

Graphical abstract
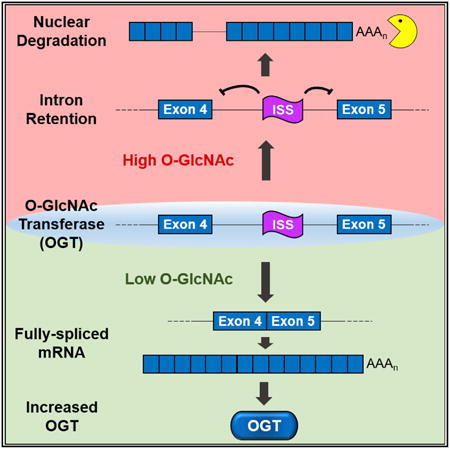
Summary
Modification of nucleocytoplasmic proteins with O-GlcNAc regulates a wide variety of cellular processes and has been linked to human diseases. The enzymes O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) add and remove O-GlcNAc, but the mechanisms regulating their expression remain unclear. Here, we demonstrate that retention of the fourth intron of OGT is regulated in response to O-GlcNAc levels. We further define a conserved intronic splicing silencer (ISS) that is necessary for OGT intron retention. Deletion of the ISS in colon cancer cells leads to increases in OGT, but O-GlcNAc homeostasis is maintained by concomitant increases in OGA protein. However, the ISS-deleted cells are hypersensitive to OGA inhibition in culture and in soft agar. Moreover, growth of xenograft tumors from ISS-deleted cells is compromised in mice treated with an OGA inhibitor. Thus, ISS-mediated regulation of OGT intron retention is a key component in OGT expression and maintaining O-GlcNAc homeostasis.
Introduction
Nucleocytoplasmic proteins are reversibly modified by addition of O-linked β-N-acetylglucosamine (O-GlcNAc) on serine or threonine hydroxyl groups (Torres and Hart, 1984). While over 1,000 proteins are modified by O-GlcNAcylation (Hahne et al., 2012; Nandi et al., 2006; Teo et al., 2010), only two enzymes add and remove O-GlcNAc (Kreppel et al., 1997; Lubas et al., 1997; Gao et al., 2001). O-GlcNAc transferase (OGT) transfers GlcNAc to proteins using the substrate UDP-GlcNAc, an end product of the hexosamine biosynthetic pathway (HBP), whereas O-GlcNAcase (OGA) removes O-GlcNAc from proteins.O-GlcNAc-modified proteins are involved in a wide variety of cellular processes, but how O-GlcNAc regulates protein function is only beginning to be understood (Bond and Hanover, 2015; Hanover et al., 2012; Hart et al., 2011). Perturbation of O-GlcNAc homeostasis is associated with human diseases including diabetes, Alzheimer's disease, and cardiovascular disease (Bond and Hanover, 2013; Brownlee, 2001; Hart et al., 2011; Yuzwa and Vocadlo, 2014). Moreover, OGT and O-GlcNAcylation are upregulated in a wide variety of tumor types and this appears to influence tumor biology by modulating critical regulators of cell proliferation (de Queiroz et al., 2014; Lynch and Reginato, 2011; Ma and Vosseller, 2014; Singh et al., 2015; Slawson and Hart, 2011). Thus, loss of O-GlcNAc homeostasis has serious consequences to normal cell function.
Given that O-GlcNAcylation plays a role in many cellular processes, but it is driven by only two enzymes, OGT and OGA must be tightly regulated. In fact, their activities are coordinately regulated to maintain O-GlcNAc homeostasis. Increases in OGT activity lead to concomitant increases in OGA activity and vice versa, thereby buffering cells from drastic shifts in O-GlcNAcylation. For example, OGA protein levels are downregulated upon OGT inhibition, OGT knockdown, or OGT knockout (Burén et al., 2016; Kazemi et al., 2010; Ortiz-Meoz et al., 2015). Conversely, upon OGA inhibition, OGT protein levels are downregulated (Slawson et al., 2005; Zhang et al., 2014). Although transcriptional control has been reported (Muthusamy et al., 2015; Zhang et al., 2014), mechanisms regulating the coordination of OGT and OGA activities remain largely undefined.
Recent studies identified thousands of transcripts in mammals that undergo intron retention, a relatively understudied form of alternative splicing (Boutz et al., 2015; Braunschweig et al., 2014; Yap et al., 2012). Polyadenylated transcripts containing one specifically retained intron are often retained in the nucleus, where they can be targeted for degradation by PABPN1 and PAPα/γ-mediated RNA decay (PPD) (Bresson et al., 2015). Alternatively, the posttranscriptional splicing of the nuclear transcripts with retained introns can be induced in response to extracellular signals to rapidly produce mRNAs (Boutz et al., 2015; Ninomiya et al., 2011). Importantly, the efficiency of splicing of retained introns is governed by gene-specific regulatory cues that respond to the cell environment and/or developmental state (Boutz et al., 2015; Ni et al., 2016; Ninomiya et al., 2011; Pendleton et al., 2017; Pimentel et al., 2016; Yap et al., 2012). Thus, unlike forms of alternative splicing in which distinct protein isoforms are generated, intron retention in mammals generally controls the levels and timing of the production of a mature mRNA.
A fraction of cellular OGT transcripts retain the fourth intron, suggesting that intron retention contributes to the regulation of OGT expression and O-GlcNAc homeostasis (Boutz et al., 2015; Bresson et al., 2015; Hanover et al., 2003). Here, we show that OGT intron retention is dynamically regulated in response to changes in O-GlcNAc levels. Under conditions of high O-GlcNAcylation, the nuclear OGT retained-intron (OGT-RI) isoform increases while inhibition of OGT decreases OGT-RI. We identify a conserved OGT intronic splicing silencer (ISS) that is necessary for OGT intron retention. Deletion of the ISS abolishes OGT intron retention and its responsiveness to metabolic conditions that alter O-GlcNAc levels in the cell. Importantly, loss of the ISS induces OGT expression, but it has little effect on overall O-GlcNAc levels or cell growth under normal conditions due to compensatory increases in OGA protein. However, inhibition of OGA is more toxic to cell lines that have ISS deletions when compared to wild-type cells. Similarly, anchorage-independent growth in soft agar and tumor growth in vivo are compromised in ISS-deletion lines upon OGA inhibition. We conclude that cells regulate OGT expression by intron retention through the activity of the OGT-ISS. Moreover, this regulatory mechanism is essential for cells to coordinate OGT and OGA activities and maintain O-GlcNAc homeostasis.
Results
OGT Expression Is Regulated by Intron Retention
Previous studies showed that the OGT RNA accumulates primarily as two isoforms: a fully spliced cytoplasmic mRNA and OGT-RI, a nuclear RNA that retains the fourth intron (Figure 1A; orange) (Boutz et al., 2015; Bresson et al., 2015; Hanover et al., 2003). In addition, this retained intron is considerably more conserved among vertebrates than other OGT introns (Figure 1A). To test whether OGT intron retention is regulated, we examined OGT isoform changes under several different treatments that alter bulk O-GlcNAcylation (Figure 1B). We treated cells with the OGT inhibitor OSMI-1 (Ortiz-Meoz et al., 2015) or with the OGA inhibitors thiamet-G (TG) or O-(2-acetamido-2-deoxy-D-glycopyranosylidene) amino-N-phenylcarbamate (PUGNAc) (Yuzwa et al., 2008). We also modulated O-GlcNAcylation indirectly by altering several key steps in the HBP. We deprived cells of glucose (Glc), treated cells with the GFAT inhibitor 6-diazo-5-oxo-L-norleucine (DON), or supplied cells with exogenous glucosamine (GlcN) (Figure 1B). We observed robust changes in intron retention upon these treatments in two cell lines, the HEK293 derivative 293A-TOA (Sahin et al., 2010) and in the colorectal carcinoma line HCT116 (Figure 1C). In most cases, the treatments that decrease O-GlcNAcylation (Figures 1B, red, and 1C, anti-O-GlcNAc) led to simultaneous increases in OGT mRNA and decreases in OGT-RI (Figure 1C). The only exception was that DON had little effect on 293A-TOA cells whereas it strongly stimulated OGT mRNA production in HCT116 cells. The reason for this is unknown, but DON has previously been reported to have cell-specific effects (Slawson et al., 2005). Conversely, the treatments that increase O-GlcNAcylation (Figures 1B, green, and 1C, anti-O-GlcNAc) led to loss of OGT mRNA and increases in OGT-RI. The changes in OGT isoform usage were rapid: TG, OSMI-1, and glucose depletion caused changes in isoform usage within a few hours in both cell lines (Figures 1D, 1E, and S1). Interestingly, HCT116 cells consistently showed higher intron retention than 293A-TOA cells in untreated conditions (∼40% versus 20%, Figure 1E), potentially reflecting increases in O-GlcNAcylation observed in colorectal cancers (Mi et al., 2011). These results show that cells regulate OGT intron retention and support a role for this regulation in the maintenance of O-GlcNAc homeostasis.
