

Abstract
Stromal cell-derived factor-1 (SDF-1) and its key receptor CXCR4 have been implicated in directing cellular recruitment for several pathological/disease conditions thus also gained considerable attention for regenerative medicine. One regenerative approach includes sustained release of SDF-1 to stimulate prolonged stem cell recruitment. However, the impact of SDF-1 sustained release on the endogenous SDF-1/CXCR4 signaling axis is largely unknown as auto-regulatory mechanisms typically dictate cytokine/receptor signaling. We hypothesize that spatiotemporal presentation of exogenous SDF-1 is a key factor in achieving long-term manipulation of endogenous SDF-1/CXCR4 signaling. Here in the present study, we sought to probe our hypothesis using a transgenic mouse model to contrast the spatial activation of endogenous SDF-1 and CXCR4 in response to exogenous SDF-1 injected in bolus or controlled release (PLGA nanoparticles) form in the adult rodent cortex. Our data suggests that the manner of SDF-1 presentation significantly affected initial CXCR4 cellular activation/recruitment despite having similar protein payloads over the first 24 hrs (∼30ng for both bolus and sustained release groups). Yet, one week post-injection, this response was negligible. Therefore, the transient nature CXCR4 recruitment/activation in response to bolus or controlled release SDF-1 indicated that cytokine/receptor auto-regulatory mechanisms may demand more complex release profiles (i.e. delayed and/or pulsed release) to achieve sustained cellular response.
Graphical abstract
Temporal control over SDF-1 release via PLGA nanoparticles differentially affects the SDF-1/CXCR4 signaling axis across the adult cortex.
Introduction
The signaling axis between stromal cell-derived factor-1 (SDF-1) and its receptor, CXCR4, has gained considerable attention as a therapeutic target in diverse areas of study such as immune-modulation, trafficking of stem cells and cancer metastasis. Among them is the role of SDF-1 in the migration of neural progenitor/stem cells (NPSCs) during pathological conditions of the central nervous system (CNS) 1,2. After neural injury (traumatic brain injury, TBI, or stroke), NPSCs in the neurogenic niches, exhibit a remarkable ability for directed migration to reach the injury penumbra 3,4. Migrated NPSCs not only maintain neurogenic capacity, but they also secrete neurotropic factors and assist in preserving synaptic connectivity in the injury area 1,5. As such, ablation of endogenous NPSC populations before induction of neural injury significantly hampers the endogenous neurogenic potential contribution to increased cognitive impairments 6. However, this NPSC migratory response is transient, peaking at 3-7 days post injury (in rodent models) and decreases dramatically, although not completely, by two weeks post injury 7. Mechanisms controlling this endogenous injury response are not yet fully understood, but the SDF-1/CXCR4 signaling cascade is thought to play an important role due to the following: 1) NPSCs are CXCR4+ and respond chemotactically to SDF-1 gradients in vitro 8, 2) NPSCs migrate to local sources of SDF-1 in vivo after neural injury 3,4, 3) local administration of the CXCR4 antagonist, AMD3100 attenuates NPSC migration 9, and 4) decreasing local concentrations of SDF-1 coincides with the aforementioned decrease in the number of migrating NPSCs in vivo after neural injury 3,7. Thus, control over the local bioavailability of SDF-1 serves as a potential means for amplifying/sustaining the innate NPSC homing response after neural injury.
Growing evidence indicates that the spatiotemporal presentation of bioactive factor(s) alters the overall biochemical cellular response. For example, sustained release of proteins differentially affects stem cell proliferation, migration and differentiation both in vitro and in vivo when compared to bolus administration 10–12. Additional studies demonstrate that sustained release of proteins may not lead to an improvement in therapeutic efficacy 13,14. Instead, differing protein release profiles (bolus, sustained, delayed and pulsed) may activate distinct biochemical cascades that determines overall therapeutic outcome 15,16. Additionally, receptor desensitization/downregulation due to overstimulation is another well-known phenomenon that may occur with sustained controlled release devices 17,18. Specifically, for SDF-1/CXCR4, it is unclear how or if autocrine/paracrine signaling affects regulation of SDF-1/CXCR4 expression. Some in vitro evidence points to downregulation of CXCR4 after continuous exposure to SDF-1 in neonatal E14.5 telencephalic neurons, but its relation to native cortical tissues in the mature forebrain is not certain, where multiple cell-types express CXCR4 and secrete SDF-1 19,20. Although a number of studies have focused on endogenous SDF-1/CXCR4 signaling in the developing brain and after neural injury, none have directly assessed endogenous SDF-1/CXCR4 signaling in response to different spatiotemporal presentations of exogenous SDF-1. We believe that this represents a fundamental barrier in the development of efficacious strategies for manipulating the SDF-1/CXCR4 signaling cascade, a ubiquitous therapeutic target for diverse applications.
Poly(lactic-co-glycolic) acid (PLGA) is FDA-approved and PLGA-based nanoparticles (NPs) have the added benefits of: 1) direct injection into target tissues, 2) biodegradability, 4) metabolizable degradation products, 5) tunable release profiles and, 6) lowering risks of infections compared to conventional osmotic pumps 8,21,22. Moreover, PLGA matrices maintain prolonged, localized bioavailability and may aid in protecting the encapsulated cargo from degradation, a critical parameter for protein delivery 23–25.
The goal of this study was to begin to elucidate the SDF-1/CXCR4 signal propagation in the adult rodent cortex in response to either bolus or sustained release of exogenous SDF-1α form our previously developed PLGA NPs that encapsulate and release bioactive SDF-1 over a period of 60 days 8. To facilitate a direct correlation between administration of exogenous SDF-1 and endogenous SDF-1/CXCR4 signaling, we performed in vivo studies in the intact mouse cortex 1. Critical tools employed include: 1) transgenic (CXCR4-EGFP) 20, 2) bioactive fluorophore-conjugated SDF-1 (AlexaFluoro-647nm SDF-1; AFSDF-1) and, 3) PLGA nanoparticles loaded with bioactive AFSDF-1 8. Using this toolset, we tracked spatiotemporal profile of endogenous SDF-1 and CXCR4+ cells over 7 days in response to intracortical injections of either bolus AFSDF-1 or AFSDF-1-loaded NPs. We hypothesized that endogenous SDF-1/CXCR4 expression will increase acutely after exposure to both bolus and controlled release of exogenous AFSDF-1. However, at the later time points we expected differential SDF-1/CXCR4 activation between the bolus and sustained release that may partly be attributed to alterations in the signaling milieu (i.e. autocrine/paracrine signaling, gene regulation, cell phenotype etc.) caused by the AFSDF-1 release profile. The data showed bolus administration of AFSDF-1 led to a transient and localized response from the endogenous SDF-1/CXCR4 signaling axis. In contrast, sustained release of exogenous AFSDF-1 initiated spatially dispersed activation/recruitment of CXCR4 cells for up to 3 days after injection, which subsequently decreased markedly by day 7. The results of these experiments will inform design and testing of future biologically relevant release devices capable to modulating the SDF-1/CXCR4 signaling axis over extended periods of time.
Experimental Methods and Materials
Materials
Poly(lactic-co-glycolic) acid (PLGA; 50:50 ester-terminated; inherent viscosity = 0.55-0.75dL/g) was purchased from Lactel (Birmingham, USA). Recombinant mouse stromal cell-derived factor-1α (SDF-1α) were acquired from PeproTech (Rocky Hill, USA). Recombinant human-derived SDF-1α conjugated with AlexaFluor-647 at the C-terminus (AFSDF-1) was acquired from Almac (Craigavon, UK). The organic solvent ethyl acetate was acquired from Alfa Aesar (Ward Hill, USA) and dimethyl sulfoxide (DMSO) from American bioanalytical (Natick, USA). All other materials and chemicals were purchased from Sigma-Aldrich (St. Louis, USA) and used without further modification or purification.
