

Abstract
Abiraterone, a potent inhibitor of the human enzyme CYP17A1 (cytochrome P450c17), provides a last line of defense against ectopic androgenesis in advanced prostate cancer. Herein we report an unprecedented off-target interaction between abiraterone and oncogenic hedgehog proteins. Our experiments indicate that abiraterone and its structural congener, galeterone, can replace cholesterol as a substrate in a specialized biosynthetic event of hedgehog proteins, known as cholesterolysis. The off-target reaction generates covalent hedgehog–drug conjugates. Cell-based reporter assays indicate that these conjugates activate hedgehog signaling when present in the low nanomolar range. Because hedgehog signaling is implicated in prostate cancer progression, and abiraterone is administered to treat advanced stages of the disease, this off-target interaction may have therapeutic significance.
Keywords: androgens, cholesterol, CYP17A1, hedgehog, prostate cancer
Graphical Abstract
Wrong way! Biochemical experiments indicate that steroidal anti-androgens, abiraterone and galeterone, undergo off-target covalent reactions with hedgehog proteins to produce hedgehog– drug conjugates. Cell-based assays indicate that these conjugates stimulate hedgehog signaling in the low nanomolar range.
Hedgehog (Hh) proteins serve as cell-signaling ligands involved in embryo development, whereas deregulated signaling by Hh is implicated in cancer.[1] Multiple studies link aberrant signaling by hedgehog to prostate cancer progression.[2] One attractive target to modulate the activity of Hh is the protein’s unique biosynthesis.[3] Hh proteins are expressed in the form of a self-catalytic, multidomain precursor protein. The Hh signaling ligand, HhN, is released from this precursor by peptide bond cholesterolysis, a cleavage/lipidation event unique to the Hh family[4] (Figure 1A). Cholesterolysis occurs in the secretory pathway,[5] before signaling, and represents one of two Hh-specific lipidations.[6] The reaction is brought about by the precursor’s C-terminal segment, HhC. Mutations in HhC that deactivate cholesterolysis result in endoplasmic reticulum (ER)-associated degradation of the precursor, effectively shutting off downstream signaling.[7]
Figure 1.
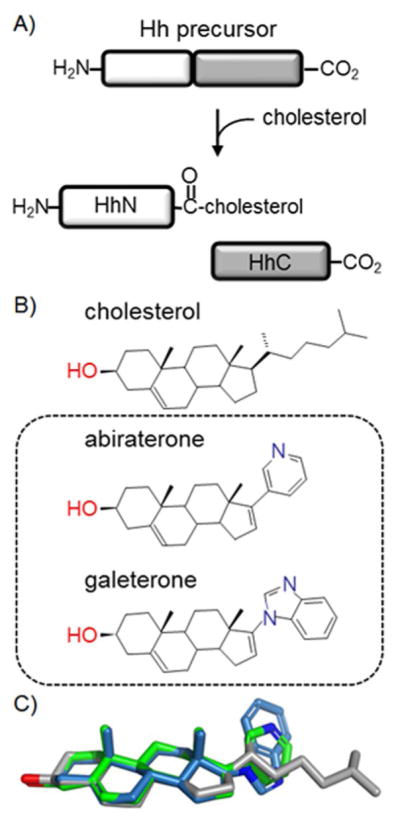
Hedgehog protein biogenesis. A) Cholesterolysis: Hh precursors react autonomously with substrate cholesterol, generating cholesterol-modified signaling ligand, HhN, and the cholesterolysis domain, HhC. B) Comparison of the native substrate with steroidal anti-androgens, abiraterone and galeterone, depicted in simple bond line format. C) Structure overlay of cholesterol (grey), abiraterone (green), galeterone (blue).
During small-molecule screens intended to find inhibitors of Hh cholesterolysis, we noticed an unexpected activity enhancement in the presence of abiraterone, a powerful antagonist of the steroidogenic enzyme, CYP17A1. Abiraterone (A) is an active metabolite of abiraterone acetate (Zytiga™), currently prescribed for the treatment of advanced prostate cancer.[8] Similar activation of Hh cholesterolysis was observed with galeterone (G), a structural analogue of abiraterone and clinical candidate for prostate cancer therapy. Results herein indicate that both steroidal agents are accepted by the Hh precursor as alternative substrates for cholesterolysis. The reaction generates covalent HhN–drug conjugates (HhN–A and HhN–G) in place of native, Hh–cholesterol (HhN–chol). The potential adverse effects of this off-target interaction are compounded by our observation that HhN–A and HhN–G activate Hh signaling in the low nanomolar range, similar to HhN–chol. Along with identifying a new, potentially oncogenic activity of Hh in drug metabolism, these findings expand the polypharmacological profile of two clinically significant anticancer agents.