Figure 1. OGT Intron Retention Is Regulated in Response to O-GlcNAcylation.
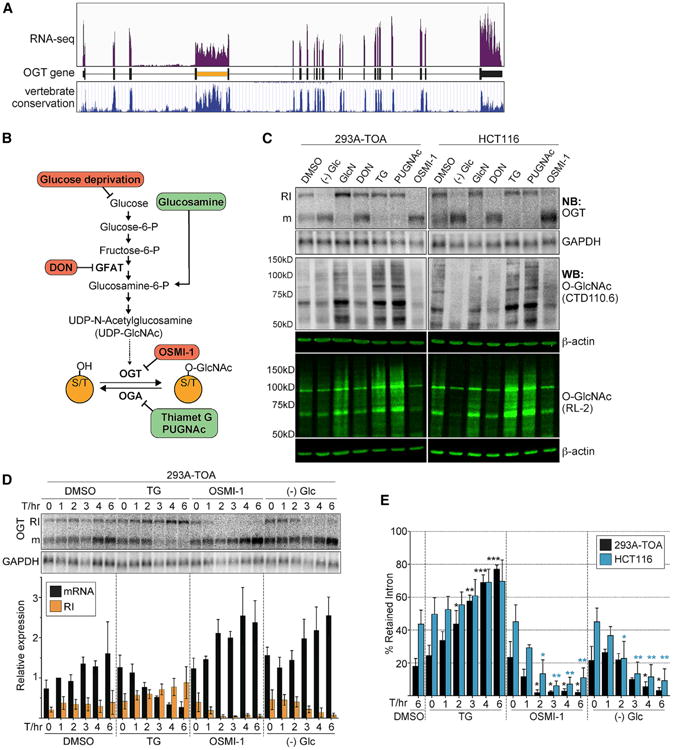
(A) RNA sequencing (RNA-seq) trace of reads mapping to the OGT gene (Bresson et al., 2015). The bottom panel is the vertebrate conservation track from UCSC genome browser.
(B) Simplified scheme of HBP and O-GlcNAcylation pathway. Treatments predicted to increase or decrease overall O-GlcNAcylation are shown in green or red, respectively.
(C) Analysis of treatments on OGT intron retention and O-GlcNAcylation. The top two panels are northern blots for OGT and GAPDH (control). RI and m mark OGT-RI and mRNA isoforms, respectively. O-GlcNAc was monitored using two different anti-O-GlcNAc antibodies (RL-2, CTD110.6), each of which has broad but distinct specificities (Tashima and Stanley, 2014). b-actin serves as a control.
(D) Representative northern blot and quantification of a time course of TG, OSMI-1, and glucose depletion in 293A-TOA cells. Data were normalized to the DMSO, 1-hr mRNA signal. Data are mean ± SD (n = 3).
(E) Comparison of the percent intron retention of HCT116 and 293A-TOA; same data as (D) and Figure S1. The blue and black asterisks are color coded as the bars. All statistical analyses are unpaired Student's t tests, and significance is annotated as *p < 0.05, **p < 0.01, or ***p < 0.001. Here the comparisons were referenced to the T = 0 value for each treatment. See also Figure S1.
Our data suggest that upon sensing low O-GlcNAc levels, cells induce efficient splicing of OGT to produce a cytoplasmic, translated mRNA. Conversely, OGT intron four splicing is inefficient in high O-GlcNAc conditions leading to nuclear-retained, untranslated RNAs. To further test this model, we examined OGT protein levels upon TG and OSMI-1 treatments. As expected, we observed that OGT protein levels increase upon OSMI-1 treatment, decrease in TG (Figure 2A), and the changes in protein levels tended to lag behind the changes of isoform usage (Figure 1D). As previously observed, OGA protein levels decrease upon OSMI-1 treatment (Ortiz-Meoz et al., 2015) (Figure 2A), supporting the existence of additional mechanisms that modulate OGA production in response to OGT activity.
Figure 2. OGT Intron Retention Regulates Cytoplasmic mRNA and Protein Production.
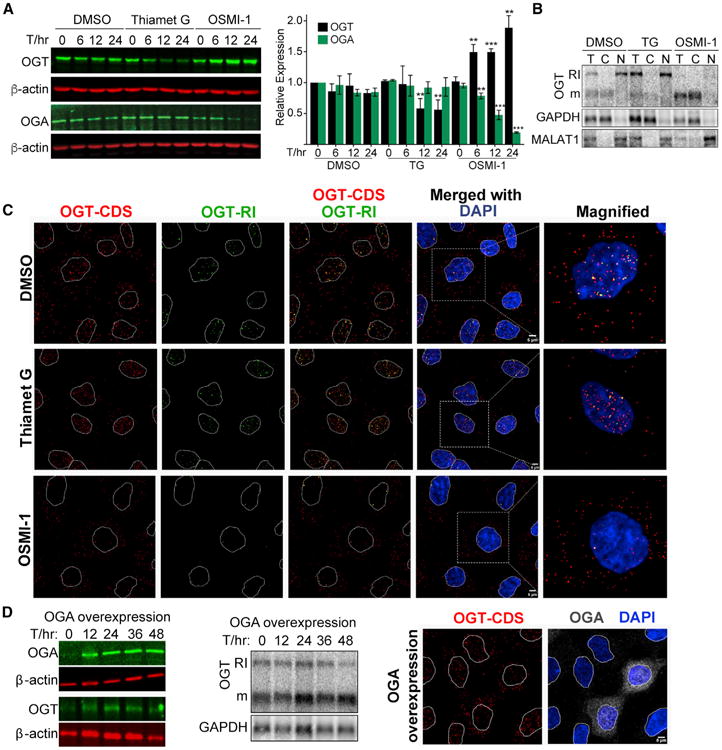
(A) Western blot and quantification of OGT and OGA protein levels after treatments in 293A-TOA cells. Data are represented as mean ± SD (n = 3). Statistical comparisons were referenced to the T = 0 value for each treatment.
(B) Northern blot of cell fractionation experiment with RNA from DMSO-, TG-, or OSMI-1-treated 293A-TOA cells. GAPDH mRNA and MALAT-1 noncoding RNA are cytoplasmic and nuclear markers, respectively. T, total; C, cytoplasmic; N, nuclear.
(C) FISH using probes to OGT coding sequence (CDS) or retained intron (RI) after the indicated treatments (6 hr) in 293A-TOA cells. Scale bar, 5 μm.
(D) Left: western blot using total lysate from 293A-TOA cells at the given times post-transfection. Middle: northern blot of the same samples. Right: FISH and indirect immunofluorescence were applied to the same cells using OGT-CDS probes and anti-OGA antibodies to mark the cells overexpressing OGA. See also Figure S2.