Fabrication of AFSDF-1 Loaded Nanoparticles
AFSDF-1 loaded PLGA nanoparticles were fabricated using a water/oil/water (W/O/W) emulsion technique using a previously published protocol that established prolonged release of bioactive SDF-1 past 60 days 8. Briefly, the first emulsion (W/O) was obtained by vortexing the oil phase (100 mg/mL PLGA in ethyl acetate) with PBS buffer solution (pH = 7.4) containing 20.0mg/ml bovine serum albumin (BSA; 2.0% w/w of PLGA) and 2.0mg/mL AFSDF-1 (0.2% w/w of PLGA). The above mixture was added dropwise to a 3.6× volume excess of an aqueous 5.0% (w/v) d-α tocopheryl polyethylene glycol 1000 succinate (TPGS) and the second emulsion (W/O/W) was produced by ultrasonicating (Omni Ruptor 4000; Omni International; Kennesaw, USA) the solution for two consecutive 15sec periods in an ice bath. The emulsion was then quickly transferred to a stirring (300RPM) aqueous bath containing 0.5% TPGS + 1.25% (w/v) NaCl (10× volume excess) and left undisturbed for 3hrs for solvent evaporation. The particle suspension was washed three times with deionized water and recovered through lyophilization. A blank NP control was generated with the exact same procedure without the addition of AFSDF-1.
Transgenic Mouse Model
The CXCR4-EGFP transgenic mice were kindly donated to us by Dr. Richard Miller of Northwestern University. All studies were conducted in accordance with approved protocols reviewed by the Institutional Animal Care and Use Committee at Arizona State University. The CXCR4-EGFP mice are well characterized and utilized in studies characterizing the developing and adult rodent CNS 20,26,27. The CXCR4–EGFP bacterial artificial chromosome (BAC) transgenic mouse was originally developed by the gene expression nervous system atlas (GENSAT; NINDS contract N01Nso2331 to Rockefeller University, NY). Expression of EGFP in these mice is expected to be identical to endogenous gene expression as examined by in situ hybridization (http://www.gensat.org/index.html).
Intracortical Injections
Adult CXCR4-EGFP transgenic mice (n=5 per group/time point) were anesthetized and a 1.5mm craniotomy was performed centered over 1.5mm anterior of bregma and 1.5mm lateral of midline. Injections (3μL) were performed at a depth of 0.8mm into the cortical tissue using a 26G needle for the following groups: 1) bolus AFSDF-1, 2) bolus vehicle, 3) AFSDF-1 NPs and, 4) blank NPs. Separate Hamilton syringes and needles were used for all groups (Hamilton, Reno, NV). The syringe and needle was stereotaxically placed, in the cortical tissue by lowering the needle at a rate of 0.15 mm/min and upon reaching the target depth of 0.8mm, the needle was kept stationary for 1 min. The needle was then retracted back up to 0.5 mm before the injections were initiated at 0.5 μL/min, pausing every 1μL for 30s until 3μL dose was delivered. The needle was subsequently held in place for 1 min before being retracted at 0.15 mm/min. For the particle groups, lyophilized NPs were resuspended at 140mg/ml, subjected to water-bath sonication on ice for 2mins immediately before following the same the injection procedure. After needle retraction, the sutures were used to close soft tissue incisions and the mice were allowed to recover in an incubator before returning to their home cage.
Immunohistochemistry
At specified times (1, 3, and 7 days post-injection), mice were anaesthetized and sacrificed by pericardial perfusion and brains extracted for 24hr post-fixation in 4% paraformaldehyde. Following fixation, brains were incubated in 30% sucrose followed by cryo-embedding in OCT and stored at -80 °C prior to serial cryosectioning at 25μm thickness. Sections were blocked, permeabilized and incubated with the appropriate primary antibody overnight at 4 °C followed by 2 hr incubation with the appropriate secondary antibody at room temperature. Cell nuclei were stained with DAPI (Thermo Fisher Scientific, Waltham, MA) prior to mounting sections with VectaShield (Vector Laboratories, Burlingame, CA) and coverslips. Primary antibodies included polyclonal rabbit IgG anti-SDF-1 (Abcam, Cambridge, MA), polyclonal rabbit IgG anti-doublecortin (Dcx; Abcam), polyclonal chicken IgY anti-nestin (Novus Biologics), polyclonal goat IgG anti-glial fibrillary acidic protein (GFAP; Abcam), and polyclonal rabbit IgG anti-Iba1 (Wako Pure Chemical Industries, Ltd); see supplementary table for full antibody information. Secondary antibodies included goat anti-rabbit IgG (H+L) Alexa Fluor® 555, goat anti-chicken IgY (H+L) Alexa Fluor® 555, and donkey anti-Goat IgG (H+L) cross-adsorbed Alexa Fluor® 555 (all from ThermoFisher Scientific). Stained sections were visualized using fluorescence microscopy (DMI6000B, Leica) with 20× magnification mosaic tiles scans to produce cortical representations.
Image Processing
For each group/time point, 4-5 animals with 4-6 tissue sections per animal were quantified. Tissue sections were selected based on identified constraint boundaries of the outer most coronal sections containing CXCR4+ cells; a distance that typically spanned 500 μm total (∼20 sections of 25μm thickness). Within this defined boundary, 4-6 tissue sections evenly spaced across the injection region were selected for analysis. Fluorescence-based quantitative analyses were completed with ImageJ to determine spatial distribution of CXCR4+ cell bodies, as well as percent immunopositive area for total SDF-1 (exogenous + endogenous) and exogenous AFSDF-1. Specifically, the apparent “on/off” nature of the endogenous CXCR4 expression reporter allowed for thresholding, followed by particle count algorithms to determine total count and spatiotemporal localization of CXCR4+ cells. For total SDF-1 and exogenous AFSDF-1 analysis, contralateral-adjusted thresholded values on micrographs acquired with consistent settings (same gain and exposure) were used to determine the area fraction of SDF-1 positive stain a 3.2mm × 1mm ROI centred around the injection tract to minimize subjectivity (see Figure 1). Spatial distribution of immunostained SDF-1, AF-SDF-1 and CXCR4+ cells was presented in 400 μm × 1mm binned regions of interest relative to the center of the injection tract (see Figure 1).
Figure 1.
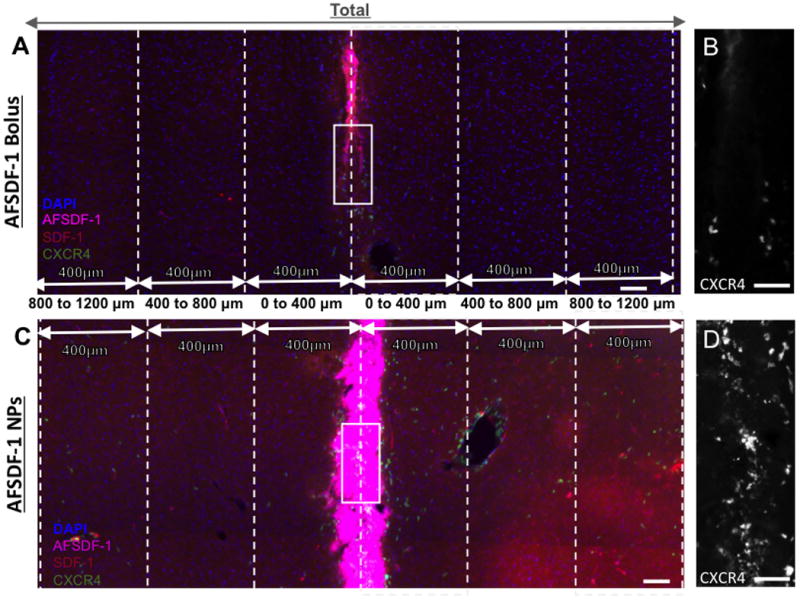
Representative fluorescent images of cortical tissue sections and the regions of interest (ROIs) used to quantify fluorescent signals. (A) Cortical reconstruction centered at the injection tract after a bolus injection of AFSDF-1 and (B) AFSDF-1 loaded PLGA NPs. All tissue sections were immunostained for SDF-1 (red) and cell nuclei (DAPI; blue) with exogenous AFSDF-1 magenta) and EGFP-CXCR4 (green). All cortical reconstructions (2.8mm×1mm) were divided into a 400 μm bins stemming from the center of the needle tract. The figure depicts truncated regions of the distal ROI for illustration purposes and extends further medially and laterally. (B) & (D) are magnified representations of the needle tract outlined in white for bolus and NP implant groups, respectively. Scale bars = 100 μm for A & C; scale bar = 30 μm for B & D.