We were drawn to abiraterone and galeterone on the basis of clinical significance and molecular structure. As mentioned, A is in clinical use for treating castration-resistant prostate cancer, a generally incurable stage of the disease; G is under clinical study for the same condition.[9] By inhibiting CYP17A1, these compounds block ectopic androgen biosynthesis, post-castration.[10] Androgen signaling is a long-recognized driver of prostate cancer,[11] and ~70 % of patients respond to A, with an average life extension of 4 to 6 months.[12] The mechanisms of chemoresistance to anti-androgens remain a subject of active debate.[13] As can be seen in Figure 1B, both A and G possess a steroidal ring system with a pyridyl or benzimidazole moiety appended to the C17 atom, replacing the native isooctyl “tail” of cholesterol. Given their structural similarity (Figure 1C), we asked whether these compounds might compete with cholesterol for binding by HhC.
We used an activity assay to evaluate interactions of A and G with the cholesterolysis-active HhC segment from Drosophila melanogaster. The assay continuously monitors activity by changes in the fluorescence of FRET-active proteins attached to HhC.[3c] Cyan fluorescent protein, serving as the FRET donor, is fused to the N terminus of HhC, replacing the signaling ligand; yellow fluorescent protein, the FRET acceptor, is fused to C terminus of HhC (Figure 2A). The construct, C–H–Y, exhibits FRET that decays at a saturable rate with added cholesterol, owing to donor–acceptor separation.[3b,c]
Figure 2.

Abiraterone and galeterone can replace cholesterol in hedgehog protein cholesterolysis. A) Optical reporter, C–H–Y, to monitor Hh activity. B) Kinetic traces showing signal from C–H–Y in buffered solution ± sterols (25 μM). C) Michaelis–Menten plot of C–H–Y initial velocity plotted as function of increasing concentration of cholesterol, abiraterone, and galeterone. Solid lines represent the expected kinetic behavior using the following KM values: cholesterol, 1×10−6 M; abiraterone, 12×10−6 M; galeterone, 3×10−6 M. D) Abiraterone and galeterone are active as substrates with chimeric hedgehog precursor, SHhN–DHhC. SDS-PAGE based assay showing time-dependent processing of SHhN–DHhC precursor processing in the presence of the indicated sterols (250 μM), or buffer alone at 22°C. Mr : SHhN–DHhC, 46 kDa; SHhN, 20 kDa; DHhC, 26 kDa.
In preliminary experiments, A was tested as an inhibitor of C–H–Y cholesterolysis. Prior to initiating the reaction with cholesterol, we monitored the FRET ratio of C–H–Y in the presence of added A for a period of 20 min; we found in previous work that subtle changes to the FRET ratio during pre-incubation can serve as a marker of Hh compound interaction.[3b] Unlike those earlier observations, however, we noticed that the addition of A induced a dramatic change in the FRET ratio of C–H–Y solutions, eventually reaching a baseline value (Figure 2B). This behavior, also observed during pre-incubation with G, suggested that C–H–Y was cleaving to form products, C–sterol and H–Y. Analysis of the reaction mixture by SDS-PAGE indeed showed a loss of precursor protein and accumulation of products (Supporting Information Figure S1). In subsequent experiments, we found that C–H–Y reacted with A in a concentration-dependent manner; a similar result was obtained with G (Figure 2C). The apparent affinity (KM value) and maximum rates of reaction with the compounds are in the neighborhood of substrate cholesterol (Supporting Information Table S1). Remarkably, the substrate activity of galeterone appears to exceed that of cholesterol in our assay. Thus, A and G appear to compete with cholesterol, not as inhibitors but as alternative substrates.