Next, we examined the localization of OGT isoforms by fractionation and fluorescence in situ hybridization (FISH) under basal, TG, and OSMI-1 treatments. We observed OGT-RI in the nuclear fraction and OGT mRNA in the cytoplasmic fraction under both basal and treated conditions (Figure 2B). We additionally performed FISH with probe sets that hybridize to the OGT coding sequence (OGT-CDS) or retained intron (OGT-RI) (Figure 2C). In control cells (DMSO), the signals from OGT-CDS were found in both the nucleus and cytoplasm, while OGT-RI was restricted to the nucleus. Upon TG treatment, the signal was primarily nuclear for both the CDS and RI probes. In contrast, OSMI-1 treatment led to increased cytoplasmic signal and reduced nuclear signal. In many cells, approximately two intense nuclear spots were detected, which presumably represent sites of transcription. Finally, we overexpressed OGA to test whether increases in its activity mimic OGT inhibition (Figure 2D). OGA overexpression was efficient (Figure 2D, left), but only subtle increases in OGT splicing were observed at the bulk level (Figure 2D, middle). However, when we identified cells specifically overexpressing OGA by indirect immunofluorescence, an increase in cytoplasmic signal of OGT-CDS was observed (Figure 2D, right) suggesting that the lack of bulk effects was due to low transfection efficiency. Indeed, co-transfection of the OGA overexpression construct with a puromycinselectable plasmid led to decreases in intron retention in puromycin-selected cells (Figure S2). We conclude that cells regulate OGT production and activity by regulating the splicing efficiency of OGT intron four.
A Conserved Intronic Element Is Necessary for OGT Intron Retention
To gain insights into the mechanism of intron retention, we sought to identify a candidate cis-acting regulator of OGT intron retention. To do so, we generated a reporter construct that includes β-globin exonic sequences (Figure 3A, yellow) and the efficiently spliced second intron of β-globin (Figure 3A, black line) flanking exon 4, intron 4, and exon 5 of OGT (Figure 3A, black and orange). As expected, the β-globin reporter (βΔ1) without any OGT sequence was efficiently spliced, but the reporter containing the full-length retained intron showed little fully spliced product (Figure 3A, lanes 1 versus 2). The presence of the large OGT retained intron also affected splicing of adjacent β-globin sequences (Figure 3A bottom, lanes 8–21), which is not apparent in the case of the endogenous gene and likely reflects an artifact of heterologous overexpression. Nonetheless, we tested deletions of the retained intron to determine whether specific regions were necessary for intron retention. Deletions including the ∼1,500 nucleotides (nt) between 798–2297 restored accumulation of fully spliced product (Figure 3A, lanes 3–5). Sub-deletions of this region showed that an upstream fragment (nt 798–1285) was dispensable for intron retention, whereas a 526-nt fragment (nt 1771–2297) was necessary (Figure 3A, lanes 6 and 7). Therefore, the 526-nt fragment is a candidate cis-acting OGT intronic splicing silencer (ISS). Importantly, OSMI-1 increased the efficiency of β-OGT splicing, although a considerable amount of retained-intron transcript remains, presumably due to overexpression of the reporter (Figure 3B, lane 3 versus 1). We saw only a subtle loss of mRNA in response to TG, and no increase in intron retention (Figure 3B, lane 2). Most importantly, the candidate ISS-deleted reporter (Figure 3A, #7) was constitutively spliced and not responsive to TG or OSMI-1, supporting its role as cis-acting regulator of OGT intron retention (Figure 3B, lanes 4–6). Finally, insertion of the ISS into β-globin intron 2 was sufficient to decrease splicing compared to reverse orientation or no insert controls (Figure 3C, left). However, the βΔ1-ISS-F construct was not responsive to drug treatments (Figure 3C, right), suggesting that additional cis-acting sequences are required for regulated de-repression of ISS activity. Thus, we have identified a 526-nt ISS within OGT intron four that is necessary and sufficient to repress splicing in a heterologous reporter gene and is a candidate cis-acting element to promote OGT intron retention. Interestingly, a region of human OGT intron four that is conserved to jawless fish overlaps the candidate ISS (Figure S3). Thus, the OGT-ISS may be an ancient cis-acting regulator of OGT expression.
Figure 3. Identification of a Candidate OGT Intronic Splicing Silencer.
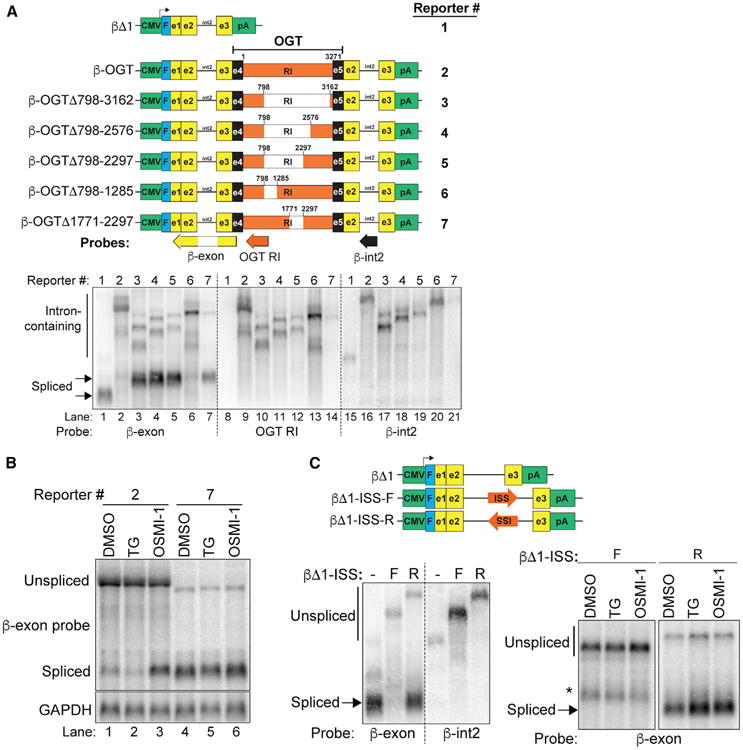
(A) Top: diagrams of reporters. Green, promoter and polyadenylation signals; blue, Flag-tag; yellow, β-globin exons; black line, β-globin intron 2; black, OGT exons; orange, OGT intron 4. Numbering is relative to the OGT intron sequence only; diagrams are not to scale. Positions of northern blot probes are also shown. Bottom: northern blot of RNA from cells transfected with the indicated reporter. The samples were loaded in parallel on the same gel. The membrane was subsequently cut (dashed lines) and hybridized to the indicated probe.
(B) Northern blot of RNA from 293A-TOA cells transfected with reporters and treated for 6 hr as indicated.
(C) Top: schematic of β-globin reporters with ISS in the forward (F) or reverse (R) orientation. Bottom: northern blots of reporter assays with transfected constructs, probes, and treatments as indicated. *Indicates an unknown transcript, likely a cryptic splice product. See also Figure S3.
The ISS Is Essential for Basal and Induced Intron Retention
We next investigated whether the ISS is essential for intron retention of the endogenous OGT RNA. To do so, we used CRISPR to create double-stranded DNA breaks on either side of the ISS in HCT116 cells and screened clones that produced ISS-deletions by non-homologous end joining (Figure 4A). We used HCT116 cells because they are stable diploid cells that are derived from a male colorectal cancer patient thereby increasing our chances of deleting the ISS in the X-linked OGT gene. We isolated 11 independent hemizygous ISS-deletion clones and fortuitously recovered one in which the ISS sequence was reversed (ΔISS #14). Herein, we refer to these strains collectively as ISS-deletion or ΔISS clones. We also maintained three clones with wild-type ISS sequence as controls. Strikingly, the 12 ΔISS clones, but none of the wild-type clones, completely lost expression of OGT-RI isoform (Figure 4B). Thus, under normal cell culture growth conditions, deletion of the ISS is sufficient to completely abrogate intron retention during steady-state cell growth. This observation validates the conclusion that the ISS is a functional cis-acting suppressor of splicing of OGT intron four.