Statistical Analysis
Statistical analysis was performed on all quantitative assays. All results are depicted as the mean ± standard error of the mean (SEM), unless otherwise stated; the data represents an average of 4-5 animals per group with 4-6 sections analyzed per animal. Statistical analyses (GraphPad Prism, La Jolla, CA) evaluated differences between groups using the appropriate one- or two-way analysis of variance (ANOVA) followed by Tukey post-hoc tests to determine statistical significance with p < 0.05 considered significant. Multiplicity adjusted p-values are reported for Tukey post-hoc comparisons.
Results and Discussion
The SDF-1/CXCR4 signaling axis is implicated in a host of pathological conditions for cellular recruitment, thus it is not surprising that this signaling axis is targeted in many therapeutic applications ranging from cancer metastases and tissue engineering/ regenerative medicine 1,28. Consequently, elucidating mechanisms to efficiently modulate the SDF-1/CXCR4 signaling axis is of great value. Specifically in the CNS, bolus delivery is the most common method used to study the effects of exogenous SDF-1 29,30. Bolus injections in to target tissue have inherent advantages over systemic administration, however, it negates temporal control over concentration or payload degradation leading to only transient therapeutic benefits 31. Bioengineered approaches for sustained SDF-1 bioavailability include a number of hydrogel-based devices for various applications with diffusion-limited release periods of 7-14 days 32–35. Others have proposed polyester-based systems to prolong release over period of weeks to months 36,37. We have previously developed and characterized PLGA nanoparticles (NPs) that sustain release of bioactive SDF-1α over 60 days 8. Yet, given the complexity and possible auto-regulatory processes that modulate SDF-1 signal transduction, the optimal mode of SDF-1 delivery locally to modulate the SDF-1/CXCR4 signaling axis has yet to be identified. Indeed recent reports suggest downregulation of CXCR4 when continuously stimulated with SDF-1 19. Others are shedding light on a new receptor, CXCR7 that modulates sensitivity to extracellular SDF-1 38. There is a lack of studies devoted to understanding how the SDF-1/CXCR4 signaling axis responds to various spatiotemporal presentations of exogenous SDF-1 in vivo. We thus used the CXCR4-EGFP transgenic mouse model to study the effects of bolus and controlled release of exogenous AFSDF-1 on the endogenous spatiotemporal localization of SDF-1 and CXCR4-expressing cells over a period of 7 days.
AFSDF-1 Delivery and Diffusion in the Cortex
Fluorescently tagged SDF-1 enabled us to track the distribution of the exogenous AFSDF-1 throughout study. The only commercially available fluorescently-tagged SDF-1 was human-derived recombinant SDF-1 with AlexaFluor-647 conjugated specifically to the C-terminus from Almac Chemokines (Craigavon, UK). Previous studies have noted cross-reactivity between diverse species, yet we verified that mouse NPSCs respond to human AFSDF1 prior to conducting the in vivo studies (Supplemental Figure 1). Dosing for the intracortical injections for each group was based on our previously published release profile for SDF-1α PLGA NPs 8. Specifically, 420μg of PLGA NPs was expected to release approximately 30 ng of AFSDF-1 in the first 24hrs and an additional 9 ng over the next 6 days 8. Subsequently, the dosage for the bolus AFSDF-1 group was 30 ng to match the estimated cumulative AFSDF-1 release in the first 24hrs from the NPs. Notably, regardless of the mode of delivery and/or time point, AFSDF-1 was confined to within 400 μm from the center of the needle tract (Figures 1-4). Representative images illustrate a concentrated source of AFSDF-1 with more diffuse signal at the edges of the implant/injection indicating modest local diffusion of exogenous AFSDF-1 in the cortical parenchyma (Figures 1-3). The main difference in AFSDF-1 distribution between the bolus and SDF-1 NP groups was prominently observed at Day 3 and 7 post-injection where high levels of AFSDF-1 remained localized within 400 μm of the injection in the SDF-1 NPs (Figure 2-4). We acknowledge that fluorescent signal from AFSDF-1 does not equate to bioactivity; yet, the fluorescent tag enables mapping of the spatial distribution. Notably, our observed short diffusion lengths agree with previous reports of limited protein diffusion in the brain parenchyma 39–41. Once in the interstitial space, protein diffusion, rather than convection, is the main mechanism of transport in the brain extracellular space (ECS). Diffusion over short distances (<0.1mm) is relatively efficient, however, the densely packed architecture of the ECS means diffusion in the order of millimeters is unlikely to occur in therapeutically-relevant time scales 42. In addition to a small void fraction, cortical extracellular matrix is composed of negatively charged components such as hyaluronic acid, heparan sulfate and chondroitin sulfate proteoglycans among others. Thus, diffusion of highly basic proteins such as SDF-1 (isoelectric point ∼9.6 for the α-isoform) will be hindered through electrostatic interactions further supporting the limited diffusion of AFSDF-1 we observed here.
Figure 4.

AFSDF-1 localized to injection site. (A) The total positive AFSDF-1 area for AFSDF-1 NPs was significantly greater than AFSDF-1 bolus at day 1 and 3, but not day 7 (# p < 0.01). (B-D) Evaluating the spatial distribution of AFSDF-1 away from the injection tract demonstrated the marked AFSDF-1 signal from the NPs compared bolus injection most prominently within 400 μm of the injection site across all time points (# p < 0.01). Box and whisker plots used in B-D to demonstrate the span of data points within each group; n = 4-5 animals per group.
Figure 3.
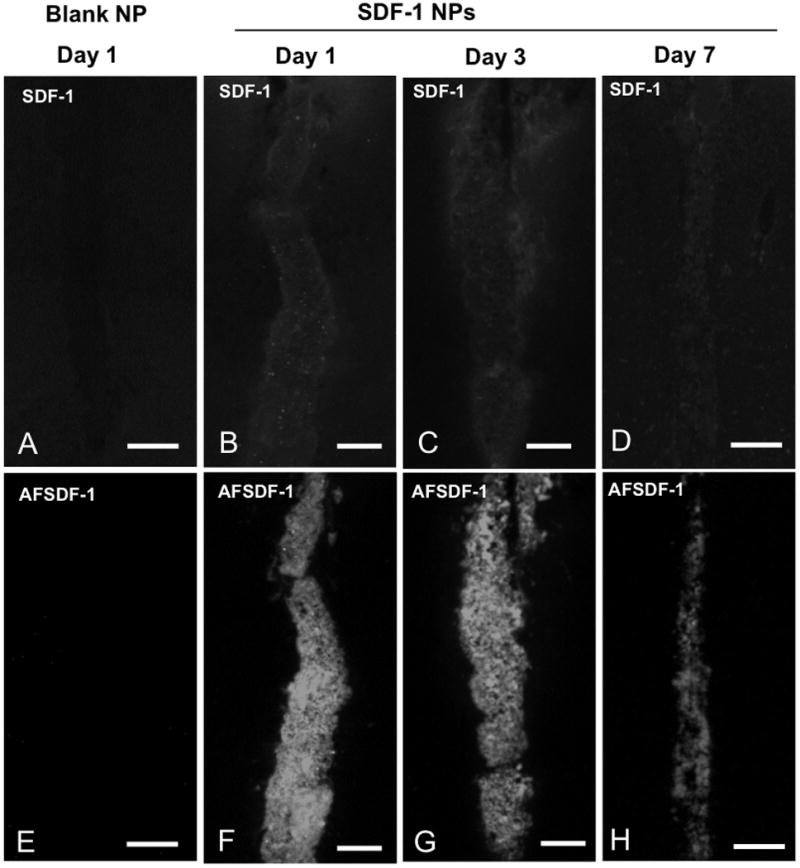
Representative AFSDF-1 and SDF-1 Immunostained images for blank NP (day 1, A, E) and SDF-1 NPs at day 1, 3, and 7; B-D and F-H). The top row consists of representative SDF-1 immunostained images, whereas the bottom row depicts corresponding AFSDF-1 images. Scale bar = 100 μm. It is important to note that these representative images in the comprise only ∼400 μm × 800 μm centred around the injection tract and constitute only a small fraction of the larger 3.2mm × 1.0 mm ROI used for the entire analysis.
Figure 2.