We validated the substrate activity of A and G through secondary assays involving a chimeric Hh precursor, where the human sonic hedgehog ligand (20 kDa) is fused to the cholesterolysis-active HhC of D. melanogaster, more closely mimicking a native Hh precursor. We devised this chimeric precursor, SHhN–DHhC, following difficulties we and others encountered expressing recombinant full-length human (and Drosophila) Hh precursor.[3b] Activity of SHhN–DHhC toward cholesterol has been established by SDS-PAGE analysis and mass spectrometry.[3b,14] Reactions of SHhN–DHhC in solutions containing A, G, or cholesterol monitored by SDS-PAGE are shown in Figure 2D. Consistent with the kinetic studies above, results indicate that both synthetic sterols stimulate processing of the precursor into SHhN–sterol and DHhC. Moreover, results of substrate competition experiments, in which cholesterol and A (or G) were added together to solutions of SHhN–DHhC, display product partitioning in ratios expected by the kinetic analysis (Supporting Information Figure S2). Thus, A is slightly less active as an alternative substrate compared with cholesterol, while G appears to surpass the substrate activity of cholesterol. Inspection of the gel also shows that the resulting SHhN conjugates exhibit varying mobility depending on the presence and identity of the attached sterol. Aberrant migration of SHhN is also consistent with covalent modification by a sterol molecule.[15] To establish conjugation of A and G to the SHhN C terminus, molecular masses of the trypsin-digested proteins were determined. Mass increases of the C-terminal peptide of 332.2 Da for SHhN–A, and 371.25 Da for SHhN–G were apparent, in accord with esterification of A and G, respectively, to the terminal glycine of SHhN (Supporting Information Figure S3).
Finally, we assessed the potential impact on downstream Hh signaling if A or G were to replace native cholesterol during Hh biosynthesis in the cell. It is known that cholesterol modification enhances but is not required for Hh signaling in vitro.[16] On the other hand, the potential influence of the appended sterol’s structure on Hh signaling has not yet been evaluated. By in vitro steroylation of the chimeric precursor, we generated SHhN–A, SHhN–G along with SHhN–chol and sterol-free SHhN in amounts suitable for signaling assays with Hh-responsive C3H10T1/2 cells[17] (Figure 3A). Two isoleucine residues at the N terminus of SHhN provided a surrogate for the protein’s native fatty acid modification.[18] As a negative control, we prepared cholesterol-modified human desert HhN ligand (DHhN–chol), which exhibits ~100-fold weaker signaling than SHhN.[19] The purity of the conjugates was determined by SDS-PAGE (Supporting Information Figure S4) and by RP-HPLC (Figure 3B).
Figure 3.

Hh–drug conjugates activate the hedgehog pathway. A) Schematic for the preparation of Hh–ligand conjugates using in vitro steroylation. B) RP-HPLC elution profiles of sterol-free and sterol-modified SHhN. Proteins were separated over a C4 column using an acetonitrile gradient; longer retention times indicate increased hydrophobicity. C) Schematic of Hh signaling assay using endogenous alkaline phosphatase as reporter. D) Sensitivity of Hh signaling pathway to Hh–drug conjugates. The plot shows averaged alkaline phosphatase activity from CH310T1/2 cells plotted as a function of increasing concentrations of the indicated Hh ligand (n >6, over three trials). Dose–response curves show expected behavior using the following EC50 values: SHhN–chol, 1×10−9 M; SHhN-A, 1×10−9 M; SHhN-G 3×10−9 M; sterol-free SHhN; 100×10−9 M.
SHhN–A and SHhN–G mimic the signaling potency of SHhN–chol. Activation of the Hh pathway in C3H10T1/2 cells promotes differentiation into osteoblasts, with an ensuing increase in alkaline phosphatase activity[20] (Figure 3C). In Figure 3D, AP activity in C3H10T1/2 cells is plotted as a function of increasing concentration of SHhN–X. In accord with earlier studies, SHhN–chol activates the pathway when present at single-digit nanomolar concentrations, whereas cholesterol-free Hh is less potent by a factor of >10.[16a] Our negative control, DHhN–chol, did not activate Hh signaling over the range we tested, consistent with earlier work.[19] When the native lipid of SHhN is replaced by A or G, pathway activation remained robust, with EC50 values in the low nanomolar range. We obtained a rank order in terms of potency of SHhN–chol ≈ SHhN–A>SHhN–G. An alternative staining assay with C3H10T1/2 cells produced similar results. Thus, a degree of functional promiscuity exists toward the sterol of SHhN both in ligand biosynthesis and in signal transduction.