Figure 4. The ISS Is Necessary for Basal and Induced OGT Intron Retention.
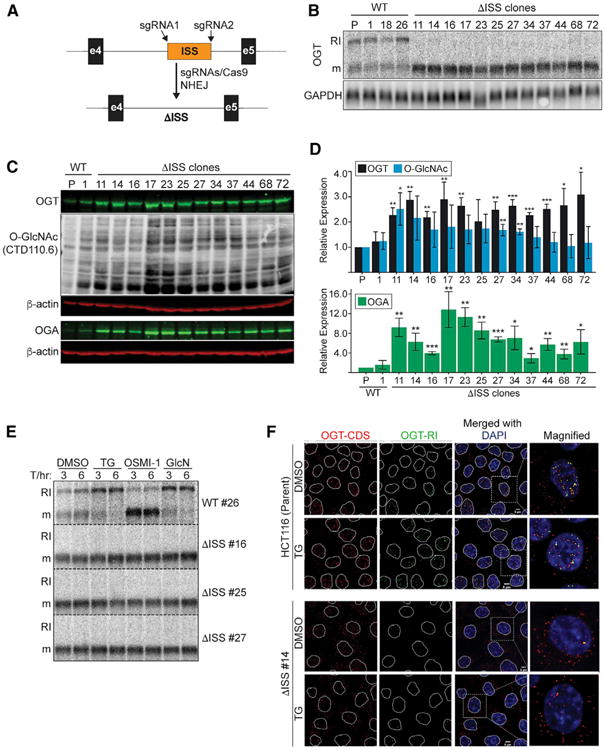
(A) CRISPR genomic deletion strategy.
(B) Northern blot using RNA from clonal wild-type and ΔISS CRISPR lines. The lane marked “P” is the parental, HCT116 line.
(C) Representative western blots of parental, wild-type #1, and all 12 ISS-deletion clones. The top three panels (OGT, O-GlcNAc, and β-actin) were the same blot, and the bottom two panels were a separate blot with the same samples.
(D) Quantification of data from westerns as in (C). Data are represented as mean ± SD (n = 3). Statistical comparisons were referenced to the wild-type #1 value for each treatment.
(E) Northern blot of four clones under the indicated treatments. Data are from the same blot, but displayed vertically for presentation (dashed lines).
(F) FISH analysis (as in Figure 2C) of HCT116 and ISS-reversed clone #14 ± TG. Scale bar, 5 μm.
We observed ∼2- to -3-fold increases in OGT protein levels in the ISS-deletion lines (Figures 4C, top, and 4D, black bars), but overall increases of O-GlcNAcylation were modest (Figures 4C and 4D, blue bars). The lack of more dramatic increases in bulk O-GlcNAc can be explained by the concomitant increases in OGA expression observed in the ISS-deletion lines (Figures 4C, bottom, and 4D, green bars). These data further highlight that O-GlcNAc homeostasis is controlled by balancing the activities of OGT and OGA, and they further suggest that the ISS is an important component for maintaining this regulatory balance.
The data presented thus far show that the ISS is essential for basal intron retention. In principle, the ISS may be dispensable for the induction of intron retention in response to increased O-GlcNAcylation. Therefore, we tested three of our ISS-deletion clones and one wild-type clone under drug treatments. The wild-type clone mirrored the parental line (Figure 1C) in response to TG, OSMI-1, and glucosamine, but ISS-deletion clones were nonresponsive (Figure 4E). In addition, the OGT transcripts in ΔISS clones were more cytoplasmic than the parental lines and were essentially undetectable with the OGT-RI probes (Figure 4F). Upon TG treatment, the parental lines displayed increasingly nuclear OGT-RI signal, while ΔISS clone RNA remained unchanged. We conclude that the ISS is necessary for the regulation of intron retention in response to changes in cellular metabolic conditions.
The ISS Is Necessary for Cell Survival upon OGA Inhibition
Our data suggest that the ISS plays a critical role in downregulation of OGT production in response to increased O-GlcNAcylation. This model predicts that OGA inhibition will be toxic to cell lines lacking the ISS, because they cannot maintain proper O-GlcNAc levels by downregulating OGT. To test this, we first treated cultured cells with DMSO or the OGA inhibitor TG and compared the growth of four ΔISS and one wild-type clone with the parental HCT116 line. We observed no major growth differences between the ΔISS and wild-type lines under control treatment (DMSO) (Figure 5A, left). In contrast, DISS clones were sensitive to TG treatment after ∼3 days, whereas wild-type cell growth was largely unaffected (Figure 5A, middle and right). Next, we tested all of our deletion and wild-type clones for growth in soft agar in the presence and absence of TG. As previously reported, the parental HCT116 cells promote anchorage-independent growth by forming colonies in soft agar, a hallmark of cellular transformation (Luo et al., 2008) (Figures 5B and 5C). We observed no major differences between wild-type and deletion clones after DMSO addition, but TG treatment robustly inhibited colony formation in ISS-deletion clones compared to wild-type cells (Figures 5B and 5C). Thus, as predicted by our model, ISS-deleted cells are hyper sensitive to TG in vitro. These data further support a biologically relevant role for the ISS in maintaining O-GlcNAc homeostasis.
Figure 5. The ISS Is Essential for O-GlcNAc Homeostasis in Cultured Colorectal Cancer Cells.
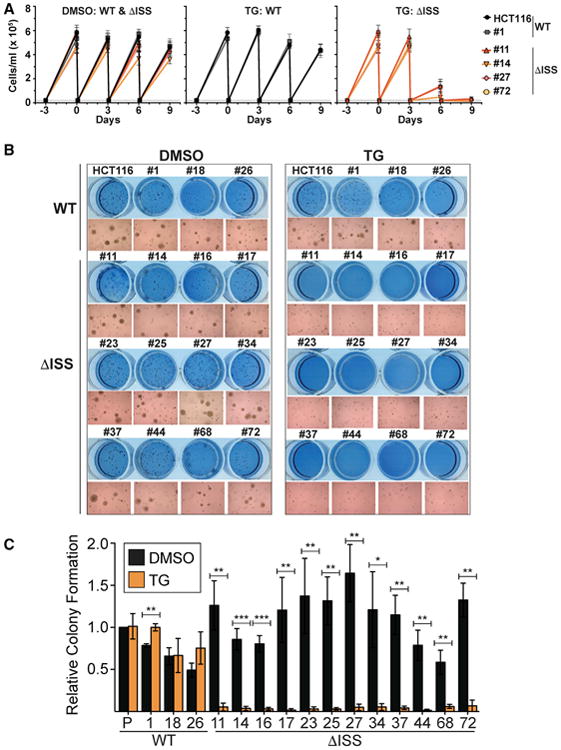
(A) Growth curves of the indicated lines after DMSO (left) or TG treatment (middle, right). Treatment was initiated at day 0. Cells were counted and re-seeded at 0.2 × 105 cells/ml (dashed line) every 3 days.
(B) Representative soft agar assay using wild-type and ISS-deletion clones shown as crystal violet staining or 10× magnified image.
(C) Quantification of soft agar assays. Values are relative to the HCT116 (P) DMSO-treated cells. Data are represented as mean ± SD (n = 3). Statistics compared DMSO to TG treatment.