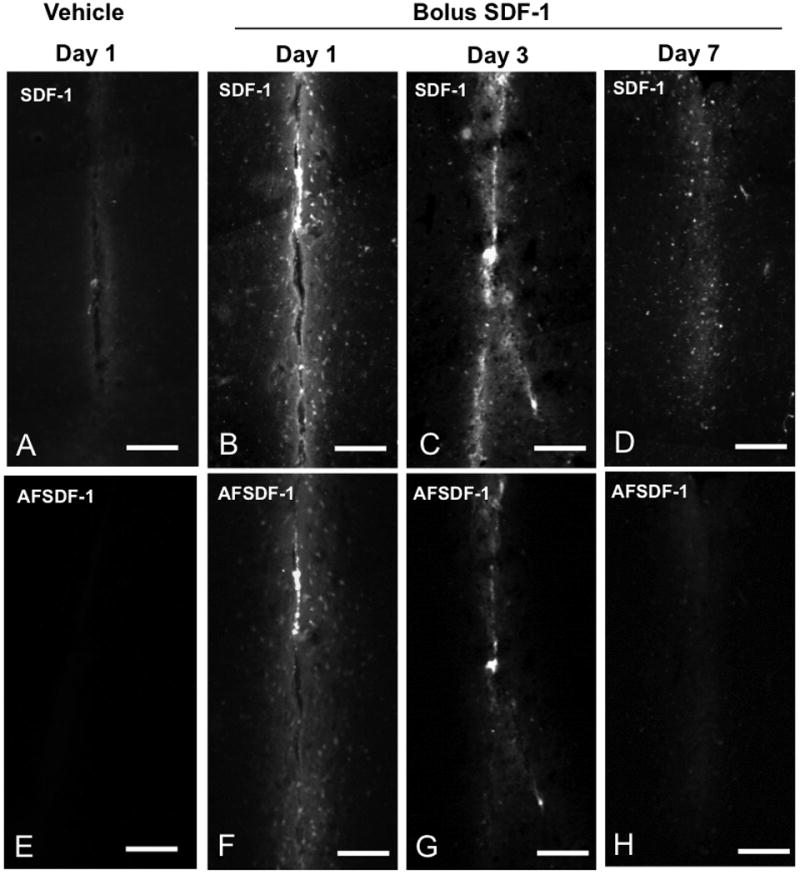
Representative AFSDF-1 and SDF-1 Immunostained images for vehicle (day 1, A, E) and SDF-1 bolus at day 1, 3, and 7; B-D and F-H). The top row consists of representative SDF-1 immunostained images, whereas the bottom row depicts corresponding AFSDF-1 images. Scale bar = 100 μm. It is important to note that these representative images in the comprise only ∼400 μm × 800 μm centred around the injection tract and constitute only a small fraction of the larger 3.2mm × 1.0 mm ROI used for the entire analysis.
Sustained Release of SDF-1 Induces SDF-1 Expression
One key question for protein delivery paradigms is how does the presence of exogenous protein affect endogenous protein expression? We probed this question by using immunohistochemistry to detect total SDF-1 expression across the cortex at 1, 3, and 7 days after injection of bolus AFSDF-1, AFSDF-1 NPs, or controls (vehicle and blank NP; Figures 2 and 3). Such immunostaining of SDF-1 has been employed in previous studies that evaluated the SDF-1 distribution in various pathologies 43–45. Quantification of total SDF-1 (endogenous and exogenous) via immunostaining revealed that the AFSDF-1 NPs elicited significantly increased total SDF-1 levels for all time points across the entire cortical ROI (Figure 5A; p < 0.01). Interestingly, soluble AFSDF-1 administration did not significantly affect total overall SDF-1 expression relative to vehicle and blank NPs controls across the entire cortical ROI (Figure 5A). Probing the spatial distribution of total SDF-1 relative to the injection site revealed nuanced differences among treatment groups (Figure 5B-D). At day 1, both AFSDF-1 bolus and AFSDF-1 NPs demonstrated significantly higher levels of total SDF-1 within 400 μm of the injection tract compared to vehicle and blank NP controls (Figure 5B; p <0.05 and p < 0.001, respectively). This result is not unexpected as a substantial source of total SDF-1 surrounding the needle tract is most likely exogenous AFSDF-1 (Figure 4 and 5). Beyond 400 μm, the AFSDF-1 NP group trended higher than the other groups, though not statistically significant in post-hoc pairwise tests (p ∼ 0.1). This trend continued out to day 3 for soluble AFSDF-1 and day 7 for AFSDF-1 NPs where total SDF-1 immunostaining was significantly upregulated and localized within 400 μm of the injection tract compared to vehicle and blank NP controls (Figure 5C, D; p < 0.05 and p < 0.01). Notably, the vehicle injection and blank NPs did not mount a significant endogenous SDF-1 response (Figures 2 and 3).
Figure 5.

AFSDF-1 NPs enable sustained SDF-1 delivery. (A) Total SDF-1 (endogenous + exogenous) immunopostitive staining for days 1, 3, and 7 after injection. Only the SDF-1 NPs demonstrated significantly higher percent positive area compared to all groups at day 1 and 3 (A; #p < 0.01) and the vehicle control at day 7 (A; *p < 0.05). (B-D) Spatial distribution of the SDF-1 immunostain relative to the injection site over days 1, 3, and 7. SDF-1 NPs displayed significantly higher percent area of immunostain within 400 μm of the injection compared to all groups at all days (B-D; #p < 0.01). SDF-1 bolus was significantly higher than vehicle control and blank NPs at day 1 (B; #p < 0.01) and only vehicle control at day 3 (C; $p < 0.05). Box whisker plots used in B-D to demonstrate the span of data points within each group; n = 4-5 animals per group.
An overlay of the spatial distribution of both AFSDF-1 and total SDF-1 (immunostain) revealed marked overlap of these two signals within 400 μm of the injection tract (Figure 2, 3, and 6). Yet, at day 1 after injection, a significant increase in total SDF-1 was observed over AFSDF-1 at 800 μm and 1200 μm from the injection tract for both AFSDF-1 bolus and AFSDF-1 NPs (Figure 6A, D). Therefore, it appears that within one day after exposure to exogenous AFSDF-1, the surrounding cells initiate autocrine/paracrine signaling to propagate SDF-1 expression. Moreover, the half-life of SDF-1 in vivo has been reported to be 26 min when administered systemically 46, therefore, the significant increase in total SDF-1 levels particularly at 3 and 7 days most likely consists of endogenously produced SDF-1. This data taken together with the absence of positive SDF-1 immunostaining in the vehicle and blank NP groups further supported the notion that the presence of exogenous SDF-1 kick starts autocrine/paracrine signaling to induce endogenous SDF-1 production.
Figure 6.

Comparison of AFSDF-1 to total SDF-1 positive area. Overlay of AFSDF-1 and total SDF-1 immunostain area in response to SDF-1 bolus (A-C) or SDF-1 NPs (D-F) revealed significantly higher levels of total SDF-1 immunostain at 400-800 μm and 800-1200 μm at day 1 (A, D; #p < 0.05). The AFSDF-1 and total SDF-1 staining pattern essentially matched for days 3 and 7 for both groups (B, C and E, F). Box and whisker plots used to demonstrate the span of data points within each group; n = 4-5 animals per group.
Sustained SDF-1 Release Induced Transient and Dispersed CXCR4 Activation/Recruitment
The transgenic CXCR4-EGFP mouse enabled spatiotemporal tracking of the cellular CXCR4 activation in response to bolus administration and sustained release of AFSDF-1 (Figures 1 and 7). Quantifying the CXCR4+ cell density across the entire cortical ROI revealed a significant increase in the AFSDF-1 NP group compared to all other groups at days 1 and 3, but not at day 7 (Figure 7A; p < 0.01). Interestingly, at day 3, the blank NP control initiated a significant increase in total CXCR4+ cell density over vehicle and AFSDF-1 bolus groups (Figure 7A; p < 0.05). Looking more closely at the spatial distribution of the CXCR4+ cells relative to the point of injection again revealed nuanced trends in cellular response (Figure 7B-D). At day 1, the AFSDF-1 NPs elicited a robust and dispersed CXCR4 expression compared to all other injection groups consistently across the entire cortical ROI (Figure 7B; p < 0.01). This trend continued at day 3, however, by day 7 the only significance noted was at the most proximal region within 400 μm of the injection tract (Figure 7C-D; p < 0.01). Both the AFSDF-1 bolus and blank NP injections elicited a significant CXCR4 response within 400 μm of the injection tract compared to vehicle control at days 1 and 3 (Figure 7B-C; p< 0.05). However, at day 7 among the controls, the blank NP group remained significantly higher than vehicle and AFSDF-1 bolus injections (Figure 7D; p < 0.05).