Binding to more than a single protein target can sometimes enhance a drug’s efficacy;[21] however, the polypharmacology of A and G identified here seem to point in the opposite direction. Our studies suggest that interactions with Hh could divert A and G from the intended therapeutic target, CYP17A1, and generate unnatural Hh conjugates competent to activate a tumorigenic pathway. From the perspective of treatment, identifying an off-target interaction could prove useful to guide the design of next-generation analogues that: a) retain CYP17A1 inhibition, and b) bypass covalent interaction with Hh. A 3-keto analogue of abiraterone,[22] Δ4-abiraterone, along with nonsteroidal anti-androgens, provide logical points of departure.[23] The present findings also support a new oncogenic role of Hh in drug metabolism, a consequence of sterol promiscuity in Hh precursor cholesterolysis.[4] Hh’s self-lipidation activity is thereby brought into sharper focus as an important target for prostate cancer. Selective inhibitors hold promise of suppressing Hh biosynthesis while rescuing tumor sensitivity to a currently approved anti-androgen.
Supplementary Material
Acknowledgments
We thank members of the Callahan research group for technical assistance and helpful comments on the manuscript. Special thanks to Dr. George Ngoje and Timothy Owen for carrying out preliminary experiments. This work was supported in part by funding from the Office of the Assistant Secretary of Defense for Health Affairs, through the Prostate Cancer Research Program under Award No. W81XWH-14-1-0155. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the United States Department of Defense.
Footnotes
Supporting information (preparation and characterization of proteins, cell signaling assays and analysis, mass spectrometry, and kinetic methods) and the ORCID identification number(s) for the author(s) of this article can be found under http://dx.doi.org/10.1002/cmdc.201600238.
References
- 1.a) Hanna A, Shevde LA. Mol Cancer. 2016;15:24. doi: 10.1186/s12943-016-0509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Briscoe J, Therond PP. Nat Rev Mol Cell Biol. 2013;14:416–429. doi: 10.1038/nrm3598. [DOI] [PubMed] [Google Scholar]
- 2.a) Shaw A, Gipp J, Bushman W. Oncogene. 2009;28:4480–4490. doi: 10.1038/onc.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Nature. 2004;431:707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]; c) Sanchez P, Hernandez AM, Stecca B, Kahler AJ, De-Gueme AM, Barrett A, Beyna M, Datta MW, Datta S, Ruiz i Altaba A. Proc Natl Acad Sci USA. 2004;101:12561–12566. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chen M, Feuerstein MA, Levina E, Baghel PS, Carkner RD, Tanner MJ, Shtutman M, Vacherot F, Terry S, de La Taille A, Buttyan R. Mol Cancer. 2010;9:89. doi: 10.1186/1476-4598-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Peng YC, Levine CM, Zahid S, Wilson EL, Joyner AL. Proc Natl Acad Sci USA. 2013;110:20611–20616. doi: 10.1073/pnas.1315729110. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Li H, Liu W, Chen W, Zhu J, Deng CX, Rodgers GP. Sci Rep. 2015;5:16974. doi: 10.1038/srep16974. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Peng YC, Joyner AL. Dev Biol. 2015;400:94–104. doi: 10.1016/j.ydbio.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Jiang SQ, Paulus H. J Biomol Screening. 2010;15:1082–1087. doi: 10.1177/1087057110377498. [DOI] [PubMed] [Google Scholar]; b) Owen TS, Xie XJ, Laraway B, Ngoje G, Wang C, Callahan BP. Chem Bio Chem. 2015;16:55–58. doi: 10.1002/cbic.201402421. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Owen TS, Ngoje G, Lageman TJ, Bordeau BM, Belfort M, Callahan BP. Anal Biochem. 2015;488:1–5. doi: 10.1016/j.ab.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ciepla P, Magee AI, Tate EW. Biochem Soc Trans. 2015;43:262–267. doi: 10.1042/BST20150032. [DOI] [PubMed] [Google Scholar]