The ISS Is Necessary for Tumor Growth upon OGA Inhibition In Vivo
To validate that the ISS is important for cell growth in vivo, we compared the growth of tumors produced by the WT and ISS-deletion clones in a mouse xenograft assay. Seventeen days following subcutaneous injection of cells, mice were administered daily intraperitoneal injections with 20 mg/kg TG or PBS as a control (Figure 6A). Tumor volume was estimated every 3 days after initiation of treatment. On day 24 post-treatment, mice were sacrificed, and the tumors were weighed. We analyzed the parental line, three wild-type clones, and nine independent ISS-deletion lines. Growth of the tumors from wild-type clones showed no significant differences upon TG treatment, whereas tumors derived from all ISS-deletion clones were TG-sensitive (Figures 6B and S4A–S4C). Comparison of estimated tumor volumes from pre-treatment (day 0) to day 21 post-treatment yielded significant differences for the TG-treated ΔISS clones (Figures 6C and S4B). Similarly, the final weights of the ISS-deletion clones ± TG were significantly less than those in the wild-type clones (Figures 6D and S4D). Specifically, the wild-type clones weighed an average of 1.2-fold more in TG-treated mice, whereas the TG-treated ΔISS clones were on average 51% of the weight of the PBS-treated counterparts. Not surprisingly, the independent clones displayed different growth characteristics presumably due to random changes that occur during single-cell selection and clonal expansion. Importantly, this heterogeneity contributes rigor to our strategy as the ISS-dependent differences can confidently be attributed to the loss of ISS activity rather than stochastic differences among selected clones.
Figure 6. The ISS Is Important for O-GlcNAc Homeostasis in Colorectal Cancer Cell Tumors Grown In Vivo.
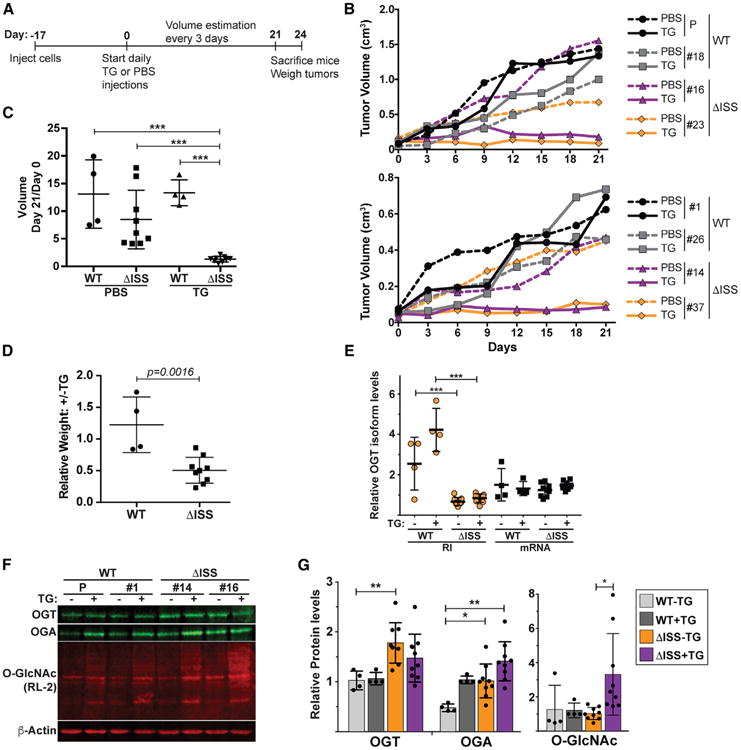
(A) Time line of xenograft experiments.
(B) Representative growth curves of wild-type and ΔISS tumor volumes in vivo. Wild-type clones are in black and gray and mutants in purple and orange. Growth of control (PBS) and TG-treated mice are in dashed and solid lines, respectively.
(C) Plot comparing the estimated tumor volumes at day 21 relative to day 0 post-treatment. The vertical line is the mean and the error bars are SD.
(D) Ratio of the ± TG tumor weights derived from each individual clone.
(E) qRT-PCR results of OGT isoforms in tumor samples. Primers amplify a sequence within the OGT-RI or across the exon 4–5 junction (mRNA). The OGT data were normalized to GAPDH and are presented relative to the mRNA signal for the parental clone. See Figure S4E for data on each individual tumor.
(F) Representative western blots using protein extracted form tumor samples.
(G) Quantification of protein from western blots as in (F). Bars represent the mean, error bars are SD, and each data point is shown (n = 3). If no asterisks are shown, the comparisons were not statistically significant. WT+TG samples were excluded from statistical tests as one of these values was set to 1 for normalization in each western blot. See also Figure S4.
We also analyzed RNA and protein from the tumor samples. As expected, the OGT-RI isoform was elevated in tumors derived from wild-type cells compared to those derived from ISS-deletion clones (Figures 6E and S4E). In addition, OGT protein levels increased ∼1.8-fold in the untreated ΔISS lines, and we observed increases in OGA in ΔISS lines and upon TG treatment in wild-type lines (Figures 6F and 6G). Furthermore, bulk O-GlcNAc levels were highest in TG-treated ΔISS tumors on average, although this only reached statistical significance when comparing the treated and untreated ΔISS lines due to high variability (Figure 6G).
A few observations suggest differences in the regulation of OGT in tumors compared to cell culture. Most surprisingly, while we observed increases of OGT protein in ΔISS tumors, the mRNA isoform was not statistically significantly upregulated in vivo (Figure 6E). In addition, OGA protein levels increased upon TG treatment in vivo, but not in the cultured cells (Figures 2A, 6F, and 6G). Moreover, we did not observe an increase in OGT-RI for two of the four wild-type lines upon TG treatment (Figures 6E and S4E). However, these two lines displayed >70% OGT-RI in the control mice (HCT116 and #1; Figure S4E), which approaches the highest levels observed in cells (Figure 1E). These observations point to additional complexities in the regulation of O-GlcNAc in vivo that may arise from the tumor environment or length of treatment. Moreover, many of the ISS-deleted cells that overexpressed OGT mRNA and protein are likely to have stopped growing over the 24-day course. Therefore, examination of the final tumors may be biased against the cells that substantially overproduce OGT mRNA, protein, and O-GlcNAc. Nonetheless, the TG-dependent changes in tumor growth and the differences in OGT intron retention in ISS-deleted cells clearly indicate that the ISS is an important regulator of OGT activity in vivo. They further suggest uncontrolled O-GlcNAc activity is detrimental to cell growth, even in cancer cells that generally upregulate O-GlcNAc levels.
Discussion
The activities of OGT and OGA are governed by multilayered feedback mechanisms that finely tune the overall levels of O-GlcNAcylation in the cell. Multiple studies have established that alterations in OGA activity affect OGT activity and vice versa (Burén et al., 2016; Kazemi et al., 2010; Ortiz-Meoz et al., 2015; Slawson et al., 2005; Zhang et al., 2014). While the mechanism used to modulate OGA protein levels in response to O-GlcNAc remain unknown, our data show that cells employ a posttranscriptional mechanism to coordinate OGT expression with cellular O-GlcNAcylation. We observed robust effects on OGT intron retention using strong modulators of overall O-GlcNAc levels (Figure 1), but we speculate that cells rarely experience such dramatic alterations of O-GlcNAcylation in natural contexts. Instead, it seems more likely that this pathway is used as an ongoing surveillance mechanism that fine-tunes OGT mRNA levels in response cellular O-GlcNAcylation. Using this mechanism, cells can maintain constant transcription rates of the essential OGT gene, but also preserve O-GlcNAc homeostasis by modulating production of the mature mRNA.