Figure 7.

Sustained release of SDF-1 incites transient dispersed CXCR4 activation. (A) CXCR4 cell density (cell/mm2) across the entire cortical ROI significantly increased with SDF-1 NPs compared to all groups at day 1 and 3 (#p < 0.01), then returned to control levels by day 7. At day 3, the blank NPs also elicited an increased CXCR4 cellular response compared to vehicle and bolus SDF-1 (A; *p < 0.05). (B-D) Spatial analysis demonstrated significantly increased and dispersed CXCR4 response with the SDF-1 NPs compared to all other groups at day 1 and 3 (B,C; #p < 0.01) and only at the most proximal region to the injection at day 7 (D; #p < 0.01). Both bolus SDF-1 and blank NPs elicited significantly increased CXCR4 within 400 μm of the injection compared to vehicle at day 1 (B; $p < 0.05) and compared to all other groups at day 3 (C; #p < 0.01). The blank NP cell density was significantly higher than vehicle at day 7 (D; $p < 0.05). Note: y-axis in B-D is log scale. Box and whisker plots used in B-D to demonstrate the span of data points within each group; n = 4-5 animals per group.
Densely populated CXCR4+ cell bodies were closely associated with the needle tract at day 1 post bolus injection suggesting that exogenous AFSDF-1 induced CXCR4 activation and/or robust CXCR4+ cellular recruitment, agreeing with in vitro and in vivo trends reported in literature 19,30,47. Sustained release of AFSDF-1 induced significantly higher total CXCR4+ cell populations compared to bolus AFSDF-1 that was more dispersed across the cortex. Most notably, AFSDF-1 NPs was the only group to significantly increase CXCR4 response in cells located distally compared to controls and bolus AFSDF-1 (>400 μm away from the needle tract). We saw this result even though AFSDF-1 was not detected more the 400μm away from the needle tract, raising the question of how exogenous SDF-1 with minimal direct diffusion into the brain interstitial space affected cellular activation at such large distances. One hypothesis is that relatively slow release of exogenous SDF-1 in the NP groups and the modest yet significant increase in endogenous SDF-1 expression propagated the signaling axis much further than SDF-1 can physically diffuse. Our results and previous studies with mesenchymal stem cells (MSCs) support this notion as MSCs treated with SDF-1 upregulated SDF-1 mRNA 47. Additionally, in contrast to AFSDF-1 bolus injection where the entire delivered dose is bioavailable immediately and subject to proteolytic degradation, the sustained release group is expected to release AFSDF-1 in a more controlled fashion even in the first 24hrs. This slower rate of protein release from the NPs may have provided insulation of released AFSDF-1 from environmental factors. Thus, increased CXCR4+ cell density may also be a product of improved AFSDF-1 half-life and bioavailability 24.
Aside from a strict feed-forward autocrine/paracrine signaling loop for SDF-1, we cannot rule out the involvement of other propagating signal mediators. Several soluble factors (e.g. vascular endothelial growth factor, VEGF; basic fibroblast growth factor, bFGF), immune modulators, and others are known to crosstalk with the SDF-1/CXCR4 signaling axis 47,48. The VEGF interaction with SDF-1/CXCR4 signaling is of particular interest since SDF-1 treatment of MSCs has shown increased VEGF secretion in vitro 47, while others indicate that VEGF upregulates expression of both SDF-1 and CXCR4 49. VEGF and SDF-1 also overlap in their gene regulation through hypoxia inducible factor-1 and endothelial progenitor cells express both simultaneously under hypoxia 50,51. bFGF is another signaling mediator that may be involved in transducing AFSDF-1 signal distally. CXCR4 and SDF-1 expression increases significantly in CXCR4+ endothelial cells after exposure to bFGF 52,53. More interestingly, VEGF and bFGF both appear to increase only CXCR4 expression and do not modulate expression of other CXC or CC chemokine receptors suggesting that SDF-1, VEGF and bFGF take part in a positive feedback loop to propagate SDF-1/CXCR4 signaling 54. Immune modulators such as, tumor necrosis factor-α (TNF-α) and interleukin 1-β (IL-1β) also affect the SDF-1/CXCR4 signaling axis indirectly by inducing release of VEGF and/or bFGF 53,55. TNF-α also has a reported biphasic effect on CXCR4 expression where, CXCR4 expression is downregulated initially within in 3 hrs, and upregulated subsequently after 24 hrs 53,55. Thus, secondary signaling mediators may play an important role in propagating SDF-1/CXCR4 signaling to the distal areas of the cortex. Another mechanism for activation of distal cells may be due to exogenous SDF-1 bypassing diffusion through the brain interstitium and utilizing the cerebrovasculature. Although it is not empirically apparent that intracortical injections lead to breakage of the BBB, neural injuries such as focal TBI are well known to cause BBB dysfunction 56. In the study presented here, escape of AFSDF-1 and/or NPs into the systemic circulation through leaky vasculature may allow for alternate means of transport and interaction with endothelial cells of the BBB in distal regions of the brain. Endothelial cells strongly express both SDF-1 and CXCR4 and as mentioned before, may relay signals that directly or indirectly affect endogenous SDF-1 expression in distal portions of the brain 52–55,57.
Probing Cellular Players in the Response to Exogenous SDF-1
We performed a series of immunohistological stains to begin elucidating key cellular players in the SDF-1/CXR4 signaling axis; several phenotypic markers were probed including astrocytes (glial fibrillary acidic protein; GFAP), neuronal committed progenitor cells/neuroblasts (doublecortin; Dcx), immature neural progenitors (nestin), and microglia/macrophages (ionized calcium-binding adapter molecule 1; Iba-1). Even though we probed for several cellular phenotypes, we did not detect significant colocalization of phenotypic markers with CXCR4+ cells, and therefore did not quantitate this data. Moreover, our staining revealed a complex temporal cellular response that ultimately warrants further characterization outside of the scope of this study. Nonetheless, we noted some key observation in this study that informs future studies. While we conducted immunostaining for all groups and time points, we chose to present and discuss representative images from one day for the majority of immunostains to highlight the key host response observations. First, we were interested in whether the injections elicited recruitment of NPSCs from the SVZ. We stained for Dcx to probe for the presence of committed neuronal progenitor/migrating neuroblasts. In all groups, evidence of Dcx+ staining was observed near the SVZ at day 1 following injection for all groups (Figure 8). These results suggest that the injury sustained from the injection itself may have impacted neuroblast migration; however, additional studies are warranted to compare to staining patterns to naïve animals. Recent studies reported robust Dcx+ cellular recruitment following AFSDF-1 bolus or NP injections 58,59. Yet, we observed very limited to no Dcx+ cells near the injection tract by day 7 across all groups (Figure 9). However, stark differences in our study compared to these two studies may have contributed to differing observations including administration of SDF-1 in conjunction with a traumatic brain injury model and the amount of SDF-1 delivered in the bolus injection was two orders of magnitude greater than used in our study 58,59. Additionally, stroke and brain injury literature report the presence of Dcx+ cells to a site of neural injury is nearly 2-4 weeks post injury 60,61.
Figure 8.
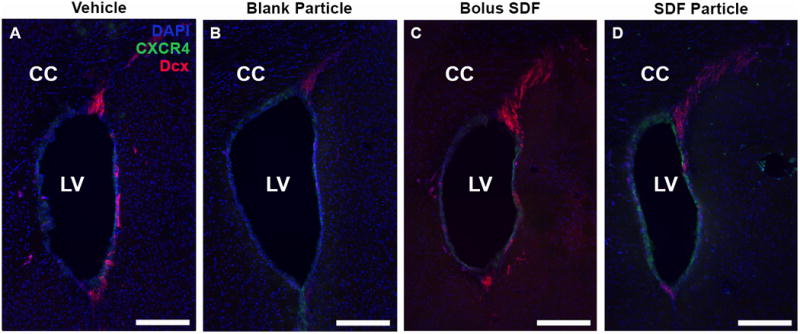
Neuroblast migration from SVZ. Representative day 1 images revealed prominent neuroblast migration in response to all injection conditions as visualized with Dcx staining (red) stemming from the SVZ. (A) Vehicle, (B) Blank NP, (C) SDF-1 bolus, and (D) SDF-1 NPs. Note the CXCR4+ cells (green) surrounding the lateral ventricles, but the migrating Dcx+ cells are not CXCR4+. LV = Lateral ventricle. Red = Dcx, Green = CXCR4, Blue = DAPI; Scale bar = 200 μm.