- 4.Mann RK, Beachy PA. Annu Rev Biochem. 2004;73:891–923. doi: 10.1146/annurev.biochem.73.011303.073933. [DOI] [PubMed] [Google Scholar]
- 5.Chen X, Tukachinsky H, Huang CH, Jao C, Chu YR, Tang HY, Mueller B, Schulman S, Rapoport TA, Salic A. J Cell Biol. 2011;192:825–838. doi: 10.1083/jcb.201008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Buglino JA, Resh MD. J Biol Chem. 2008;283:22076–22088. doi: 10.1074/jbc.M803901200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Long J, Tokhunts R, Old WM, Houel S, Rodgriguez-Blanco J, Singh S, Schilling N, Capobianco AJ, Ahn NG, Robbins DJ. Cell Rep. 2015;10:1280–1287. doi: 10.1016/j.celrep.2015.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Roessler E, Belloni E, Gaudenz K, Vargas F, Scherer SW, Tsui LC, Muenke M. Hum Mol Genet. 1997;6:1847–1853. doi: 10.1093/hmg/6.11.1847. [DOI] [PubMed] [Google Scholar]; b) Guy RK. Proc Natl Acad Sci USA. 2000;97:7307–7312. doi: 10.1073/pnas.97.13.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin LN, Hu QZ. Nat Rev Urol. 2014;11:32–42. doi: 10.1038/nrurol.2013.274. [DOI] [PubMed] [Google Scholar]
- 9.Bryce A, Ryan CJ. Clin Pharmacol Ther. 2012;91:101–108. doi: 10.1038/clpt.2011.275. [DOI] [PubMed] [Google Scholar]
- 10.a) Attard G, Belldegrun AS, de Bono JS. BJU Int. 2005;96:1241–1246. doi: 10.1111/j.1464-410X.2005.05821.x. [DOI] [PubMed] [Google Scholar]; b) Page ST, Lin DW, Mostaghel EA, Hess DL, True LD, Amory JK, Nelson PS, Matsumoto AM, Bremner WJ. J Clin Endocrinol Metab. 2006;91:3850–3856. doi: 10.1210/jc.2006-0968. [DOI] [PubMed] [Google Scholar]; c) Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Balk SP. Cancer Res. 2011;71:6503–6513. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huggins C, Stevens RE, Jr, Hodges CV. Arch Surg. 1941;43:209–223. [Google Scholar]
- 12.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson PA, Arora VK, Sawyers CL. Nat Rev Cancer. 2015;15:701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie J, Owen T, Xia K, Singh AV, Tou E, Li L, Arduini B, Li H, Wan LQ, Callahan B, Wang C. J Biol Chem. 2015;290:11591–11600. doi: 10.1074/jbc.M114.623264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter JA, Young KE, Beachy PA. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 16.a) Baker DP, Taylor FR, Pepinsky RB. Methods Mol Biol. 2007;397:1–22. doi: 10.1007/978-1-59745-516-9_1. [DOI] [PubMed] [Google Scholar]; b) Grover VK, Valadez JG, Bowman AB, Cooper MK. PLOS ONE. 2011;6:e21353. doi: 10.1371/journal.pone.0021353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K, Taylor FR, Wang EA, Galdes A. J Biol Chem. 1998;273:14037–14045. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- 18.Taylor FR, Wen D, Garber EA, Carmillo AN, Baker DP, Arduini RM, Williams KP, Weinreb PH, Rayhorn P, Hronowski X, Whitty A, Day ES, Boriack-Sjodin A, Shapiro RI, Galdes A, Pepinsky RB. Biochemistry. 2001;40:4359–4371. doi: 10.1021/bi002487u. [DOI] [PubMed] [Google Scholar]
- 19.Pathi S, Pagan-Westphal S, Baker DP, Garber EA, Rayhorn P, Bumcrot D, Tabin CJ, Blake Pepinsky R, Williams KP. Mech Dev. 2001;106:107–117. doi: 10.1016/s0925-4773(01)00427-0. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura T, Aikawa T, Iwamoto-Enomoto M, Iwamoto M, Higuchi Y, Pacifici M, Kinto N, Yamaguchi A, Noji S, Kurisu K, Matsuya T. Biochem Biophys Res Commun. 1997;237:465–469. doi: 10.1006/bbrc.1997.7156. [DOI] [PubMed] [Google Scholar]
- 21.Bolognesi ML, Cavalli A. Chem Med Chem. 2016;11:1190–1192. doi: 10.1002/cmdc.201600161. [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N. Nature. 2015;523:347–351. doi: 10.1038/nature14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.