In normal culture conditions and in vivo, cells produce considerable amounts of the OGT-RI isoform (Figures 1, 6E, and S4E). While our data show that intron 4 splicing is regulated, the OGT-RI itself could be subject to a number of distinct cellular fates. First, some transcripts with retained introns serve as nuclear reservoirs of pre-mRNA that can be quickly induced to produce spliced mRNA (Boutz et al., 2015; Ninomiya et al., 2011). This is an attractive hypothesis for OGT-RI given the rapid response to different stimuli (Figure 1), but a precursor-product relationship between OGT-RI and mRNA has yet to be established. Second, transcripts with retained introns could be nonfunctional “dead-ends” that are degraded in the nucleus. Indeed, we previously showed that PPD degrades OGT-RI, albeit less efficiently than other PPD targets (Bresson et al., 2015). Third, OGT-RI could be subject to slow posttranscriptional splicing. In general, mammalian introns are spliced co-transcriptionally, but some introns are spliced after polyadenylation (Ameur et al., 2011; Bhatt et al., 2012; Brugiolo et al., 2013; Girard et al., 2012; Shalgi et al., 2014; Tilgner et al., 2012; Vargas et al., 2011; Windhager et al., 2012). Inhibition of PPD increases OGT-RI abundance without increasing OGT mRNA suggesting that OGT-RI is not a precursor to OGTmRNA under normal growth conditions (Bresson et al., 2015). In some cases, slow splicing contributes to AS potential as it allows the splicing machinery to choose among alternate exons after they have all emerged from the transcriptional machinery (Ameur et al., 2011; Tilgner et al., 2012; Vargas et al., 2011). For OGT-RI, there is no choice between alternate exons, so the regulation does not need to occur subsequent to transcription of all alternate exons. Therefore, it seems unlikely that accumulation of OGT-RI is solely due to slow splicing. Fourth, a speculative model posits that OGT-RI accumulates in the nucleus and functions as a nuclear noncoding RNA. Intriguingly, we observed significant changes in tumor growth and OGT-RI levels in ΔISS tumors in vivo, but the mRNA levels were not significantly changed (Figure 6). Thus, it is formally possible that the OGT-RI RNA itself modulates O-GlcNAc homeostasis by an unknown nuclear mechanism. Fifth, OGT-RI has been suggested to be a protein-coding mRNA (Hanover et al., 2003). Further experimentation is necessary to distinguish among these non-mutually exclusive mechanisms, but the results here provide a framework for examination of the fate of a retained intron transcript in biologically relevant regulatory pathway.
Our data strongly support the model that cells regulate OGT expression by control of OGT-RI and mRNA isoforms, which are the two most highly expressed OGT RNA isoforms (Bresson et al., 2015; Hanover et al., 2003). However, OGT activity is highly regulated and additional protein and RNA isoforms likely contribute to its function in vivo (Hanover et al., 2003; Kreppel et al., 1997; Lubas et al., 1997). Moreover, the conservation of the ISS suggests that intron retention is conserved in vertebrates and jawless fish. In contrast, Drosophila OGT does not have an ISS-like element, but its expression appears to be regulated posttranscriptionally by controlling the splicing of a long intron (Ashton-Beaucage et al., 2010; Hanover et al., 2012). Thus, we are only beginning to define the multiple mechanisms that contribute to posttranscriptional control of OGT expression across cell types and species.
O-GlcNAcylation has been linked to a number of disease states including cancer, diabetes, and Alzheimer's disease (AD) (Bond and Hanover, 2013; Brownlee, 2001; Hart et al., 2011; Ma and Vosseller, 2014; Marshall, 2006; Singh et al., 2015). The compensatory mechanisms controlling OGT and OGA activities have important implications for potential therapeutic interventions that target O-GlcNAcylation. For example, in ovarian cancer cells, expression of p53 is depleted, but increased O-GlcNAcylation can induce p53 stabilization. Accordingly, combinatorial treatment of the chemotherapeutic cisplatin with TG decreased tumor cell growth in a p53-dependent fashion (de Queiroz et al., 2016). In addition, O-GlcNAcylation decreases protein aggregations associated with Alzheimer's disease and TG treatment of a mouse model led to increased O-GlcNAcylation and prevention of neuronal loss (Yuzwa et al., 2012). Thus, it has been suggested that modulation of O-GlcNAc levels could potentially provide a strategy for novel therapeutic approaches (de Queiroz et al., 2014; Yuzwa and Vocadlo, 2014). The TG-dependent increases in OGT intron retention reported here have implications for therapeutic elevation of O-GlcNAc as a strategy. On one hand, the compensatory downregulation of OGT by intron retention could mute increases in O-GlcNAcylation thereby diminishing the effectiveness of treatments. On the other hand, the compensation by intron retention may maintain sufficient control of O-GlcNAc levels in normal cells to reduce the toxicity resulting from unchecked O-GlcNAcylation. In either case, it will be important to determine whether manipulation of intron retention and ISS function modulates the effectiveness of O-GlcNAc treatments in vivo.
Nearly all cancer types upregulate bulk O-GlcNAcylation, and the elevated O-GlcNAc levels appear to be important for cancer cell proliferation, epigenetics, and metastasis (de Queiroz et al., 2014; Lynch and Reginato, 2011; Ma and Vosseller, 2014; Singh et al., 2015). Consistent with this, OGT and O-GlcNAcylation are upregulated in colon cancer tissues compared to adjacent normal tissues and in colon cancer lines including HCT116 (Bhatt et al., 2012; Itkonen et al., 2013; Phueaouan et al., 2013; Singh et al., 2015; Steenackers et al., 2016; Yehezkel et al., 2012). Interestingly, HCT116 cells can tolerate OGA inhibition or ISS deletion, but ISS-deleted cells cannot survive under OGA inhibition (Figures 5 and 6). This is consistent with previous studies showing that overexpression of OGT or increased O-GlcNAcylation disrupts the cell cycle (Slawson et al., 2005). Thus, while cancer cells appear to be selected for elevated GlcNAc levels, they also require negative regulation of overall O-GlcNAcylation.
We have shown that the ISS plays a role in the posttranscriptional regulation of OGT production, however, future studies are warranted to identify the trans-acting factor(s) that bind the ISS. Many RNA-binding proteins (RBP) are modified by O-GlcNAc (Hahne et al., 2012; Nandi et al., 2006; Teo et al., 2010), so we speculate that the O-GlcNAc status of a specific RBP(s) regulates OGT intron retention. For example, under high GlcNAc conditions, the O-GlcNAcylated RBP binds to the ISS resulting in intron retention. When O-GlcNAc levels drop, the RBP is no longer O-GlcNAcylated leading to splicing of the retained intron. We postulate that RBP O-GlcNAcylation decreases RNA-binding affinity or changes the interactions with additional proteins that dictate splicing efficiency. Importantly, the changes in intron retention occur within a few hours (Figure 1), so the RBP in question would need to have a rapid turnover in its O-GlcNAc status in order to drive these changes. Ongoing efforts focused on testing this model will provide additional insight into regulation of O-GlcNAc homeostasis by intron retention.
Experimental Procedures
Cell Culture
293A-TOA cells were grown in DMEM medium (Sigma) supplemented with 10% tetracycline-free fetal bovine serum (Clontech), 1 × penicillin/streptomycin (Sigma), 2 mM L-glutamate, and 100 μg/mL G418. HCT116 cells were grown in McCoy's 5A medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Sigma) and 1 × Antibiotic-Antimycotic (Thermo Fisher Scientific). DON (100 μM), TG (1 μM), glucosamine (10 mM), glucose-free DMEM, and DMSO were purchased from Sigma. OSMI-1 (10 μM) was initially provided by Dr. Suzanne Walker (Harvard Medical School) and subsequently was purchased from Glixx Laboratories. PUGNAc (50 μM final) was obtained from Santa Cruz Biotechnology. For DNA transfection, cells were transfected with TransIT-293 (Mirus) for 293A-TOA or FuGENE HD (Promega) for HCT116 following the manufacturer's protocol. In OGA overexpression experiments (Figure S2), pcDNA-OGA and pX459 were co-transfected at a 1:1 ratio. The next day, the cells were split, replated at 50% density, and 5 hr later puromycin (1 μg/μL) was added. Fresh puromycin containing media was added ∼14 hr prior to harvesting at 72 hr post transfection.
RNA Methods, Western Blotting, and Plasmid Construction
Standard molecular procedures for RNA analyses, western blotting, and plasmid construction are described in the Supplemental Experimental Procedures. Oligonucleotides are listed in Table S1. Antibodies used were: anti-OGT (Sigma, O6264), anti-OGA (Sigma, SAB4200267), anti-O-GlcNAc (CTD110.6; Santa Cruz Biotechnologies, SC-59623 or RL-2; Thermo Fisher Scientific, MA1-072), and/or anti-β-actin (Abcam, ab6276). IRDye-680 or -800 conjugated secondary antibodies (LI-COR Bioscience) and HRP conjugated anti mouse IgM antibody (CTD110.6 only) were used to visualize the detected protein bands through an Odyssey Fc Imaging System and quantitative analyses were performed using ImageStudio software (LI-COR Bioscience).