Figure 9.
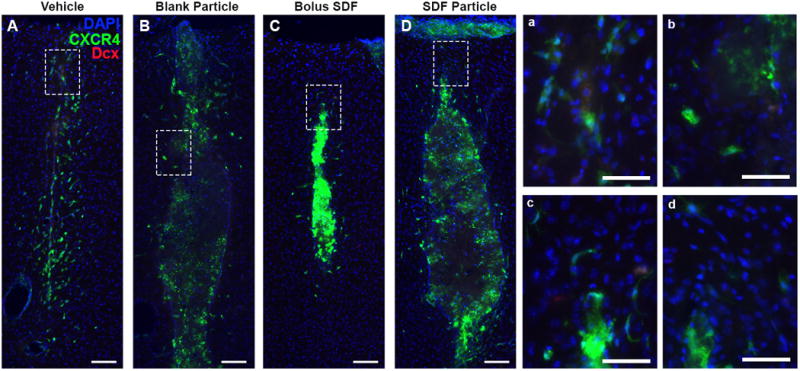
Minimal neuroblast/neuronal progenitor cells localized to injection. At day 7 post injection, little to no Dcx+ co-localization with CXCR4 was observed in response to any of the injection conditions, Vehicle (A, a), blank NPs (B, b), SDF-1 bolus (C, c), and SDF-1 NPs (D, d). Red = Dcx, Green = CXCR4, Blue = DAPI. (A-D) Scale bar = 100 μm; (a-d) scale bar = 20 μm.
Next, we aimed to evaluate the presence of neural progenitor cells through nestin staining; however, we acknowledge that nestin expression is not limited to neural progenitors involved in neurogenesis, but also astrogliogenesis 61–63. Focusing on the most prominent response at day 7, nestin+ cells appear prominently adjacent and within the injection tract for all groups (Figure 10). Again, no co-localization of nestin with CXCR4+ cells was observed across all groups (Figure 10). Subsequently, we probed for the astrocytic marker GFAP to compare with the nestin staining pattern. GFAP staining revealed a unique pattern over the 7-day time course for all groups. At day 1, GFAP+ stain was noted prominently in the cortical region and corpus callosum directly below the injection site stemming from the SVZ (Figure 11). By day 3 and 7, the GFAP+ signal expanded to include encompass the cortical region surrounding the injection (Figure 11). This series of GFAP staining pattern suggests that both local resident astrocytes as well as newly generated astrocytes originating from the SVZ are actively recruited predominately in response to the injury sustained by the injection regardless of injection group (Figures 11 and 12) 60,61,63. Moreover, though we cannot rule out potential co-expression of GFAP/nestin by a subset of cells, the GFAP+ staining at day 7 was prominently adjacent to the injection tract while nestin+ cells were also observed within the injection tract (Figures 10 and 12). These results collectively support the notion that cortical injury via the injection alone stimulates both robust astrogliogenesis and modest neurogenesis 60,62,63.
Figure 10.
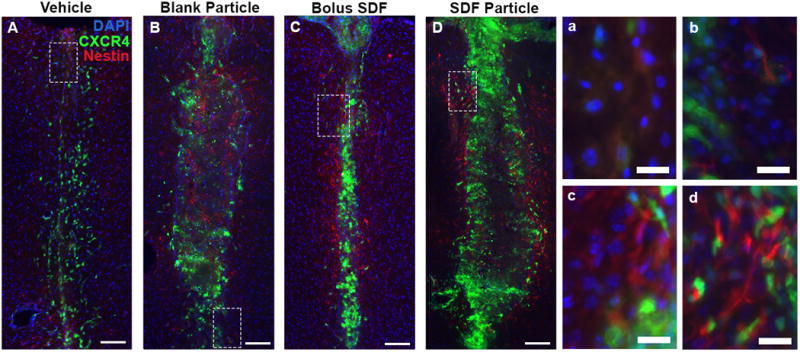
Prominent neural progenitor/glial cell presence 7 days post injection. Nestin+ staining (red) in and surrounding the injection site was observed for all groups at day 7 (A-D). Little to no co-localization of nestin with CXCR4+ cells (green) was noted at day 7 for any of the conditions, Vehicle (A, a), blank NPs (B, b), SDF-1 bolus (C, c), and AFSDF-1 NP (D, d). Red = nestin, Green = CXCR4, Blue = DAPI. (A-D) Scale bar = 100 μm; (a-d) scale bar = 20 μm.
Figure 11.
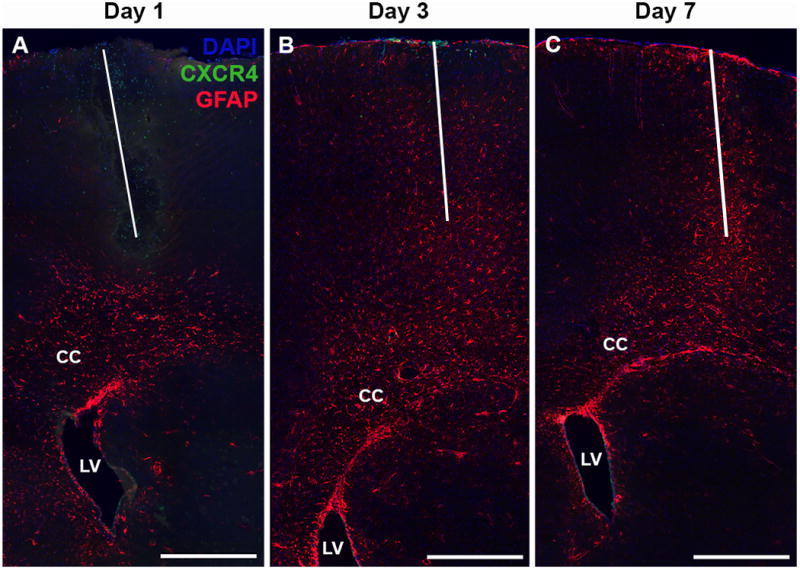
Recruitment and activation of astrocytes. Representative GFAP immunostain images from the vehicle injection group at days 1 (A), 3 (B), and 7 (C) illustrating the combination of local GFAP activation and cellular recruitment from the SVZ (through the corpus callosum). CC = Corpus callosum, LV = Lateral ventricle. Red = GFAP, Green = CXCR4, Blue = DAPI. Scale bar = 500 μm. White line designates injection track.
Figure 12.
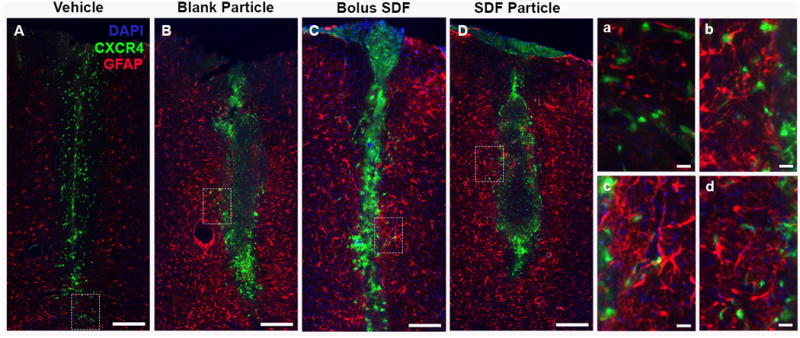
Pronounced astrocytic response following intracortical injections. (A-D) Representative day 7 images demonstrated pronounced GFAP response adjacent to the inject tract for all groups with little to no co-localization with CXCR4 (a-d). (A, a) Vehicle, (B, b) Blank NPs, (C, c) SDF-1bolus, and (D, d) SDF-1 NPs. Red = GFAP, Green = CXCR4, Blue = DAPI. (A-D) Scale bar = 200 μm, (a-d) scale bar = 20 μm.
Finally, we probe the inflammatory response with Iba-1 to evaluate the presence of activated microglial and systemic macrophages. Here, the most prominent observation was that injection initiated a prominent microglial/macrophage localized to the injection site by day 7 (Figure 13). Nuanced variations within the NP groups warrant further investigation into the contribution of NP inciting an inflammatory response versus the SDF-1. Previous reports have shown that PLGA microparticles implanted in the striatum are surrounded by activated microglia/macrophage as early as one day post-injection 64. Since activated microglia and infiltrating systemic macrophages employ the SDF-1/CXCR4 signaling cascade, the EGFP-CXCR4+ signal near the NP implants is a possible indicator of a systemic host response 65,66.
