M, Wang H, Carico Z, Hopper K, Shin J et al TSC1 promotes B cell maturation but is dispensable for germinal center formation. PLoS One 2015; 10:e0127527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Donahue AC, Fruman DA. Distinct signaling mechanisms activate the target of rapamycin in response to different B‐cell stimuli. Eur J Immunol 2007; 37:2923–36. [DOI] [PubMed] [Google Scholar]
- 24. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol 2015; 15:137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dogan I, Bertocci B, Vilmont V, Delbos F, Mégret J, Storck S et al Multiple layers of B cell memory with different effector functions. Nat Immunol 2009; 10:1292–9. [DOI] [PubMed] [Google Scholar]
- 26. McCausland MM, Crotty S. Quantitative PCR technique for detecting lymphocytic choriomeningitis virus in vivo . J Virol Methods 2008; 147:167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kelly AP, Finlay DK, Hinton HJ, Clarke RG, Fiorini E, Radtke F et al Notch‐induced T cell development requires phosphoinositide‐dependent kinase 1. EMBO J 2007; 26:3441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iwata TN, Ramirez JA, Tsang M, Park H, Margineantu DH, Hockenbery DM et al Conditional disruption of raptor reveals an essential role for mTORC1 in B cell development, survival, and metabolism. J Immunol 2016; 197:2250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM et al mTOR has distinct functions in generating versus sustaining humoral immunity. J Clin Invest 2016; 126:4250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ye L, Lee J, Xu L, Mohammed AUR, Li W, Hale JS et al mTOR promotes anti‐viral humoral immunity by differentially regulating CD4 helper T cell and B cell responses. J Virol 2016; 91:e02279–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang J, Lin X, Pan Y, Wang J, Chen P, Huang H et al Critical roles of mTOR Complex 1 and 2 for T follicular helper cell differentiation and germinal center responses. eLife 2016; 5:e17936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA et al mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity 2016; 45:540–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibodies and reagents used in flow cytometry.