FISH
For multi-color RNA FISH, oligonucleotide probe sets (48 probes each) were designed by Stellaris probe designer (Biosearch Technologies) with Quasar 670 (OGT-CDS) or CAL Fluor Red 610 (OGT-RI). Procedures were based on the manufacturer's protocols, with minor revisions as described in the Supplemental Experimental Procedures.
Generation of OGT-ISS-Deleted Cell Lines Using CRISPR-Cas9
Single-guide RNAs (sgRNAs) specific to up- or down-stream region of OGTISS were designed using Target Finder (http://crispr.mit.edu). HCT116 cells were transfected with pX458-OGT-ISS-upstream and pX459-OGT-ISS-downstream. After 24 hr, we selected co-transfected cells for GFP expression by MoFlo Cell Sorter (Flow Cytometry Core at UT Southwestern Medical Center) and then selected by puromycin treatment (1 μg/mL). The surviving cells were cloned in 96-well plates by limiting dilution. OGT-ISS deletions were identified by PCR genotyping using primers NC2260 and NC2261 (Table S1) and sequence verified (NC2260).
Cell Viability Assay
Cells were trypsinized and counted with a hemocytometer using Trypan blue (0.4%) exclusion to assess cell viability. Counts were performed in duplicate and averaged. Each clone was counted and seeded at 0.2 × 105 cells/mL with or without TG in a 12-well plate every 3 days.
Anchorage-Independent Growth Assay
HCT116 or its clonal derivatives were mixed with 1 mL of 0.37% top agar (SeaKem agarose, Lonza) in M25 (McCoy's 5A medium supplemented with 25% FBS) plus 1× Antibiotic-Antimycotic ± TG (2 μM final) were plated in one well of 12-well plate pre-coated with 1 mL of 0.5% bottom agar in the same medium as the top agar. The colonies were grown by changing media with or without TG every 3 days and then visualized by staining with 0.005% crystal violet solution on 15 days after plating or by capturing 10× magnified image using Zeiss Axio Observer Z1. Three biological replicates were performed. In one case, 104 cells were plated, while in the subsequent two experiments, 103 were plated. The stained colonies were counted and the values were normalized to the HCT116 DMSO-treated sample.
Animals and Xenograft Assay
All procedures involving mice were approved by the Institutional Animal Care and Use Committee (IACUC) of UT Southwestern Medical Center. Four-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) purchased from the Jackson Laboratories were used in xenograft experiments. HCT116 or its clonal derivative cells (1.0 × 106) were injected subcutaneously into the left and right flanks of the mice. Drug treatment by intraperitoneal (i.p.) injection was started on the 17th day after tumor inoculation. In three cases, the matched duplicates showed >5-fold variation prior to drug treatment and were therefore excluded from the analysis. One group of mice received 20 mg/kg TG in 13 PBS by i.p. injection every day, and the other group received 13 PBS as a control. Tumor volume was estimated using digital calipers every 3 days and calculated with the formula (length × width2)/2. All mice were euthanized on the day 24 post drug treatment, and the tumors were resected and the tumor weight was recorded. Each of the tumors was homogenized in Buffer RLT of RNeasy mini kit (QIAGEN) using Precellys homogenizer (Bertin Instruments) at 4°C. RNA or proteins were isolated from the supernatant of each ruptured tumors after centrifugation with high speed using TRI re-agent following the manufacturer's protocols.
Supplementary Material
Highlights.
O-GlcNAc transferase (OGT) expression is regulated by intron retention
OGT intron retention dynamically responds to O-GlcNAc levels
A conserved intronic splicing silencer (ISS) regulates OGT expression
The OGT-ISS is necessary for maintaining O-GlcNAc homeostasis
In Brief.
O-GlcNAc transferase (OGT) modifies cellular proteins, but the mechanisms that regulate OGT expression remain unclear. Park et al. show intron retention of the OGT transcript is responsive to cellular O-GlcNAc levels. They further define an intronic splicing silencer that is necessary to maintain O-GlcNAc homeostasis in cells and tumors.
Acknowledgments
We thank Dr. Suzanne Walker for generously providing OSMI-1. We thank Drs. Julio Ruiz and Aparna Anantharaman for helpful comments on the manuscript. The work was supported by the American Cancer Society (RSG-14-064-01-RMC to N.K.C.), the Sidney Kimmel Foundation (SKF-15-067 to K.A.O), the Welch Foundation (I-1881 to K.A.O.), and the NIH (GM113874 to N.K.C. and R21DK112733 to J.J.K.). N.K.C. is a Southwestern Medical Foundation Scholar in Biomedical Research. K.A.O. is a CPRIT Scholar in Cancer Research.
Footnotes
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures, four figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.07.017.
Author Contributions: N.K.C. and S.-K.P. conceived the study and wrote the manuscript. S.-K.P., X.Z., K.E.P., J.J.K., K.A.O., and N.K.C. designed the experiments, analyzed the results, and edited the manuscript. S.-K.P., X.Z., K.E.P., and O.V.H. performed the experiments.
References
- Ameur A, Zaghlool A, Halvardson J, Wetterbom A, Gyllensten U, Cavelier L, Feuk L. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat Struct Mol Biol. 2011;18:1435–1440. doi: 10.1038/nsmb.2143. [DOI] [PubMed] [Google Scholar]
- Ashton-Beaucage D, Udell CM, Lavoie H, Baril C, Lefrançois M, Chagnon P, Gendron P, Caron-Lizotte O, Bonneil E, Thibault P, Therrien M. The exon junction complex controls the splicing of MAPK and other long intron-containing transcripts in Drosophila. Cell. 2010;143:251–262. doi: 10.1016/j.cell.2010.09.014. [DOI] [PubMed] [Google Scholar]
- Bhatt DM, Pandya-Jones A, Tong AJ, Barozzi I, Lissner MM, Natoli G, Black DL, Smale ST. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell. 2012;150:279–290. doi: 10.1016/j.cell.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, Hanover JA. O-GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr. 2013;33:205–229. doi: 10.1146/annurev-nutr-071812-161240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. 2015;208:869–880. doi: 10.1083/jcb.201501101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Bhutkar A, Sharp PA. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015;29:63–80. doi: 10.1101/gad.247361.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014;24:1774–1786. doi: 10.1101/gr.177790.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresson SM, Hunter OV, Hunter AC, Conrad NK. Canonical poly(A) polymerase activity promotes the decay of a wide variety of mammalian nuclear RNAs. PLoS Genet. 2015;11:e1005610. doi: 10.1371/journal.pgen.1005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Brugiolo M, Herzel L, Neugebauer KM. Counting on co-transcriptional splicing. F1000Prime Rep. 2013;5:9. doi: 10.12703/P5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burén S, Gomes AL, Teijeiro A, Fawal MA, Yilmaz M, Tummala KS, Perez M, Rodriguez-Justo M, Campos-Olivas R, Megías D, Djouder N. Regulation of OGT by URI in response to glucose confers c-MYC-dependent survival mechanisms. Cancer Cell. 2016;30:290–307. doi: 10.1016/j.ccell.2016.06.023. [DOI] [PubMed] [Google Scholar]
- de Queiroz RM, Carvalho E, Dias WB. O-GlcNAcylation: the sweet side of the cancer. Front Oncol. 2014;4:132. doi: 10.3389/fonc.2014.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Queiroz RM, Madan R, Chien J, Dias WB, Slawson C. Changes in O-linked N-acetylglucosamine (O-GlcNAc) homeostasis activate the p53 pathway in ovarian cancer cells. J Biol Chem. 2016;291:18897–18914. doi: 10.1074/jbc.M116.734533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- Girard C, Will CL, Peng J, Makarov EM, Kastner B, Lemm I, Urlaub H, Hartmuth K, Lührmann R. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat Commun. 2012;3:994. doi: 10.1038/ncomms1998. [DOI] [PubMed] [Google Scholar]