Figure 13.
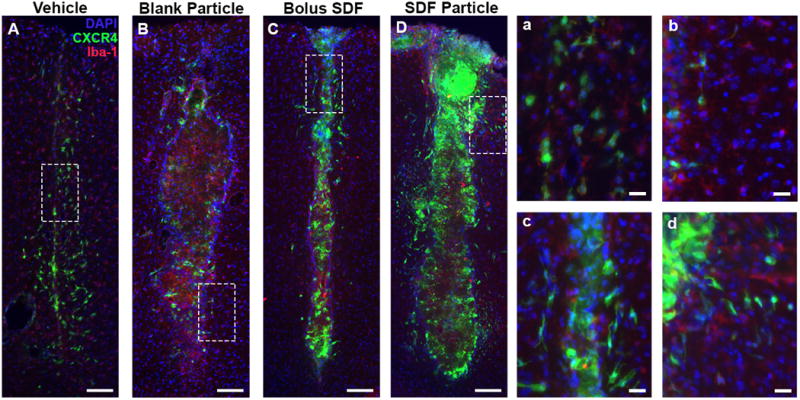
NPs incite localized inflammatory response by day 7. Representative images for (A) Vehicle, (B) Blank NPs, (C) SDF-1 bolus, and (D) SDF-1 NPs at day 7 immunostained with Iba-1 demonstrated the prominent and robust inflammatory response local to injections (B and D). Red = Iba-1, Green = CXCR4, Blue = DAPI. Scale bar = 200 μm.
Other CNS resident cell types such as, mature neurons, oligodendrocytes, endothelial cells of the BBB are not only CXCR4+, have been shown to regulate CXCR4 expression when exposed to SDF-1 19,66–69. Future studies will include probing these phenotypes to understand their contribution in this signaling paradigm. Moreover, companion in vitro studies will help determine how SDF-1, CXCR4 and potentially CXCR7 expression relates to sustained exposure to SDF-1. Additionally, if desensitization of SDF-1 plays an important role due to a sustained release, it remains to be seen whether other release profiles (delayed or pulsed) is a better fit for modulating the SDF-1/CXCR4 signaling axis in the week-long period tested here, and beyond.
Conclusion
Exogenous intracortical administration of soluble SDF-1 led to localized endogenous SDF-1 expression and subsequent activation of CXCR4 for three days post injection, attenuating completely by 7 days post injection. Conversely, sustained release of SDF-1 from PLGA NPs elicited a more prolonged and significant SDF-1 expression pattern that persisted for seven days post injection. This altered SDF-1 temporal presentation led to significant increases in CXCR4+ cell densities locally and in regions far more distal to the injection site for three days and adjacent to the injection for seven days. Immunohistochemical staining revealed a complex temporal response to cortical injections with most trends related the injection itself and not specific AFSDF-1 administration. Future studies will focus on exploring the role of vascular and neuronal contributions to the SDF-1/CXCR4 signaling cascade.
Supplementary Material
Acknowledgments
The authors acknowledge Amanda Witten for assistance with graphic design. This work was supported by NSF CBET 1454282 (SES) and NIH NICHD 1DP2HD084067 (SES).
Footnotes
Footnotes relating to the title and/or authors should appear here.
Conflicts of Interest: There are no conflicts of interest to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Addington CP, Roussas A, Dutta D, Stabenfeldt SE. Biomark Insights. 2015;10:43–60. doi: 10.4137/BMI.S20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christie K, Turnley A. Front Cell Neurosci. 2013;6 doi: 10.3389/fncel.2012.00070. Article 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh T, Satou T, Ishida H, Nishida S, Tsubaki M, Hashimoto S, Ito H. Neurol Res. 2009;31:90–102. doi: 10.1179/174313208X332995. [DOI] [PubMed] [Google Scholar]
- 4.Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD, Frenkel D, Li J, Sidman RL, Walsh CA, Snyder EY, Khoury SJ. Proc Natl Acad Sci U S A. 2004;101:18117–18122. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon KJ, Theus MH, Nelersa CM, Mier J, Travieso LG, Yu TS, Kernie SG, Liebl DJ. J Neurotrauma. 2015;32:753–764. doi: 10.1089/neu.2014.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun C, Sun H, Wu S, Lee CC, Akamatsu Y, Wang RK, Kernie SG, Liu J. J Neurosci. 2013;33:17314–17325. doi: 10.1523/JNEUROSCI.2129-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yi X, Jin G, Zhang X, Mao W, Li H, Qin J, Shi J, Dai K, Zhang F. PLoS ONE. 2013;8:e70306. doi: 10.1371/journal.pone.0070306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutta D, Fauer C, Mulleneux HL, Stabenfeldt SE. J Mater Chem B. 2015;3:7963–7973. doi: 10.1039/C5TB00935A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thored P, Arvidsson A, Cacci E, Ahlenius H, Kallur T, Darsalia V, Ekdahl CT, Kokaia Z, Lindvall O. Stem Cells Dayt Ohio. 2006;24:739–747. doi: 10.1634/stemcells.2005-0281. [DOI] [PubMed] [Google Scholar]
- 10.Sun D, Bullock MR, Altememi N, Zhou Z, Hagood S, Rolfe A, McGinn MJ, Hamm R, Colello RJ. J Neurotrauma. 2010;27:923–938. doi: 10.1089/neu.2009.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cooke MJ, Wang Y, Morshead CM, Shoichet MS. Biomaterials. 2011;32:5688–5697. doi: 10.1016/j.biomaterials.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 12.Huang F, Yin Z, Wu D, Hao J. NeuroReport. 2013;24:101–107. doi: 10.1097/WNR.0b013e32835c93c5. [DOI] [PubMed] [Google Scholar]
- 13.Cai Y, Xu M, Yuan M, Liu Z, Yuan W. Int J Nanomedicine. 2014;9:3527–3538. doi: 10.2147/IJN.S63507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lefèbvre PJ, Paolisso G, Scheen AJ, Henquin JC. Diabetologia. 1987;30:443–452. doi: 10.1007/BF00279610. [DOI] [PubMed] [Google Scholar]
- 15.Dutta D, Fauer C, Hickey K, Salifu M, Stabenfeldt SE. J Mater Chem B. doi: 10.1039/C7TB00138J. Advance Article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laursen T. Growth Horm IGF Res. 2004;14:16–44. doi: 10.1016/j.ghir.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Finch AR, Caunt CJ, Armstrong SP, McArdle CA. Am J Physiol - Cell Physiol. 2009;297:C591–C600. doi: 10.1152/ajpcell.00166.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olanow CW, Obeso JA. Ann Neurol. 2000;47:S167–176. discussion S176-178. [PubMed] [Google Scholar]
- 19.Abe P, Mueller W, Schutz D, MacKay F, Thelen M, Zhang P, Stumm R. Development. 2014;141:1857–1863. doi: 10.1242/dev.104224. [DOI] [PubMed] [Google Scholar]
- 20.Tran PB, Banisadr G, Ren D, Chenn A, Miller RJ. J Comp Neurol. 2007;500:1007–1034. doi: 10.1002/cne.21229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aprili D, Bandschapp O, Rochlitz C, Urwyler A, Ruppen W. Anesthesiology. 2009;111:1346–1355. doi: 10.1097/ALN.0b013e3181bfab9a. [DOI] [PubMed] [Google Scholar]
- 22.Dutta D, Salifu M, Sirianni RW, Stabenfeldt SE. J Biomed Mater Res A. 2016;104:688–696. doi: 10.1002/jbm.a.35608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu G, Mallery SR, Schwendeman SP. Nat Biotechnol. 2000;18:52–57. doi: 10.1038/71916. [DOI] [PubMed] [Google Scholar]
- 24.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. J Controlled Release. 2012;161:505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 25.Moghimi SM, Hunter AC, Murray JC. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 26.Bhattacharyya BJ, Banisadr G, Jung H, Ren D, Cronshaw DG, Zou Y, Miller RJ. J Neurosci. 2008;28:6720–6730. doi: 10.1523/JNEUROSCI.1677-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belmadani A, Jung H, Ren D, Miller RJ. Differ Res Biol Divers. 2009;77:395–411. doi: 10.1016/j.diff.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. Clin Cancer Res. 2011;17:2074–2080. doi: 10.1158/1078-0432.CCR-10-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun W, Liu J, Huan Y, Zhang C. Inflamm Res. 2014;63:287–297. doi: 10.1007/s00011-013-0699-8. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Wei M, Zhou Z, Wang B, Zhao X, Zhang J. Brain Res. 2012;1444:76–86. doi: 10.1016/j.brainres.2011.12.055. [DOI] [PubMed] [Google Scholar]