- Hahne H, Gholami AM, Kuster B. Discovery of O-GlcNAcmodified proteins in published large-scale proteome data. Mol Cell Proteomics. 2012;11:843–850. doi: 10.1074/mcp.M112.019463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JA, Yu S, Lubas WB, Shin SH, Ragano-Caracciola M, Kochran J, Love DC. Mitochondrial and nucleocytoplasmic isoforms of O-linked GlcNAc transferase encoded by a single mammalian gene. Arch Biochem Biophys. 2003;409:287–297. doi: 10.1016/s0003-9861(02)00578-7. [DOI] [PubMed] [Google Scholar]
- Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13:312–321. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, Svindland A, Schlomm T, Mills IG. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 2013;73:5277–5287. doi: 10.1158/0008-5472.CAN-13-0549. [DOI] [PubMed] [Google Scholar]
- Kazemi Z, Chang H, Haserodt S, McKen C, Zachara NE. O-linked beta-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3beta-dependent manner. J Biol Chem. 2010;285:39096–39107. doi: 10.1074/jbc.M110.131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–9315. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- Lubas WA, Frank DW, Krause M, Hanover JA. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem. 1997;272:9316–9324. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- Luo W, Liu J, Li J, Zhang D, Liu M, Addo JK, Patil S, Zhang L, Yu J, Buolamwini JK, et al. Anti-cancer effects of JKA97 are associated with its induction of cell apoptosis via a Bax-dependent and p53-independent pathway. J Biol Chem. 2008;283:8624–8633. doi: 10.1074/jbc.M707860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TP, Reginato MJ. O-GlcNAc transferase: a sweet new cancer target. Cell Cycle. 2011;10:1712–1713. doi: 10.4161/cc.10.11.15561. [DOI] [PubMed] [Google Scholar]
- Ma Z, Vosseller K. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem. 2014;289:34457–34465. doi: 10.1074/jbc.R114.577718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall S. Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional perspective of diabetes, obesity and cancer. Sci STKE. 2006;346:re7. doi: 10.1126/stke.3462006re7. [DOI] [PubMed] [Google Scholar]
- Mi W, Gu Y, Han C, Liu H, Fan Q, Zhang X, Cong Q, Yu W. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta. 2011;1812:514–519. doi: 10.1016/j.bbadis.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Muthusamy S, Hong KU, Dassanayaka S, Hamid T, Jones SP. E2F1 transcription factor regulates O-linked N-acetylglucosamine (O-GlcNAc) transferase and O-GlcNAcase expression. J Biol Chem. 2015;290:31013–31024. doi: 10.1074/jbc.M115.677534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi A, Sprung R, Barma DK, Zhao Y, Kim SC, Falck JR, Zhao Y. Global identification of O-GlcNAc-modified proteins. Anal Chem. 2006;78:452–458. doi: 10.1021/ac051207j. [DOI] [PubMed] [Google Scholar]
- Ni T, Yang W, Han M, Zhang Y, Shen T, Nie H, Zhou Z, Dai Y, Yang Y, Liu P, et al. Global intron retention mediated gene regulation during CD4+ T cell activation. Nucleic Acids Res. 2016;44:6817–6829. doi: 10.1093/nar/gkw591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya K, Kataoka N, Hagiwara M. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J Cell Biol. 2011;195:27–40. doi: 10.1083/jcb.201107093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S. A small molecule that inhibits OGT activity in cells. ACS Chem Biol. 2015;10:1392–1397. doi: 10.1021/acschembio.5b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK. The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169:824–835. doi: 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phueaouan T, Chaiyawat P, Netsirisawan P, Chokchaichamnankit D, Punyarit P, Srisomsap C, Svasti J, Champattanachai V. Aberrant O-GlcNAc-modified proteins expressed in primary colorectal cancer. Oncol Rep. 2013;30:2929–2936. doi: 10.3892/or.2013.2794. [DOI] [PubMed] [Google Scholar]
- Pimentel H, Parra M, Gee SL, Mohandas N, Pachter L, Conboy JG. A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res. 2016;44:838–851. doi: 10.1093/nar/gkv1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin BB, Patel D, Conrad NK. Kaposi's sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 2010;6:e1000799. doi: 10.1371/journal.ppat.1000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalgi R, Hurt JA, Lindquist S, Burge CB. Widespread inhibition of posttranscriptional splicing shapes the cellular transcriptome following heat shock. Cell Rep. 2014;7:1362–1370. doi: 10.1016/j.celrep.2014.04.044. [DOI] [PubMed] [Google Scholar]
- Singh JP, Zhang K, Wu J, Yang X. O-GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett. 2015;356(2 Pt A):244–250. doi: 10.1016/j.canlet.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer. 2011;11:678–684. doi: 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, Hart GW. Perturbations in O-linked beta-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J Biol Chem. 2005;280:32944–32956. doi: 10.1074/jbc.M503396200. [DOI] [PubMed] [Google Scholar]
- Steenackers A, Olivier-Van Stichelen S, Baldini SF, Dehennaut V, Toillon RA, Le Bourhis X, El Yazidi-Belkoura I, Lefebvre T. Silencing the nucleocytoplasmic O-GlcNAc transferase reduces proliferation, adhesion, and migration of cancer and fetal human colon cell lines. Front Endocrinol (Lausanne) 2016;7:46. doi: 10.3389/fendo.2016.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashima Y, Stanley P. Antibodies that detect O-linked β-D-Nacetylglucosamine on the extracellular domain of cell surface glycoproteins. J Biol Chem. 2014;289:11132–11142. doi: 10.1074/jbc.M113.492512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo CF, Ingale S, Wolfert MA, Elsayed GA, Nöt LG, Chatham JC, Wells L, Boons GJ. Glycopeptide-specific monoclonal antibodies suggest new roles for O-GlcNAc. Nat Chem Biol. 2010;6:338–343. doi: 10.1038/nchembio.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, Curado J, Snyder M, Gingeras TR, Guigó R. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 2012;22:1616–1625. doi: 10.1101/gr.134445.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem. 1984;259:3308–3317. [PubMed] [Google Scholar]
- Vargas DY, Shah K, Batish M, Levandoski M, Sinha S, Marras SA, Schedl P, Tyagi S. Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell. 2011;147:1054–1065. doi: 10.1016/j.cell.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windhager L, Bonfert T, Burger K, Ruzsics Z, Krebs S, Kaufmann S, Malterer G, L'Hernault A, Schilhabel M, Schreiber S, et al. Ultra-short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. 2012;22:2031–2042. doi: 10.1101/gr.131847.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap K, Lim ZQ, Khandelia P, Friedman B, Makeyev EV. Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev. 2012;26:1209–1223. doi: 10.1101/gad.188037.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehezkel G, Cohen L, Kliger A, Manor E, Khalaila I. O-linked β-N-acetylglucosaminylation (O-GlcNAcylation) in primary and metastatic colorectal cancer clones and effect of N-acetyl-β-D-glucosaminidase silencing on cell phenotype and transcriptome. J Biol Chem. 2012;287:28755–28769. doi: 10.1074/jbc.M112.345546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Vocadlo DJ. O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer's disease and beyond. Chem Soc Rev. 2014;43:6839–6858. doi: 10.1039/c4cs00038b. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Tan EP, VandenHull NJ, Peterson KR, Slawson C. O-GlcNAcase expression is sensitive to changes in O-GlcNAc homeostasis. Front Endocrinol (Lausanne) 2014;5:206. doi: 10.3389/fendo.2014.00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.