- 31.Overstreet DJ, Dutta D, Stabenfeldt SE, Vernon BL. J Polym Sci Part B Polym Phys. 2012;50:881–903. [Google Scholar]
- 32.Henderson PW, Singh SP, Krijgh DD, Yamamoto M, Rafii DC, Sung JJ, Rafii S, Rabbany SY, Spector JA. Wound Repair Regen. 2011;19:420–425. doi: 10.1111/j.1524-475X.2011.00687.x. [DOI] [PubMed] [Google Scholar]
- 33.Rabbany SY, Pastore J, Yamamoto M, Miller T, Rafii S, Aras R, Penn M. Cell Transplant. 2010;19:399–408. doi: 10.3727/096368909X481782. [DOI] [PubMed] [Google Scholar]
- 34.Fujio M, Yamamoto A, Ando Y, Shohara R, Kinoshita K, Kaneko T, Hibi H, Ueda M. Bone. 2011;49:693–700. doi: 10.1016/j.bone.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 35.Prokoph S, Chavakis E, Levental KR, Zieris A, Freudenberg U, Dimmeler S, Werner C. Biomaterials. 2012;33:4792–4800. doi: 10.1016/j.biomaterials.2012.03.039. [DOI] [PubMed] [Google Scholar]
- 36.Zamani M, Prabhakaran MP, Thian ES, Ramakrishna S. J Colloid Interface Sci. 2015;451:144–152. doi: 10.1016/j.jcis.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Cross DP, Wang C. Pharm Res. 2011;28:2477–2489. doi: 10.1007/s11095-011-0474-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh AK, Arya RK, Trivedi AK, Sanyal S, Baral R, Dormond O, Briscoe DM, Datta D. Cytokine Growth Factor Rev. 2013;24:41–49. doi: 10.1016/j.cytogfr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolak DJ, Thorne RG. Mol Pharm. 2013;10:1492–1504. doi: 10.1021/mp300495e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pencea V, Bingaman KD, Wiegand SJ, Luskin MB. J Neurosci. 2001;21:6706–6717. doi: 10.1523/JNEUROSCI.21-17-06706.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mufson EJ, Kroin JS, Liu YT, Sobreviela T, Penn RD, Miller JA, Kordower JH. Neuroscience. 1996;71:179–191. doi: 10.1016/0306-4522(95)00431-9. [DOI] [PubMed] [Google Scholar]
- 42.Yi X, Manickam DS, Brynskikh A, Kabanov AV. J Controlled Release. 2014;190:637–663. doi: 10.1016/j.jconrel.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kokovay E, Goderie S, Wang Y, Lotz S, Lin G, Sun Y, Roysam B, Shen Q, Temple S. Cell Stem Cell. 2010;7:163–173. doi: 10.1016/j.stem.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holt JE, Jackson A, Roman SD, Aitken RJ, Koopman P, McLaughlin EA. Dev Biol. 2006;293:449–460. doi: 10.1016/j.ydbio.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 45.Weiss JM, Cufi P, Bismuth J, Eymard B, Fadel E, Berrih-Aknin S, Le Panse R. Immunobiology. 2013;218:373–381. doi: 10.1016/j.imbio.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Misra P, Lebeche D, Ly H, Schwarzkopf M, Diaz G, Hajjar RJ, Schecter AD, Frangioni JV. J Nucl Med. 2008;49:963–969. doi: 10.2967/jnumed.107.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Duan B, Cheng Z, Jia X, Mao L, Fu H, Che Y, Ou L, Liu L, Kong D. Protein Cell. 2011;2:845–854. doi: 10.1007/s13238-011-1097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCandless EE, Budde M, Lees JR, Dorsey D, Lyng E, Klein RS. J Immunol Baltim Md 1950. 2009;183:613–620. doi: 10.4049/jimmunol.0802258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang Z, Brooks J, Willard M, Liang K, Yoon Y, Kang S, Shim H. Biochem Biophys Res Commun. 2007;359:716–722. doi: 10.1016/j.bbrc.2007.05.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong WX, Hu MS, Esquivel M, Liang GY, Rennert RC, McArdle A, Paik KJ, Duscher D, Gurtner GC, Lorenz HP, Longaker MT. Adv Wound Care. 2014;3:390–399. doi: 10.1089/wound.2013.0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Arthritis Rheum. 2002;46:2587–2597. doi: 10.1002/art.10520. [DOI] [PubMed] [Google Scholar]
- 52.Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Blood. 2002;99:2703–11. doi: 10.1182/blood.v99.8.2703. [DOI] [PubMed] [Google Scholar]
- 53.Salcedo R, Wasserman K, Young HA, Grimm MC, Howard OMZ, Anver MR, Kleinman HK, Murphy WJ, Oppenheim JJ. Am J Pathol. 1999;154:1125–35. doi: 10.1016/s0002-9440(10)65365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salcedo R, Oppenheim JJ. Microcirculation. 2010;10:359–370. doi: 10.1038/sj.mn.7800200. [DOI] [PubMed] [Google Scholar]
- 55.Gupta SK, Lysko PG, Pillarisetti K, Ohlstein E, Stadel JM. J Biol Chem. 1998;273:4282–4287. doi: 10.1074/jbc.273.7.4282. [DOI] [PubMed] [Google Scholar]
- 56.Chodobski A, Zink BJ, Szmydynger-Chodobska J. Transl Stroke Res. 2011;2:492–516. doi: 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu KKY, Dorovini-Zis K. J Neuroimmunol. 2009;215:49–64. doi: 10.1016/j.jneuroim.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 58.Zamproni LN, Mundim MV, Porcionatto MA, des Rieux A. Int J Pharm. 2017;519:323–331. doi: 10.1016/j.ijpharm.2017.01.036. [DOI] [PubMed] [Google Scholar]
- 59.Mao W, Yi X, Qin J, Tian M, Jin G. Neurochem Res. 2016;41:1315–1322. doi: 10.1007/s11064-016-1831-2. [DOI] [PubMed] [Google Scholar]
- 60.Li L, Harms KM, Ventura PB, Lagace DC, Eisch AJ, Cunningham LA. Glia. 2010;58:1610–1619. doi: 10.1002/glia.21033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sohn J, Orosco L, Guo F, Chung SH, Bannerman P, Ko EM, Zarbalis K, Deng W, Pleasure D. J Neurosci. 2015;35:3756–3763. doi: 10.1523/JNEUROSCI.3454-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Benner EJ, Luciano D, Jo R, Abdi K, Paez-Gonzalez P, Sheng H, Warner DS, Liu C, Eroglu C, Kuo CT. Nature. 2013;497:369–373. doi: 10.1038/nature12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Menei P, Daniel V, Montero-Menei C, Brouillard M, Pouplard-Barthelaix A, Benoit JP. Biomaterials. 1993;14:470–478. doi: 10.1016/0142-9612(93)90151-q. [DOI] [PubMed] [Google Scholar]
- 65.Sharp CD, Glawe JD, Huang M, Barlow SC, Kevil CG. FASEB J. 2006;20:A202–A202. [Google Scholar]
- 66.Stumm RK, Rummel J, Junker V, Culmsee C, Pfeiffer M, Krieglstein J, Höllt V, Schulz S. J Neurosci. 2002;22:5865–5878. doi: 10.1523/JNEUROSCI.22-14-05865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonavia R, Bajetto A, Barbero S, Pirani P, Florio T, Schettini G. Toxicol Lett. 2003;139:181–189. doi: 10.1016/s0378-4274(02)00432-0. [DOI] [PubMed] [Google Scholar]
- 68.Würth R, Bajetto A, Harrison JK, Barbieri F, Florio T. Front Cell Neurosci. doi: 10.3389/fncel.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ji JF, He BP, Dheen ST, Tay SSW. Neurosci Lett. 2004;355:236–240. doi: 10.1016/j.neulet.2003.11.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.